

Abstract
Intramolecular interactions between the amino and carboxy termini of apolipoprotein A-I (apoAI) are believed to stabilize the helix bundle conformation of the protein. During lipid assembly the protein undergoes conformational changes that result in an exposure of the carboxy terminus and its insertion into the lipid phase. To determine the role of the two termini in the energetics of unfolding, we studied the guanidine-hydrochloride-induced unfolding and refolding of apoAI as well as its N-terminal deletion (del[1–43]), C-terminal deletion (del[186–243]), and the double deletion containing only the central residues 44–185. Thermodynamic analysis of the equilibrium unfolding measured by fluorescence spectroscopy revealed the presence of an intermediate unfolded state (Iequil) in addition to the native (N) and unfolded states. Refolding kinetics of apoAI, measured by stopped-flow circular dichroism, revealed two kinetic intermediates, Iburst and Irecovery. Computer modeling suggested that the first resembles the partially unfolded protein, whereas the second overlaps with the native state of the protein. The free energy changes for the N → Iequil transition of the N-terminal and double deletions were lower then that of the full-length form, whereas that for the C-terminal deletion was higher. Our findings suggest that the N-terminus of apoAI stabilizes the native state of the protein by increasing the Eyring energy barrier for the N → Iequil unfolding transition; whereas the carboxyl terminus destabilizes that state.
INTRODUCTION
The 243-amino-acid apolipoprotein A-I (apoAI) is the main protein component in high-density lipoprotein (HDL) and thus plays an important role in reverse cholesterol transport and reduction in risk for cardiovascular and atherosclerotic diseases (1). HDL is the main mediator of the “reverse cholesterol transport” pathway by acting as an acceptor of cellular cholesterol and transporting this cholesterol to the liver where it can be excreted directly or indirectly after conversion to bile acids (2). Lipid-free apoAI can also act as an acceptor of cellular cholesterol in a process that is mediated by the membrane protein ABCA1 (3,4). The primary sequence of apoAI from residues 44–243 can be divided into 22- and 11-residue tandem repeats that form type A amphipathic α-helices (5). Although the crystal structure of the entire lipid-free apoAI has not been determined, the C-terminal α-helices appear to have a less organized structure than the rest of the protein (6,7). Secondary-structure predictions suggest that the amino-terminal 43 amino acids form a G* class amphipathic α-helix (8). Deletion of these 43 amino acids (N-terminal deletion, or del[1–43]) allowed crystals of the lipid-free protein to be obtained, and the x-ray structure of this protein has been used as a model for the conformation that resembles the one conferred on the intact protein upon binding to lipid in its lipid-bound state (6). This result is consistent with the finding by Borhani et al. (6), who reported, using x-ray crystallography, that the del[1–43] mutant of apoAI forms an extended α-helix horseshoe-shaped belt, with the N- and C-termini at adjacent ends of this horseshoe, and a tetramer of these stacked in an offset antiparallel manner to a size consistent with apoAI lipid disks. This view is also supported by the finding that in recombinant discoidal HDL, apoAI forms an extended series of α-helices that circumscribe the perimeter of the disk-shaped phospholipid bilayer such that the α-helix segments align perpendicular to the fatty acid chains of the phospholipids (9–11). The amino-terminal deletion has lipid binding and ABCA1-dependent cholesterol acceptor activity equivalent to that of full-length apoAI (12,13). 1-Anilinonaphthalene-8-sulfonate binding experiments and circular dichroism spectroscopy have demonstrated that an extended portion of the amino terminus (amino acids 1–90) is critical for maintaining and stabilizing the folded, lipid-free structure of apoAI (14). In contrast, carboxy terminal deletions ([1–192]apoA1) exhibit severely impaired lower lipid binding and ABCA1-dependent cholesterol acceptor activity (13,15). Interestingly, double deletion of both the N- and C-termini of apoAI leads to a restoration of these apoAI activities, showing that the central domain is sufficient for these functions of apoAI (13,15).
Davidson and colleagues have recently proposed a detailed folded helical bundle model of the lipid-free form of apoAI, with the hydrophobic faces binding adjacent surface and the N- and C-termini in close proximity with each other (11,16). Lipid binding is thought to first occur in the C-terminus, which undergoes a transition from an unordered to an α-helical conformation and acts as the trigger to allow exposure of the remainder of apoAI hydrophobic domains to the lipid phase (7). The idea that the carboxy terminus of the protein is associated with or embedded within the lipid phase is further supported by the finding that, when bound to lipid, the carboxy terminus of human apoAI was less susceptible to proteolytic cleavage than the amino terminus (17,18). An earlier study indicated that lipid binding of apoAI proceeds via a molten globule (MG) state (19). The MG state is a partially folded, nonnative-state conformation of the protein that plays a key role in a variety of physiological functions such as lipid-binding or penetration of the protein into the lipid bilayer in a variety of globular proteins (20). The MG state appears to be intermediate between the native and denatured states, is relatively compact, and is characterized by a well-preserved secondary structure and a significant reduction in tertiary structure (21,22).
In this study, we used circular dichroism (CD) spectroscopy in combination with stopped-flow techniques to demonstrate that lipid-free apoAI forms at least two kinetic folding intermediate states (Iburst and Irecovery). In another set of experiments, using fluorescence spectroscopy under equilibrium conditions, we also found a denatured state of the protein that is stable in 3 M GdnHCl. This unfolded intermediate state, which we refer to as an equilibrium intermediate state (Iequil), has a CD spectrum that closely resembles the denatured state of the protein and a fluorescence spectrum that is distinct from either that of the native protein state (N, observed at 0 M GdnHCl) or the unfolded state (U, observed at 5 M GdnHCl). Deletion of the amino terminus of the protein stabilizes Iequil by lowering the Eyring energy barrier for the N → Iequil transition. Deletion of the carboxy terminus, on the other hand, stabilizes the N state of the protein by elevating that barrier. These findings have implications for lipid binding.
MATERIALS AND METHODS
ApoA1 mutant constructs
cDNA encoding 6-his-tagged recombinant human apoAI (rh-apoAI) in the pET-20b bacterial expression vector was a gift from Dr. Michael Oda (23). Deletions and stop codon point mutations were made by polymerase chain reaction and use of the Quick-change Mutagenesis Kit (Stratagene, La Jolla, CA) to create del[1–43], the C-terminal deletion (del[186–243]), and the N- and C-terminal double deletion, containing only the central residues 44–185. All mutations were confirmed by DNA sequencing. The plasmids were transformed into Escherichia coli strain BL21(DE-3) pLysS, and apoAI expression was induced with 0.5 mM isopropyl-β-D-thiogalactopyranoside by overnight incubation at room temperature. The cell pellet was dissolved in B-PER lysis buffer (BioRad, Hercules, CA) followed by centrifugation to sediment cell debris. The supernatant was diluted into phosphate-buffered saline containing 3 M GdnHCl, pH 7.0, and then loaded onto a nickel-chelating histidine-binding resin column and specifically eluted with imidazole as previously described (23). Recombinant apoAI was dialyzed extensively in 100 mM sodium phosphate buffer, pH 7.0, containing 100 μM DTPA. ApoAI samples were analyzed by electrophoresis on a 14% sodium dodecyl sulfate polyacrylamide gel under reducing conditions. The gels were stained for protein with 0.25% Coomassie blue. All samples were >95% pure by this method (data not shown). All constructs in this study contained the same amino-terminal 6-his tag. Pilot studies with purified native human apoAI revealed CD and fluorescence spectra as well as folding kinetics that were very similar to the 6-his-tagged recombinant human apoAI used in this study (data not shown).
Equilibrium spectra
Steady-state fluorescence spectra were measured with a Perkin-Elmer (Wellesley, MA) LS-50B spectrofluoremeter. Samples of 25 μg/ml apoAI were prepared in PBS (10 mM phosphate buffer, pH 7.4, 150 mM NaCl) and the indicated concentration of GdnHCl. Steady-state CD spectra were acquired with an Applied Photophysics (Leatherhead, Surrey, UK) piStar180 spectrophotometer equipped with a thermostated equilibrium sample handling unit.
Free energy of GdnHCl-induced unfolding (ΔGNU) was calculated by three independent methods as follows:
Method I
By fitting the equilibrium CD data (see Fig. 2 b) to the relationship ΔGNU = −RTlnKD, where the equilibrium constant KD is calculated from the mean residual ellipticity (MRE) using the relation:
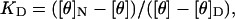 |
(1) |
where [θ] is the observed MRE at a given concentration of GdnHCl and [θ]N and [θ]D are the MREs for the native and fully denatured forms of the protein.
FIGURE 2.

(a) CD spectra of full-length apoAI in the far-UV region. Shown are spectra of the protein in the native (N, solid line, 0 M GdnHCl), unfolded (U, dotted line, 5 M GdnHCl), and intermediate (I, dashed line) forms calculated with Eq. 15, as explained in the text. Open squares represent the CD spectrum of the burst phase intermediate and the solid squares represent the spectrum of the recovery phase intermediate, as explained in the text. (b) MRE at 222 nm as a function of concentration of GdnHCl, measured at equilibrium, for the following forms of apoAI (corresponding EC50 values are also indicated): full-length (squares, 1.17 M), del[1–43] (triangles, 0.77 M), del [186–243] (circles, 1.19 M), and [44–185] (diamonds, 1.11 M).
Method II
By fitting the equilibrium wavelength of maximum fluorescence (WMF) data (see Fig. 3 b) to the relationship ΔGNU = −RTlnKD, where the equilibrium constant KD is calculated from the WMF (peak fluorescence emission for 295-nm excitation) using the relation
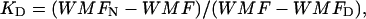 |
(2) |
where WMF is the observed WMF at a given concentration of GdnHCl and WMFN and WMFD are the WMFs for the native and fully denatured forms of the protein.
FIGURE 3.

(a) Fluorescence spectra of full-length apoAI at 0 and 5 M GdnHCl, as indicated on the graphs. λexc = 295 nm. (b) WMF and EC50 values as a function of GdnHCl concentration measured at equilibrium for full-length apoAI (squares, 1.38 M), del[1–43] (triangles, 0.88 M), del[186–243] (circles, 1.51 M), and [44-185] apoAI (diamonds, 1.29 M). λexc = 295 nm. (c) Fluorescence intensity at the peak of emission spectrum as a function of GdnHCl concentration measured at equilibrium for full-length apoAI (squares), del[1–43] apoAI (triangles), del[186–243] apoAI (circles), and [44–185] apoAI (diamonds). Symbols represent the experimental data and lines represent the theoretical curves predicted by Eq. 13. λexc = 295 nm.
Method III
By fitting Eq. 13 (see Results section) to the fluorescence peak intensity data (as depicted in Fig. 3 c).
Kinetics of refolding/unfolding
All fast kinetics studies were carried out with an Applied Photophysics piStar180 spectrophotometer equipped with an xv18 stopped-flow mixing chamber. The observed kinetic curves were fitted by the nonlinear least-squares method with the multiexponential relation:
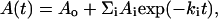 |
(3) |
where A(t) and Ao are the signal values at time t and at infinite time, respectively; and Ai and ki are the amplitude and the rate constant of phase i.
Computer modeling
Free energy changes, m values, and the molar fractions of the three protein folding states—native, unfolded, and intermediate—were calculated by globally fitting all equilibrium peak fluorescence data (see Fig. 3 c) to Eq. 13 (see Results section) using the SCoP modeling package (24,25).
Reagents
Ultrapure GdnHCl was from USB Chemicals (Cleveland, OH). All other salts were from Sigma-Aldrich (St. Louis, MO).
RESULTS
In the first set of experiments, we used CD to measure the kinetics of the change in MRE at 222 nm ([θ]222) upon refolding of apoAI initiated by a GdnHCl concentration jump from 5 M to 0.45M using a stopped-flow apparatus. Fig. 1 a illustrates the outcome of such an experiment performed with full-length apoAI. As can be seen, [θ]222 rapidly decreases (α-helix content increases) with an overshoot before relaxing toward the value characteristic of the native protein. We thus labeled the fast, or burst, phase intermediate Iburst, and the recovery phase intermediate Irecovery. The kinetics of signal recovery upon refolding as shown in Fig. 1 a was best fitted by a double exponential function (see Table 1). Table 1 also lists the corresponding values for the three deleted forms of apoAI. Comparing the rate constants for the N → Iburst transition of protein refolding in Table 1 reveals the following order of folding kinetics (starting from the fastest): del[186–243] > apoAI ∼ apoAI[44–185] > del[1–43]. Accordingly, the Eyring free energy barrier for the Iburst → N transition is largest for the del[1–43] mutant and smallest for the del[186–243] mutant. We also studied the unfolding of apoAI and its deleted forms. This was done by measuring θ222 immediately after a GdnHCl concentration jump from 0 to 4.55 M. In contrast to the refolding experiments, the kinetic traces of the unfolding process for all four proteins could be fitted with a single exponential function (Table 1). Furthermore, the kinetic traces did not exhibit an overshoot (Fig. 1 b).
FIGURE 1.

(a) Kinetic refolding curves for full-length apoAI measured by far-UV CD at 222 nm and converted to MRE units. Refolding reaction was initiated by a GdnHCl concentration jump from 5 M to 0.45 M. The experimental data (dots) were fitted with Eq. 3 (solid line). The inset shows the difference between the theoretical curve and the experimental data. The single data point represented by a solid square at the top left corner of the graph is the MRE value at 222 nm of the native protein. (b) Kinetic unfolding curves for full-length apoAI measured by far-UV CD at 222 nm and converted to MRE units. Unfolding reaction was initiated by a GdnHCl concentration jump from 0 M to 4.55 M. The experimental data (dots) were fitted with Eq. 3 (solid line). The inset shows the difference between the theoretical curve and the experimental data. The single data point represented by a solid triangle at the bottom left corner of the graph is the MRE value at 222 nm of the unfolded protein.
TABLE 1.
Rate constants (s−1) for refolding and unfolding kinetics of apoAI
| [GdnHCl]*final | apoAI
|
del[1–43] apoAI
|
del[186–243] apoAI
|
[44–185] apoAI
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refold
|
Refold
|
Refold
|
Refold
|
|||||||||
| Refold/unfold | Fast phase | Recovery phase | Unfold | Fast phase | Recovery phase | Unfold | Fast phase | Recovery phase | Unfold | Fast phase | Recovery phase | Unfold |
| 0.45/1.2 | 3.01 ± 0.3 | 0.59 ± 0.05 | 0.22 ± 0.02 | 1.21 ± 0.1 | 0.41 ± 0.04 | 0.48 ± 0.04 | 4.82 ± 0.4 | 0.47 ± 0.04 | 0.13 ± 0.01 | 2.31 ± 0.2 | 0.49 ± 0.04 | 0.32 ± 0.03 |
| 0.60/1.6 | 2.86 ± 0.2 | 0.46 ± 0.04 | 0.26 ± 0.02 | 1.14 ± 0.1 | 0.35 ± 0.03 | 0.59 ± 0.05 | 4.51 ± 0.4 | 0.38 ± 0.04 | 0.15 ± 0.01 | 2.26 ± 0.2 | 0.38 ± 0.04 | 0.41 ± 0.04 |
| 0.70/2.0 | 2.72 ± 0.2 | 0.38 ± 0.03 | 0.37 ± 0.03 | 0.98 ± 0.1 | 0.28 ± 0.02 | 0.68 ± 0.05 | 4.13 ± 0.4 | 0.31 ± 0.03 | 0.18 ± 0.02 | 2.11 ± 0.2 | 0.31 ± 0.03 | 0.53 ± 0.05 |
| 0.80/2.5 | 2.12 ± 0.2 | 0.28 ± 0.02 | 0.50 ± 0.04 | 0.69 ± 0.06 | 0.21 ± 0.02 | 0.76 ± 0.07 | 3.27 ± 0.3 | 0.22 ± 0.02 | 0.21 ± 0.02 | 1.51 ± 0.1 | 0.21 ± 0.02 | 0.65 ± 0.06 |
| 1.00/3.0 | 1.02 ± 0.1 | 0.23 ± 0.02 | 0.67 ± 0.05 | 0.38 ± 0.03 | 0.15 ± 0.01 | 0.85 ± 0.08 | 3.02 ± 0.3 | 0.15 ± 0.01 | 0.25 ± 0.02 | 0.65 ± 0.06 | 0.13 ± 0.01 | 0.78 ± 0.07 |
The refolding reaction initiated by a GdnHCl concentration jump from 5 M to the final value is indicated to the left of the slash (/), and the unfolding reaction initiated by a GdnHCl concentration jump from 0 M to the final value is indicated to the right of the slash.
The overshoot in the CD signal at 222 nm observed in the refolding experiments suggests the presence of a folding intermediate. To gain more insight into the properties of this intermediate and whether it can also be obtained at equilibrium, we performed a series of measurements under equilibrium conditions. At equilibrium, the secondary structure of apoAI is rich in α-helix. Fig. 2 a depicts the far-ultraviolet (UV) CD spectra of native apoAI with a large negative ellipticity between 210 and 230 nm indicative of the presence of α-helix. Most of the α-helix structure is lost in the unfolded state achieved in 5 M GdnHCl. The dose-dependent loss of molar ellipticity at 222 nm ([θ222]), indicative of α-helix content, with increasing GdnHCl concentration is shown in Fig. 2 b, along with the corresponding EC50 values, for full-length apoAI as well as for the N-terminal, C-terminal, and double N- and C-terminal deleted forms. The smaller EC50 of del[1–43] compared to the other three mutants suggests a lower energy barrier for the unfolding transition of the N-terminal deleted mutant. This is consistent with a role for the amino terminal in stabilizing the folded, N, state of the protein.
The fluorescence emission spectra of apoAI is shown in Fig. 3 a. In the absence of denaturant the spectrum exhibits a peak at around 340 nm. At 5 M GdnHCl, the emission peak shifts to the red. This shift in the WMF of full-length apoAI as well as of the three deleted forms, with increasing GdnHCl concentration is shown in Fig. 3 b along with the corresponding EC50 values. Comparing the EC50 values of all four mutants, the amino-terminal deletion exhibits the smallest value of all four with EC50 of 0.88 M. This is consistent with a lower energy barrier of the unfolding transition for the amino-terminal deletion as compared to the other mutants and further supports the notion that this segment of the peptide stabilizes the folded native state of the protein. It should also be noted that the EC50 values for the WMF changes are slightly larger then the EC50 values observed for the ellipticity changes as a function of GdnHCl concentration. This result suggests that conformational changes to the secondary structure of the protein precede changes in the tertiary structure. Fig. 3 c shows the fluorescence emission intensity of these apoAI forms at the peak of their respective spectra. It should be noticed that apoAI has four Trp residues, whereas the other three constructs have only three Trp residues each, and that it therefore would have a lower fluorescence quantum yield. To account for this difference in fluorescence quantum yield we normalized, for each of the four constructs, the peak fluorescence intensity at each GdnHCl concentration to the intensity at zero GdnHCl. Interestingly, all four forms of apoAI exhibit a biphasic profile with a decreasing normalized peak fluorescence intensity between 0 and ∼2 M GdnHCl that then increases between ∼3 and 5 M GdnHCl (Fig. 3 c). These biphasic behaviors cannot be described by a simple two-state unfolding transition, but can be fitted with a three-state transition scheme as described by Eq. 4 below. This scheme suggests the presence of a folding intermediate that exists in the transition range of GdnHCl concentration.
To gain more insight into the thermodynamics of the equilibrium intermediate state, depicted in Fig. 3 c as a nadir in the peak fluorescence curves, we used a mathematical model that describes the folding processes of apoAI as an equilibrium transition reaction between the native and unfolded states and an intermediate state (26–29). In this model we propose a three-state folding scheme as described below.
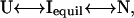 |
(4) |
where N and U are the native and unfolded states and Iequil is the equilibrium unfolding intermediate. The equilibrium constant between N and I is given by
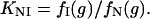 |
(5) |
Similarly, the equilibrium constant between N and U is given by
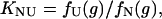 |
(6) |
where fN(g), fI(g) and fU(g) are the fractions of the three states at a denaturant (GdnHCl) concentration of g (fN + fI + fU = 1).
The measured fluorescence intensity of the protein at a given wavelength is a superposition of the contribution from all three states:
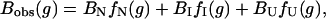 |
(7) |
where Bi represents the fluorescence intensity of the ith state. The concentration of each state is related to the equilibrium constants by the following relations (28,29):
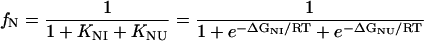 |
(8) |
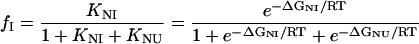 |
(9) |
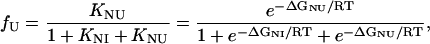 |
(10) |
where ΔGNI and ΔGNU are the free energy changes for the transition from the N to the Iequil state and from the N to the U state, respectively, and R and T have their usual meanings. We further assume that the free energy of unfolding varies linearly with g, so that
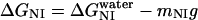 |
(11) |
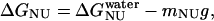 |
(12) |
where 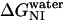 and
and 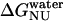 are the free energy changes,
are the free energy changes,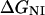 and
and 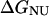 , at 0 M GdnHCl, respectively. mNI and mNU are the cooperativity indexes of the transitions. Combining Eqs. 4–12 and assuming the coefficients Bi are linearly dependent on g, we get (28)
, at 0 M GdnHCl, respectively. mNI and mNU are the cooperativity indexes of the transitions. Combining Eqs. 4–12 and assuming the coefficients Bi are linearly dependent on g, we get (28)
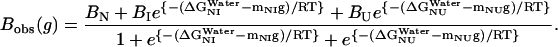 |
(13) |
The data in Fig. 3 c were analyzed with Eq. 13 using global analysis and were fitted simultaneously. The results are shown in Table 2. Examination of the data depicted in Table 2 reveals that del[186–243] exhibits the largest free energy change for the N → U transition with 5.03 kcal/mol followed by full length apoAI (4.50 kcal/mol); apoAI [44–185] (3.47 kcal/mol), and del[1–43] (2.4 kcal/mol).
TABLE 2.
Thermodynamic parameters for equilibrium unfolding of apoAI mutants
| Wild-type apoAI
|
del[1–43] apoAI
|
del[186–243] apoAI
|
[44–185] apoAI
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | I | II | III | I | II | III | I | II | III | I | II | III |
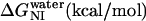 |
2.4 ± 0.2 | 1.6 ± 0.1 | 3.5 ± 0.3 | 1.8 ± 0.1 | ||||||||
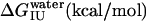 |
1.9 ± 0.2 | 1.0 ± 0.1 | 1.8 ± 0.1 | 2.0 ± 0.2 | ||||||||
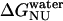 |
4.5 ± 0.4 | 4.7 ± 0.4 | 4.3 ± 0.4 | 2.4 ± 0.2 | 2.2 ± 0.2 | 2.6 ± 0.2 | 5.1 ± 0.4 | 4.7 ± 0.4 | 5.3 ± 0.4 | 3.2 ± 0.3 | 3.4 ± 0.3 | 3.8 ± 0.3 |
| mNI (kcal/mol) | 2.1 ± 0.2 | 1.2 ± 0.1 | 3.1 ± 0.3 | 1.9 ± 0.2 | ||||||||
| mIU (kcal/mol) | 1.4 ± 0.1 | 0.8 ± 0.1 | 2.1 ± 0.2 | 1.2 ± 0.1 | ||||||||
| mNU (kcal/mol) | 3.6 ± 0.3 | 3.9 ± 0.3 | 3.5 ± 0.3 | 1.9 ± 0.2 | 1.8 ± 0.2 | 2.0 ± 0.2 | 5.1 ± 0.5 | 4.7 ± 0.5 | 5.2 ± 0.5 | 2.8 ± 0.2 | 2.6 ± 0.2 | 3.1 ± 0.3 |
Chevron plots
To further evaluate the kinetic scheme of folding of apoAI and its deleted forms, we carried out a series of GdnHCl concentration jump experiments using the stopped-flow system. In these experiments, we monitored the kinetics of the protein's CD signal change at 222 nm after concentration jumps from high to various final low concentrations of GdnHCl. In addition, details of the unfolding kinetics were acquired after concentration jumps from 0 M to final concentrations in the range of 1–4 M GdnHCl. The kinetic curves from the unfolding experiments were fitted to a single exponential from which the rate constants for the unfolding kinetics were calculated. The refolding kinetic traces were fitted to a two-exponential function from which the rate constants for the fast and recovery kinetic phases were calculated. The logarithm of the rate constants (lnKobs) for the fast refolding phase measured at 222 nm were plotted as a function of the final GdnHCl concentration in the mixing chamber (Fig. 4). On the same graph we also plotted the logarithm of the rate constants for the unfolding kinetic traces. Such plots are referred to as “chevron plots” (30–32). The unfolding branch of the chevron plots did not show any detectable curvature. On the other hand, nonlinearities were observed for the refolding part of the plots (<0.5 M GdnHCl). This curvature could be due to the presence of an early folding intermediate that forms during the dead time of the instrument and becomes increasingly populated under stabilizing conditions (i.e., at low GdnHCl concentration).
FIGURE 4.

Chevron plots showing GdnHCl concentration dependence of the observed rate constants for the refolding and unfolding reactions of apoAI measured by CD at 222 nm: full-length apoAI (squares); del[1–43] apoAI (triangles); del[186–243] apoAI (circles); and [44–185] apoAI (diamonds).
CD spectra of the I state
To gain more insight into the spectral properties of the intermediate states of apoAI, we tried to reconstruct the CD spectra of these states. To achieve this task we used two approaches. 1), In the first approach, we measured the kinetics of apoAI refolding using stopped-flow techniques at different wavelengths after a decrease in GdnHCl concentration from 5 to 0.45 M as depicted in Fig. 1 a. The spectrum of the intermediate was reconstructed by extrapolating the CD signal, measured at each wavelength, to time zero. The spectrum obtained by this method represents the burst phase intermediate (Iburst) and is shown in Fig. 2 a (open squares). 2), In the second approach, we calculated the CD spectra of the intermediate state, as described below. The observed molar ellipticity, [θ]obs, at each wavelength is a superposition of the molar ellipticities of the N, I, and U states (28), i.e.,
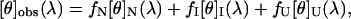 |
(14) |
where fI represents the molar fraction of the j state. fj was calculated using Eqs. 8–10. The CD spectrum of the I state is thus given by
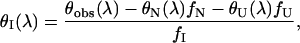 |
(15) |
where θN(λ) and θU(λ) are the experimentally measured ellipticities of apoAI at 0 and 5 M GdnHCl, respectively. The CD spectrum obtained this way for full-length apoA1 is shown in Fig. 2 a (theory). As can be seen, the spectrum of Itheory is very close to the spectrum of Iburst, suggesting that the latter represents a bona fide intermediate state.
Kinetic difference spectra
The exponential fit of the refolding kinetics curves measured at 10 different wavelengths provides kinetic difference CD spectra for the I → N transition phase (28). The difference spectra are related to fractional changes in secondary and/or tertiary structure components, which occur during the different kinetic phases. The difference spectra for this transition are given by the wavelength dependence of the amplitudes of the fast and recovery exponential functions (see Fig. 1 a). In Fig. 5, we show the kinetic difference spectra calculated from the exponential fits of the refolding curves measured at 10 different wavelengths. The spectra are for the I → N transition of apoAI.
FIGURE 5.

Kinetic difference spectrum of the refolding of full-length apoAI from I to N. Data were calculated from kinetic CD spectra acquired at 10 different wavelengths, as indicated, during refolding experiments of apoAI upon GdnHCl concentration jumps from 5 M to 0.45 M. The spectra were calculated as the difference in the preexponential amplitudes of the recovery phase and the slow decay phase as illustrated in Fig. 1 A.
DISCUSSION
Protein folding is a fundamental process in biology that is not yet fully understood. Nevertheless, it has been generally accepted that detection and characterization of intermediate conformational states between the native and fully unfolded states are useful for elucidating the mechanism of folding of globular proteins.
In the past, apoAI unfolding transitions were studied by steady-state CD spectroscopy and spectral shifts in the wavelength of maximum fluorescence (33). Unfortunately, equilibrium CD spectra and the WMF cannot capture short-lived intermediate states of the protein. The CD spectrum reflects changes in the secondary structure (i.e., α-helix), whereas the WMF reflects only the dielectric constant (electrical polarity) of the fluorophore microenvironment. Indeed, in our hand, the ellipticity parameter has already reached and completed its full range of change within several hundred milliseconds. The fluorescence intensity (which is proportional to the fluorescence quantum yield and thus the transition dipole moment) is a more complicated function that also includes large geometrical coefficients and short distances between Trp residues and other groups that can act as fluorescence quenchers. These parameters reflect the tertiary, as opposed to the secondary, structure of the protein. The secondary structure goes through a two-state transition upon folding (as illustrated in Figs. 2 b and 3 b), whereas the tertiary structure does not (Fig. 3 c).
The data obtained in our refolding kinetics experiments (Fig. 1 a) suggest that there are at least two intermediate states of apoAI, the burst phase intermediate (Iburst) and the slower intermediate formed by the recovery phase (Irecovery). The intermediate formed by the first phase exhibits an overshoot in the time-dependent ellipticity change at 222 nm. To determine whether the formation of the nonnative α-helix, represented by the overshoot in the CD ellipticity, is crucial for directing the subsequent folding process, one has to establish the kinetic role of the burst phase intermediate. If the intermediate is an on-pathway and obligatory one, there should be a lag period in the appearance of the N state. We could not observe a lag phase, however, because the formation of the burst phase is too fast to be measured by our stopped-flow apparatus. Alternatively, the overshoot may represent a parallel, rather than a sequential, reorganization (e.g., due to cis-trans isomerization of X-pro peptide bonds) of the secondary structure within the already compact state formed in the burst phase.
The kinetic CD spectra represent the time-dependent changes in the structure of apoAI during the refolding process. The difference spectrum from I to N exhibits both positive and negative bands with an isoellipticity point at 222 nm. This pattern is characteristic of an exciton coupling effect The presence of an exciton in the kinetic difference spectrum suggests a close proximity between at least two Trp residues during the folding process (34,35).
Previous studies have shown that deletion of the carboxyl terminus of apoAI results in impaired lipid binding by the protein (36). Our data on GdnHCl-induced equilibrium unfolding states suggest that the N→U free energy change (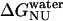 ) of the del[186–243] mutant is comparable to that of full-length apoAI (5.0 ± 0.4 vs. 4.5 ± 0.2 kcal/mol). This finding would argue against the view that lipid binding is associated with a complete unfolding of the protein. However, a closer examination of the free energy change data shown in Table 2 reveals that the N→I transition
) of the del[186–243] mutant is comparable to that of full-length apoAI (5.0 ± 0.4 vs. 4.5 ± 0.2 kcal/mol). This finding would argue against the view that lipid binding is associated with a complete unfolding of the protein. However, a closer examination of the free energy change data shown in Table 2 reveals that the N→I transition 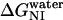 for del[186–243] is significantly larger than that for full-length apoAI (3.4 ± 0.3 vs. 2.5 ± 0.2 kcal/mol). This finding would be consistent with the idea that the intermediate state of apoAI faclilitates lipid binding by lowering the free energy barrier for the binding of the native state of apoAI to lipid (Fig. 6). Further support for the notion that the intermediate state may play a role in lipid binding emerges from our finding that
for del[186–243] is significantly larger than that for full-length apoAI (3.4 ± 0.3 vs. 2.5 ± 0.2 kcal/mol). This finding would be consistent with the idea that the intermediate state of apoAI faclilitates lipid binding by lowering the free energy barrier for the binding of the native state of apoAI to lipid (Fig. 6). Further support for the notion that the intermediate state may play a role in lipid binding emerges from our finding that 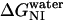 for the del[1–43] mutant is significantly smaller than that for full-length apoAI (1.5 ± 0.1 vs. 2.5 ± 0.2 kcal/mol; see Table 2). This finding is consistent with the previous finding that removal of the first 43 amino acids of apoAI allows the protein to be crystallized in a conformation that is thought to resemble the lipid-bound form of the intact protein. Our hypothesis regarding the role of the I state of apoAI in lipid binding further predicts that the [44–185] mutant would bind lipid somewhat more strongly than full-length apoAI, as the former exhibits a slightly lower
for the del[1–43] mutant is significantly smaller than that for full-length apoAI (1.5 ± 0.1 vs. 2.5 ± 0.2 kcal/mol; see Table 2). This finding is consistent with the previous finding that removal of the first 43 amino acids of apoAI allows the protein to be crystallized in a conformation that is thought to resemble the lipid-bound form of the intact protein. Our hypothesis regarding the role of the I state of apoAI in lipid binding further predicts that the [44–185] mutant would bind lipid somewhat more strongly than full-length apoAI, as the former exhibits a slightly lower 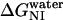 (2.0 ± 0.1 vs. 2.5 ± 0.2 kcal/mol; see Table 2). Further studies will be needed to confirm whether apoAI lipidation involves a conformation state similar to the intermediate state we describe for unfolding, and whether this state can be accurately described as a molten globule.
(2.0 ± 0.1 vs. 2.5 ± 0.2 kcal/mol; see Table 2). Further studies will be needed to confirm whether apoAI lipidation involves a conformation state similar to the intermediate state we describe for unfolding, and whether this state can be accurately described as a molten globule.
FIGURE 6.

Gibbs free energy diagram for the different states of apoAI. The free energy changes given in Table 2 for transitions between the different states of the protein are also indicated. A putative transition between the intermediate state (I) and lipid bound protein (L) is also depicted in the diagram. The existence of a folding intermediate state would lower the free energy barrier for lipid binding of apoAI.
In conclusion, several lines of evidence suggest that the refolding of the protein goes through at least two intermediate states. 1), Comparing the ellipticity at 222 nm of the unfolded protein in 5 M GdnHCl at equilibrium (Fig. 2 a, solid line) to the ellipticity of the protein extrapolated to time 0 in the refolding experiment (Fig. 1 a, solid square), revealed that the magnitude of the latter is significantly larger (ellipticity at 222 nm more negative). This suggests that a significant portion of the protein has already folded during the “dead time” of the measurement (i.e., before the first point in the kinetic trace was collected). 2), The kinetics of the refolding process as measured by protein ellipticity at 222 nm is biphasic and can be best described by a double exponential decay function. This further suggests that in addition to the fast, burst phase intermediate (Iburst), which accumulates during the dead time of the refolding experiment, there is a slow, recovery intermediate (Irecovery) that appears later in the process. 3), An overshoot of the CD signal was observed in the first phase of the folding process followed by a slower change in the signal, which converged toward a value characteristic of the native state of the protein. 4), At equilibrium, the transition curve for the fluorescence peak intensity (Fig. 3 c) strongly suggests that the unfolding process of the protein is going through an equilibrium intermediate state (Iequil). One unresolved issue of this study is that we do not know if the folding intermediates observed in the kinetic studies are related to the intermediate observed in the equilibrium experiments.
Acknowledgments
This work was supported by National Institutes of Health grant HL66082 to J.D.S. The SCoP simulation package was purchased with a grant-in-aid from the American Heart Association to E.G.
References
- 1.Linsel-Nitschke, P., and A. R. Tall. 2005. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nature Reviews. 4:193–205. [DOI] [PubMed] [Google Scholar]
- 2.Lewis, G. F., and D. J. Rader. 2005. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res. 96:1221–1232. [DOI] [PubMed] [Google Scholar]
- 3.Oram, J. F. 2003. HDL apolipoproteins and ABCA1: partners in the removal of excess cellular cholesterol. Arterioscler. Thromb. Vasc. Biol. 23:720–727. [DOI] [PubMed] [Google Scholar]
- 4.Wang, N., and A. R. Tall. 2003. Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arterioscler. Thromb. Vasc. Biol. 23:1178–1184. [DOI] [PubMed] [Google Scholar]
- 5.Segrest, J. P., L. Li, G. M. Anantharamaiah, S. C. Harvey, K. N. Liadaki, and V. Zannis. 2000. Structure and function of apolipoprotein A-I and high-density lipoprotein. Curr. Opin. Lipidol. 11:105–115. [DOI] [PubMed] [Google Scholar]
- 6.Borhani, D. W., D. P. Rogers, J. A. Engler, and C. G. Brouillette. 1997. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc. Natl. Acad. Sci. USA. 94:12291–12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oda, M. N., T. M. Forte, R. O. Ryan, and J. C. Voss. 2003. The C-terminal domain of apolipoprotein A-I contains a lipid-sensitive conformational trigger. Nat. Struct. Biol. 10:455–460. [DOI] [PubMed] [Google Scholar]
- 8.Sergest, J. P., M. K. Jones, H. De Loof, C. G. Bouillette, Y. V. Venkatachalapathi, and G. M. Anantharamaiah. 1992. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J. Lipid Res. 33:141–166. [PubMed] [Google Scholar]
- 9.Koppaka, V., L. Silvestro, J. A. Engler, C. G. Brouillette, and P. H. Axelsen. 1999. The structure of human lipoprotein A-I. Evidence for the “belt” model. J. Biol. Chem. 274:14541–14544. [DOI] [PubMed] [Google Scholar]
- 10.Li, H., D. S. Lyles, M. J. Thomas, W. Pan, and M. G. Sorci-Thomas. 2000. Structural determination of lipid-bound ApoA-I using fluorescence resonance energy transfer. J. Biol. Chem. 275:37048–37054. [DOI] [PubMed] [Google Scholar]
- 11.Davidson, W.S. and G. M. Hilliard. 2003. The spatial organization of apolipoprotein A-I on the edge of discoidal high density lipoprotein particles: a mass specrometry study. J. Biol. Chem. 278:27199–27207. [DOI] [PubMed] [Google Scholar]
- 12.Zhu, H. L., and D. Atkinson. 2004. Conformation and lipid binding of the N-terminal (1–44) domain of human apolipoprotein AI. Biochemistry. 43:13156–13164. [DOI] [PubMed] [Google Scholar]
- 13.Chroni, A., T. Liu, I. Gorshkova, H. Y. Kan, Y. Uehara, A. Von Eckardstein, and V. I. Zannis. 2003. The central helices of ApoA-I can promote ATP-binding cassette transporter A1 (ABCA1)-mediated lipid efflux. Amino acid residues 220–231 of the wild-type ApoA-I are required for lipid efflux in vitro and high density lipoprotein formation in vivo. J. Biol. Chem. 278:6719–6730. [DOI] [PubMed] [Google Scholar]
- 14.Rogers, D. P., L. M. Roberts, J. Lebowitz, G. Datta, G. M. Anantharamaiah, J. A. Engler, and C. G. Brouillette. 1998. The lipid-free structure of apolipoprotein A-I: effects of amino-terminal deletions. Biochemistry. 37:11714–11725. [DOI] [PubMed] [Google Scholar]
- 15.Beckstead, J. A., B. L. Block, J. K. Bielicki, C. M. Kay, M. N. Oda, and R. O. Ryan. 2005. Combined N- and C-terminal truncation of human apolipoprotein A-I yields a folded, functional central domain. Biochemistry. 44:4591–4599. [DOI] [PubMed] [Google Scholar]
- 16.Silva, R. A., G. M. Hilliard, J. Fang, S. Macha, and W. S. Davidson. 2005. A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry. 44:2759–2769. [DOI] [PubMed] [Google Scholar]
- 17.Roberts, L. M., M. J. Ray, T. W. Shih, E. Hayden, M. M. Reader, and C. G. Brouillette. 1997. Structural analysis of apolipoprotein A-I: limited proteolysis of methionine-reduced and -oxidized lipid-free and lipid-bound human apo A-I. Biochemistry. 36:7615–7624. [DOI] [PubMed] [Google Scholar]
- 18.Rogers, D. P., L. M. Roberts, J. Lebowitz, J. A. Engler, and C. G. Brouillette. 1998. Structural analysis of apolipoprotein A-I: effects of amino- and carboxy-terminal deletions on the lipid-free structure. Biochemistry. 37:945–955. [DOI] [PubMed] [Google Scholar]
- 19.Gursky, O., and D. Atkinson. 1996. Thermal unfolding of human high-density apolipoprotein A-1: implications for a lipid-free molten globular state. Proc. Natl. Acad. Sci. USA. 93:2991–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banuelos, S., and A. Muga. 1995. Binding of molten globule-like conformations to lipid bilayers. Structure of native and partially folded alpha-lactalbumin bound to model membranes. J. Biol. Chem. 270:29910–29915. [DOI] [PubMed] [Google Scholar]
- 21.Poklar, N., J. Lah, M. Salobir, P. Macek, and G. Vesnaver. 1997. pH and temperature-induced molten globule-like denatured states of equinatoxin II: a study by UV-melting, DSC, far- and near-UV CD spectroscopy, and ANS fluorescence. Biochemistry. 36:14345–14352. [DOI] [PubMed] [Google Scholar]
- 22.Freire, E. 1995. Thermodynamics of partly folded intermediates in proteins. Annu. Rev. Biophys. Biomol. Struct. 24:141–165. [DOI] [PubMed] [Google Scholar]
- 23.Ryan, R. O., T. M. Forte, and M. N. Oda. 2003. Optimized bacterial expression of human apolipoprotein A-I. Protein Expr. Purif. 27:98–103. [DOI] [PubMed] [Google Scholar]
- 24.Gross, E., and U. Hopfer. 1999. Effects of pH on kinetic parameters ofthe Na-HCO3 cotransporter in the renal proximal tubule. Biophys. J. 76:3066–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gross, E., and U. Hopfer. 1998. Voltage and co-substrate dependence of the Na-HCO3 cotransporter kinetics in renal proximal tubule cells. Biophys. J. 75:810–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pace, C. N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131:266–280. [DOI] [PubMed] [Google Scholar]
- 27.Eftink, M. R. 1994. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys. J. 66:482–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakao, M., K. Maki, M. Arai, T. Koshiba, K. Nitta, and K. Kuwajima. 2005. Characterization of kinetic folding intermediates of recombinant canine milk lysozyme by stopped-flow circular dichroism. Biochemistry. 44:6685–6692. [DOI] [PubMed] [Google Scholar]
- 29.Chedad, A., and H. Van Dael. 2004. Kinetics of folding and unfolding of goat α-lactalbumin. Proteins. 57:345–356. [DOI] [PubMed] [Google Scholar]
- 30.Baldwin, L. R. 1996. On-pathway versus off-pathway folding intermediates. Fold. Des. 1:R1–R8. [DOI] [PubMed] [Google Scholar]
- 31.Cahn, H. S., and K. A. Dill. 1998. Protein folding in the landscape perspective: chevron plots and non-Arrhenius kinetics. Proteins. 30:2–33. [DOI] [PubMed] [Google Scholar]
- 32.Roder, H., and W. Colon. 1997. Kinetic role of early intermediates in protein folding. Curr. Opin. Struct. Biol. 7:15–28. [DOI] [PubMed] [Google Scholar]
- 33.Saito, H., P. Dhanasekaran, D. Nguyen, P. Holvoet, S. Lund-Katz, and M. C. Phillips. 2003. Domain structure and lipid interaction in human apolipoproteins A-I and E, a general model. J. Biol. Chem. 278:23227–23232. [DOI] [PubMed] [Google Scholar]
- 34.Grishina, I. B., and R. W. Woody. 1994. Contributions of tryptophan side chains to the circular dichroism of globular proteins: exciton couplets and coupled oscillators. Faraday Discuss. 99:245–262. [DOI] [PubMed] [Google Scholar]
- 35.Superchi, S., E. Giorgio, and C. Rosini. 2004. Structural determinations by circular dichroism spectra analysis using coupled oscillator methods: an update of the applications of the DeVoe polarizability model. Chirality. 16:422–451. [DOI] [PubMed] [Google Scholar]
- 36.Ji, Y., and A. Jonas. 1995. Properties of an N-terminal proteolytic fragment of apolipoprotein AI in solution and in reconstituted high density lipoproteins. J. Biol. Chem. 270:11290–11297. [DOI] [PubMed] [Google Scholar]