

Abstract
Using total-internal-reflection fluorescence microscopy equipped with alternating-laser excitation, we were able to detect abortive initiation and promoter escape within single immobilized transcription complexes. Our approach uses fluorescence resonance energy transfer to monitor distances between a fluorescent probe incorporated in RNA polymerase (RNAP) and a fluorescent probe incorporated in DNA. We observe small, but reproducible and abortive-product-length-dependent, decreases in distance between the RNAP leading edge and DNA downstream of RNAP upon abortive initiation, and we observe large decreases in distance upon promoter escape. Inspection of population distributions and single-molecule time traces for abortive initiation indicates that, at a consensus promoter, at saturating ribonucleoside triphosphate concentrations, abortive-product release is rate-limiting (i.e., abortive-product synthesis and RNAP-active-center forward translocation are fast, whereas abortive-product dissociation and RNAP-active-center reverse translocation are slow). The results obtained using this new methodology confirm and extend those obtained from diffusing single molecules, and pave the way for real-time, single-molecule observations of the transitions between various states of the transcription complex throughout transcription.
INTRODUCTION
Transcription initiation is a multistep process (reviewed in Record et al. and others (1–5)). RNA polymerase (RNAP), together with one or more initiation factor(s): i), binds to promoter DNA to yield an RNAP-promoter closed complex, ii), unwinds ∼14 bp of DNA surrounding the transcription start site to yield an RNAP-promoter open complex, iii), begins RNA synthesis as an RNAP-promoter initial transcribing complex, and, ultimately, iv), escapes from the promoter and enters into productive RNA synthesis as an RNAP-DNA elongation complex. Typically, RNAP fails to escape from the promoter on its first attempt and, instead, engages in multiple abortive cycles of synthesis and release of short RNA products (shorter than a threshold length of ∼9–11 nucleotides (nt)). Only when RNAP succeeds in synthesizing an RNA product of a threshold length of ∼9–11 nucleotides, does RNAP irrevocably break its interactions with promoter DNA, irrevocably weaken or break its interactions with initiation factor(s), and begin to translocate along DNA, processively synthesizing RNA.
Fluorescence resonance energy transfer ((FRET); reviewed in Lilley and Wilson (6)) can be used to monitor movement of the leading edge of RNAP relative to DNA (7–10). In a “leading-edge-FRET” (LE-FRET) experiment (Fig. 1 a), a fluorescent probe serving as donor (D) is incorporated into the leading edge of RNAP, and a fluorescent probe serving as acceptor (A) is incorporated into downstream DNA (7–9); any movement of the leading edge of RNAP relative to downstream DNA is detected as changes in donor-acceptor FRET efficiency (which is proportional to the inverse sixth power of donor-acceptor distance (6)).
FIGURE 1.

Immobilized transcription complexes. (a) Leading-edge FRET. By labeling the leading edge of RNAP and the downstream end of DNA, we can monitor downstream translocation of RNAP by looking at increasing values of FRET. (b) Immobilization of the transcription complexes. Amino-silanized quartz slides are covalently modified by a layer of PEGs (1.25% biotinylated). The slides are incubated with streptavidin, rinsed, incubated again with 20–50 pM biotinylated transcription complexes, and rinsed before imaging. (c) DNA constructs: lacCONS (17) derivatives having no guanine residues on the template strand from +1 to +11. The doubly labeled DNA fragments are generated by PCR. (Boxes) Transcription start site (with arrow), promoter −10 element, and promoter −35 element; (shaded boxes) halt sites for RPitc,≤2, RPitc,≤4, RPitc,≤7, and RDe,11, respectively.
In previous work, we have used ensemble FRET to show that, contrary to the traditional view of the transcription cycle (11–15), the initiation factor σ70 is not obligatorily released upon promoter escape (8–10); using single-molecule FRET to analyze freely diffusing transcription complexes with confocal microscopy and alternating-laser excitation (ALEX (16,17)), this result has been confirmed, and extended by providing quantitative information regarding the extent, the half-life, and effects of DNA sequence, on σ70 retention in mature elongation complexes (7).
In this work, to provide information on the mechanism and kinetics of abortive initiation, we have used single-molecule LE-FRET—analyzing immobilized transcription complexes with total-internal-reflection optical microscopy, alternating-laser excitation, and millisecond-scale dynamic imaging (msALEX-DI). msALEX-DI yields: i), population distributions of donor-acceptor FRET efficiency (E) and donor-acceptor stoichiometry factor (S) (16,17), and ii), single-molecule, kinetic traces of E and S. After “sorting” of data by value of S, msALEX-DI yields both: i), filtered population distributions of E, and ii), filtered single-molecule, kinetic traces of E, both free from complications due to compositional heterogeneity and photophysical heterogeneity. The results obtained on single immobilized transcription complexes show that the initiation factor σ70 is not obligatorily released upon promoter escape, confirming our previous observations on diffusing single molecules (7). Moreover, the results indicate that abortive initiation involves translocation of the RNAP leading edge relative to DNA. They further indicate that the extent of translocation of the RNAP leading edge relative to DNA correlates with the length of the abortive RNA product. Finally, the results indicate that, at saturating ribonucleoside triphosphate (NTP) concentrations, a transcription complex engaged in iterative abortive initiation spends the majority of the time in the state with forward translocation of the RNAP leading edge relative to DNA, implying that abortive-product release and reverse translocation of the RNAP leading edge are rate-limiting.
MATERIALS AND METHODS
DNA fragments
The sequences of DNA fragments used are in Fig. 1 c. Doubly labeled (biotin- and acceptor-labeled) DNA fragments were prepared as described (8,9), using a biotinylated primer and a fluorescently labeled primer.
RPitc,≤2
The derivative of σ70 containing a single cysteine at position 366 was labeled with TMR (Molecular Probes, Eugene, OR), purified and stored as described (8,9). In a 30-μL reaction mixture, 80 nM TMR-σ70 and 100 nM RNAP core (Epicentre, Madison, WI) were incubated for 20 min at 30°C in transcription buffer (50 mM Tris-HCl (pH 8), 100 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT), 100 μg/mL bovine serum albumin (BSA), and 5% v/v glycerol), to form RNAP holoenzyme. Subsequently, 20 nM DNA was added to form an open complex, and the samples were transferred to 37°C. After 15 min, 2.2 μL of 1 mg/mL heparin-Sepharose (APB, Piscataway, NJ) was added to disrupt nonspecific complexes and to remove free RNAP. After 1 min at 37°C, the samples were centrifuged to pellet the heparin-Sepharose, 9.5 μL of the supernatant were transferred into prewarmed tubes containing 0.5 μL of 10 mM ApA (final ApA concentration, 0.5 mM), and the samples were incubated for 10 min at 37°C.
Immobilized RPitc,≤2
Custom-made quartz slides were treated with an amino-silane reagent (Vectabond, Vectorlabs, Burlingame, CA) as suggested by the manufacturer, and were incubated with PEG (Nektar Therapeutics, San Carlos, CA), containing 20% PEG-succinimidyl succinate and 0.25% biotin-PEG-OCH2CH2-CO2-NHS, in 0.1 M sodium borate, pH 8.5, for 3 h. A flow-cell chamber is prepared with a PEG-coated slide, a 50-μm spacer, and a PEG-coated round glass coverslip. The quartz slides are drilled to allow flowing of reagents. The chamber is filled with 50 mM Tris-HCl, pH 7.5, 100 mM NaCl buffer, incubated with streptavidin (0.2 mg/mL) for 10 min, and rinsed. Then, it is filled with KGGA buffer, and incubated with 20–50 pM RPitc,≤2 for 10 min, and rinsed again with KGGA in the presence of 1% oxygen scavenging system (KGGA: 40 mM HEPES-NaOH, pH 7, 100 mM potassium glutamate, 10 mM MgCl2, 1 mM DTT, 100 μg/mL BSA, 1 mM mercaptoethylamine, 0.5 mM ApA, 5% glucose, 1% β-mercaptoethanol; oxygen scavenging system: Gloxy = 1665 units glucose oxidase, (G-7016, Sigma, St. Louis, MO), ∼26,000 units catalase (Roche, Indianapolis, IN), as described (18)).
Immobilized RPitc,≤4, RPitc,≤7, and RDe,11
RPitc,≤4, RPitc,≤7, and RDe,11 were generated by flowing KGGA + 80 μM UTP, KGGA + 80 μM UTP/GTP, and KGGA + 80 μM UTP/GTP/ATP, respectively, in the flow cell with the immobilized RPitc,≤2 complexes.
msALEX-DI: data acquisition
Alternating-laser excitation was achieved using the 532-nm light from a diode-pumped doubled Nd:YAG laser (Crystalaser, Reno, NV), and the 638-nm light from a diode laser (Coherent, Santa Clara, CA); the alternation period was defined by the integration time of the camera (Photometrics Cascade 650; Tucson, AZ), typically 200 ms. Extinction of the lasers was achieved using electrooptical modulators (EOM) combined with polarizers. By rotating the polarization of each laser beam individually before directing it to the polarizer, the lasers were switched on or off. Extinction ratios (ratios of laser intensities when a laser is on or off) were >100:1 for each laser. The beams were circularly polarized using achromatic waveplates, and combined using a dichroic mirror, which also enabled us to maximize beam overlap and beam centering in the field of view. The emitted photons, collected through a 1.2-NA, 63× water immersion objective (C-Apochromat, Carl Zeiss, Jena, Germany), are split into two regions of the CCD detector using two 630DRLP dichroic mirrors (Omega, Brattleboro, VT) (dual-view format (19)), allowing for simultaneous observation of the donor and acceptor fluorescence. The synchronization of the alternation with the integration periods of the camera was achieved using the “frame readout” output signal of the camera to trigger the switching of the EOMs. The typical response time of the EOMs is 2 μs, several orders of magnitude faster than the alternation period (17).
msALEX-DI: data analysis
For data analysis, a LabVIEW-based (National Instruments, Austin, TX) software routine was developed, which includes automated superimposition of the donor and acceptor channels based on an empirical correlation coefficient. Additionally, several algorithms were used to correct for possible image distortions on the two channels (19). Spots representing single molecules were identified from the sum of several frames using a spot-recognition algorithm, which takes into account intensity, spot size, spot shape, and distance to the neighboring spots to exclude cross-talk fluorescence from nearby molecules. Local background correction was performed by subtracting the average intensity around each spot, for each laser illumination. Afterwards, intensity trajectories were extracted from the whole image stack, and separated according to the laser used for each image, thus generating four time traces of fluorescence emissions: 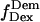 and
and 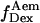 for donor excitation and
for donor excitation and 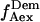 and
and 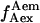 for acceptor excitation. These emissions report on donor-acceptor proximity through the calculation of ratio
for acceptor excitation. These emissions report on donor-acceptor proximity through the calculation of ratio 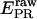 (Eq. 9). The ALEX-based ratio
(Eq. 9). The ALEX-based ratio 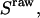 that reports on the D-A stoichiometry is calculated using Eq. 10. For each frame n corresponding to donor excitation, i.e., for each
that reports on the D-A stoichiometry is calculated using Eq. 10. For each frame n corresponding to donor excitation, i.e., for each 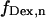 value, this ratio can be calculated using three different
value, this ratio can be calculated using three different 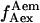 values: two coming from the closest frame corresponding to red excitation, i.e., frame n − 1 (
values: two coming from the closest frame corresponding to red excitation, i.e., frame n − 1 (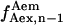 ), or frame n + 1 (
), or frame n + 1 (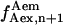 ), or one being the average of
), or one being the average of 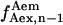 and
and 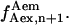 The difference between the results obtained using these three different calculations was <3%, thus the average value of
The difference between the results obtained using these three different calculations was <3%, thus the average value of 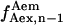 and
and 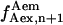 has been arbitrarily chosen for
has been arbitrarily chosen for 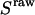 calculation. Hereafter, this value is denoted
calculation. Hereafter, this value is denoted 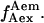 After excitation adjustment to obtain
After excitation adjustment to obtain 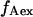 ≈
≈ 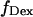 for D-A complexes, this distance-independent ratio allows to determine the stoichiometry of each complex (17).
for D-A complexes, this distance-independent ratio allows to determine the stoichiometry of each complex (17).
msALEX-DI: calculation of fraction of active complexes
For each RPitc,≤2 and RDe,11 complex the value 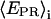 is calculated by averaging
is calculated by averaging 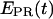 values of molecule
values of molecule 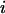 that have active D and A. For RPitc,≤2, the
that have active D and A. For RPitc,≤2, the 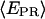 distribution is fitted by a Gaussian function (mean E1 and width W1). For RDe,11, the
distribution is fitted by a Gaussian function (mean E1 and width W1). For RDe,11, the 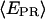 distribution is fitted by two Gaussian functions (restricted fit for one Gaussian with the E1 and W1 values determined from the RPitc,≤2 sample). The amplitudes of the two Gaussians A1 and A2 corresponding to the relative number of RPitc,≤2 and RDe,11 molecules, respectively, are determined from the fit.
distribution is fitted by two Gaussian functions (restricted fit for one Gaussian with the E1 and W1 values determined from the RPitc,≤2 sample). The amplitudes of the two Gaussians A1 and A2 corresponding to the relative number of RPitc,≤2 and RDe,11 molecules, respectively, are determined from the fit.
The apparent translocational activity A is then calculated using:
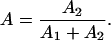 |
(1) |
However, A does not take into consideration transcription complexes that released their σ70 subunit during the transition from initiation to elongation, underestimating the number of RDe,11 complexes. Even though we have shown elsewhere that σ70 release is small upon formation of RDe,11 (7), it is necessary to consider it for accurate activity measurements on immobilized complexes. For this purpose, for both RPitc,≤2 and RDe,11, we counted nDA, the number of molecules appearing as D-A complexes (i.e., DNA-RNAP-σ70 complexes), and nA the number of molecules appearing as acceptor only (i.e., DNA only or DNA-RNAP complexes). For each sample, 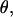 the fractional occupancy of the DNA by σ70, is calculated.
the fractional occupancy of the DNA by σ70, is calculated.
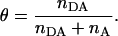 |
(2) |
Then, the retention of σ70 on the RDe,11 complex is calculated using:
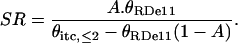 |
(3) |
This SR ratio allows us to recalculate the number of active complexes (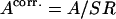 ), and by replacing
), and by replacing 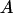 with
with 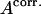 in Eq. 1, to recover the translocation activity corrected for σ70 release. It should be noted, that, in our analysis, SR was always >80%, a finding consistent with data obtained for freely diffusing molecules (7).
in Eq. 1, to recover the translocation activity corrected for σ70 release. It should be noted, that, in our analysis, SR was always >80%, a finding consistent with data obtained for freely diffusing molecules (7).
msALEX-DI: calculation of accurate FRET efficiencies and distances
The ratio 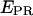 depends on donor-acceptor proximity, but it is not the FRET efficiency E that allows one to calculate accurately the distance between the two probes. The conversion from
depends on donor-acceptor proximity, but it is not the FRET efficiency E that allows one to calculate accurately the distance between the two probes. The conversion from 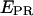 to E using ALEX has been described for diffusing molecules (20) and has been adapted here for immobilized molecules. The conversion is performed by accounting for three factors:
to E using ALEX has been described for diffusing molecules (20) and has been adapted here for immobilized molecules. The conversion is performed by accounting for three factors:
l, “leakage” of the donor emission in the acceptor emission channel. This factor is calculated using
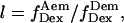 for donor-only species.
for donor-only species.d, acceptor emission due to direct excitation of the acceptor by the donor-excitation laser. This is calculated using
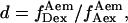 for acceptor-only species.
for acceptor-only species.γ, a factor that accounts for differences in detection efficiencies in the donor and acceptor emission channels, and the donor and acceptor quantum yields, defined as
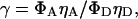 where
where 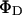 and
and 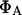 are the quantum yields of D and A, respectively, and
are the quantum yields of D and A, respectively, and 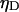 and
and 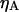 are the detection efficiencies of the D and A channels, respectively.
are the detection efficiencies of the D and A channels, respectively.
Experimentally, the l factor is the center of the 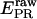 distribution for D-only molecules, and the d factor is the center of the
distribution for D-only molecules, and the d factor is the center of the 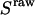 distribution for A-only molecules, as plotted in Fig. 7 a.
distribution for A-only molecules, as plotted in Fig. 7 a.
FIGURE 7.

Determination of correction factors for accurate FRET efficiency determination. (a) Determination of l, the leakage of the donor emission in the acceptor channel, and d, the acceptor emission due to the direct excitation of the acceptor by the donor-excitation laser; l is the center of the 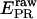 distribution for D-only molecules (top), and the d is the center of the
distribution for D-only molecules (top), and the d is the center of the 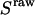 distribution for A-only molecules (right). (b) Determination of γ, the factor that accounts for differences in detection efficiencies in the donor and acceptor emission channels.
distribution for A-only molecules (right). (b) Determination of γ, the factor that accounts for differences in detection efficiencies in the donor and acceptor emission channels. 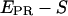 histogram for a RPitc,≤2 (dark gray) and a RDe,11 (light gray) sample. For each complex,
histogram for a RPitc,≤2 (dark gray) and a RDe,11 (light gray) sample. For each complex, 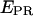 (top) and
(top) and 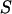 (right) distributions are fitted with a Gaussian function to determine the center of the distribution. (c) Determination of
(right) distributions are fitted with a Gaussian function to determine the center of the distribution. (c) Determination of 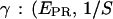 ) values are plotted for different complexes, here RPitc,≤2, RPitc,≤7, and RDe,11. The
) values are plotted for different complexes, here RPitc,≤2, RPitc,≤7, and RDe,11. The 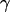 -factor is then calculated from the slope Σ and the intercept Ω of the best linear fit to the (
-factor is then calculated from the slope Σ and the intercept Ω of the best linear fit to the (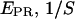 ) values, as described in the Materials and Methods section.
) values, as described in the Materials and Methods section.
The l and d factors are used to calculate the corrected proximity ratio 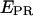 using:
using:
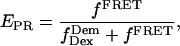 |
(4) |
and the corrected S ratio using
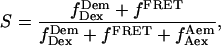 |
(5) |
where 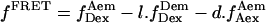 (20).
(20).
To calculate the 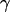 -factor, we have plotted (Fig. 7 b) an
-factor, we have plotted (Fig. 7 b) an 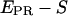 histogram for a RPitc,≤2 and a RDe,11 sample. On this histogram, two D-A complexes are seen (RPitc,≤2 (red) and RDe,11 (green)), having different
histogram for a RPitc,≤2 and a RDe,11 sample. On this histogram, two D-A complexes are seen (RPitc,≤2 (red) and RDe,11 (green)), having different 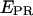 and
and 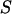 values. For each complex, and RPitc,≤7 (not shown)
values. For each complex, and RPitc,≤7 (not shown) 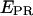 and
and 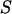 distributions are fitted with a Gaussian function to determine the center of the distribution. As described (20), plotting (
distributions are fitted with a Gaussian function to determine the center of the distribution. As described (20), plotting (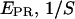 ) values allows one to recover the
) values allows one to recover the 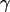 -factor from the slope Σ and the intercept Ω of the line defined by the (
-factor from the slope Σ and the intercept Ω of the line defined by the (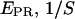 ) values (Fig. 7 c).
) values (Fig. 7 c).
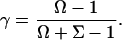 |
(6) |
It is important to note that for this calculation 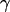 is assumed to be constant for all complexes. This means that the detection efficiencies of the D and A channel have to be constant (which is the case because the setup alignment does not change), and that the quantum yields of the dyes have to be the same in the context of the different complexes, which will be the case if their local environments do not significantly differ. The donor-probe environment is expected to be essentially identical in RPitc,≤2 and RDe,11, considering that the relevant domain of σ70 translocates as a “block” with RNAP upon formation of RDe,11 (9), and considering that the donor probe is located far from other domains of σ70, RNAP core, DNA, and RNA in modeled structures of the open and elongation complexes (21). To check for possible changes in the acceptor quantum yield, we plotted
is assumed to be constant for all complexes. This means that the detection efficiencies of the D and A channel have to be constant (which is the case because the setup alignment does not change), and that the quantum yields of the dyes have to be the same in the context of the different complexes, which will be the case if their local environments do not significantly differ. The donor-probe environment is expected to be essentially identical in RPitc,≤2 and RDe,11, considering that the relevant domain of σ70 translocates as a “block” with RNAP upon formation of RDe,11 (9), and considering that the donor probe is located far from other domains of σ70, RNAP core, DNA, and RNA in modeled structures of the open and elongation complexes (21). To check for possible changes in the acceptor quantum yield, we plotted 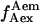 distributions for RPitc,≤2, RPitc,≤7, and RDe,11 complexes. There was no significant difference between these intensities distribution, thus we conclude that the quantum yield of the acceptor is essentially identical in the context of the two complexes. The recovered values for all these parameters depended on the setup alignment and the molecules used. Generally, we found l ≈ 0.08, d ≈ 0.1, and γ ≈ 0.45–0.76. The conversion from EPR to E is performed using the following equation (20):
distributions for RPitc,≤2, RPitc,≤7, and RDe,11 complexes. There was no significant difference between these intensities distribution, thus we conclude that the quantum yield of the acceptor is essentially identical in the context of the two complexes. The recovered values for all these parameters depended on the setup alignment and the molecules used. Generally, we found l ≈ 0.08, d ≈ 0.1, and γ ≈ 0.45–0.76. The conversion from EPR to E is performed using the following equation (20):
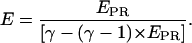 |
(7) |
The donor-acceptor distance (R) was calculated as follows using the efficiency of FRET (E), and the Förster parameter (Ro) (22):
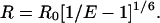 |
(8) |
Ro for our complexes, measured in KGGA buffer at 25°C, was 63.4 Å, assuming 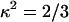 ; κ2 is the orientation factor relating the donor emission dipole and acceptor excitation dipole (approximated as 2/3—justified by fluorescence anisotropy measurements, using the same constructs, indicating donor and acceptor reorient on the timescale of the donor excited-state lifetime (21)).
; κ2 is the orientation factor relating the donor emission dipole and acceptor excitation dipole (approximated as 2/3—justified by fluorescence anisotropy measurements, using the same constructs, indicating donor and acceptor reorient on the timescale of the donor excited-state lifetime (21)).
RESULTS AND DISCUSSION
Immobilized transcription complexes
Transcription complexes were immobilized on poly(ethylene-glycol) (PEG)-coated quartz surfaces through quartz-PEG-biotin-streptavidin-biotin-DNA linkages (23) (Fig. 1 b). Custom-made microscopy quartz slides have been amino-silanized and covalently modified with PEG-succinimidyl esters. PEG, a highly hydrophilic polymer, has the ability to exclude proteins and cells from surfaces, thus reducing nonspecific binding. By using a small percentage of biotinylated PEG and a streptavidin linker, biotinylated macromolecules can bind specifically to the quartz slides in a hydrophilic environment. The DNA template is a lacCONS promoter derivative (21) having no guanine residues on the template strand from +1 to +11 (lacCONS + 2; Fig. 1 c). Different doubly labeled DNA constructs have been generated by polymerase chain reaction, with biotin at position −40 and Cy5, serving as FRET acceptor, at position +20 or +25 (Fig. 1 c); σ70 is labeled on position 366 (located in σ70 region 2) using tetramethylrhodamine-5-maleimide (TMR), serving as FRET donor (8,9). Transcription open complexes were prepared using the DNA template, RNAP core, and TMR-labeled σ70. Successive addition of NTP subsets leads to RNA transcripts of different lengths n, due to the withholding of the NTP to be incorporated at position n + 1 (Fig. 1 c). Addition of ApA (equivalent to the first dinucleotide synthesized on this promoter) to the open complex generates the RPitc,≤2 complex; addition of uridine 5′-triphosphate (UTP) generates RNA fragments up to 4 nt (RPitc,≤4 complex); addition of UTP and guanosine 5′-triphosphate (GTP) increases the maximum length to 7 nt (RPitc,≤7 complex); using these nucleotide subsets, the transcription complex is expected to generate abortive RNA products iteratively. Finally, addition of UTP, GTP, and ATP allows the RNAP to escape from the promoter to generate an early, stable elongation complex with an RNA length of 11 nucleotides (RDe,11). Immobilization of RPitc,≤2 complexes at 20–50 pM typically leads to 100–150 transcription complexes in the field of view, whereas nonspecific binding (measured using nonbiotinylated DNA, or by omitting streptavidin in the rinsing buffer) was reduced to <1%. This immobilization scheme does not perturb the transcription system under study, which retains a high translocational activity (see below).
msALEX-DI: experimental setup and data analysis
Total internal reflection (TIR) microscopy is a powerful tool for imaging single fluorescent molecules immobilized on surfaces or in cells (24,18). The emitted fluorescence of dozens of single molecules can be measured simultaneously, typically with a 100-ms temporal resolution. In the case of a FRET experiment, the emitted photons can be split into two regions of a charge-coupled device (CCD) detector, allowing for simultaneous measurement of the donor and acceptor fluorescence (24,19). We have modified a prism-based TIR microscope (25) to introduce alternating-laser excitation of the donor and acceptor probes (Fig. 2); the modification is based on a previously described microscope designed for the alternating excitation of single diffusing molecules in a confocal volume (17). Here, laser alternation was synchronized with the camera-frame acquisition, to generate four images, corresponding to four time traces per single molecule: for donor and acceptor emissions, respectively, donor excitation gives 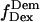 and
and 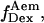 whereas acceptor excitation gives
whereas acceptor excitation gives 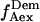 and
and 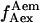 (see Fig. 3 a for an example of a D-A molecule). Typically, the term
(see Fig. 3 a for an example of a D-A molecule). Typically, the term 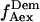 (emission signal from the donor upon acceptor excitation) equals zero, and thus is not included in the following equations and analysis. The emitted fluorescence signals are reduced to two ratios:
(emission signal from the donor upon acceptor excitation) equals zero, and thus is not included in the following equations and analysis. The emitted fluorescence signals are reduced to two ratios:
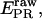
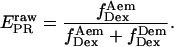
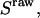
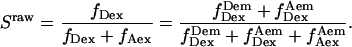
FIGURE 2.

msALEX-DI, experimental setup; EOM, electrooptic modulator; DM, dichroic mirror. The image is split into two zones on the CCD camera, corresponding to the donor (left) and the acceptor (right) emission channels. The camera is synchronized with the alternation of the lasers, resulting in four images of the illuminated area (two excitations × two emissions).
FIGURE 3.

Data analysis. (a) Representative raw data. After channels overlay and correction for optical aberrations, the molecules are identified (see Materials and Methods). The intensity corresponding to each molecule is integrated and background subtracted for each of the excitation/emission combinations, resulting in four intensities (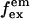 ), used to calculate the
), used to calculate the 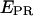 and S ratios. (b) Populations distributions. (Left) The
and S ratios. (b) Populations distributions. (Left) The 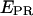 and S ratios are displayed on a two-dimensional histogram. Donor-only (S > 0.8), acceptor-only (S < 0.2), and donor-acceptor complexes (0.2 < S < 0.8) are readily identified. (Right) Example of data obtained with a low-
and S ratios are displayed on a two-dimensional histogram. Donor-only (S > 0.8), acceptor-only (S < 0.2), and donor-acceptor complexes (0.2 < S < 0.8) are readily identified. (Right) Example of data obtained with a low- 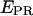 complex (RPitc,≤2; Cy5, +25), displaying all the (
complex (RPitc,≤2; Cy5, +25), displaying all the (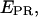 S) values at all time points, for all complexes. (c) Elimination of complications due to compositional heterogeneity. (Left) The acceptor-only population is removed by selecting the
S) values at all time points, for all complexes. (c) Elimination of complications due to compositional heterogeneity. (Left) The acceptor-only population is removed by selecting the 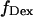 above a certain threshold (>1000 counts). (Right) The donor-only population is removed, by selecting the
above a certain threshold (>1000 counts). (Right) The donor-only population is removed, by selecting the 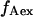 above a certain threshold (>1000 counts). As a result, only the (
above a certain threshold (>1000 counts). As a result, only the (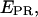 S) values at all time points corresponding to D-A complexes are displayed. The relevant low
S) values at all time points corresponding to D-A complexes are displayed. The relevant low 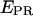 peak is separated from the D-only peak (
peak is separated from the D-only peak (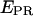 ≈ 0), and its mean
≈ 0), and its mean 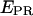 value can be accurately recovered from the projection of the histogram onto the
value can be accurately recovered from the projection of the histogram onto the 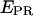 axis. (d) Elimination of complications due to inactive state of the acceptor. (Left) Using single laser excitation (donor excitation), traces corresponding to donor (
axis. (d) Elimination of complications due to inactive state of the acceptor. (Left) Using single laser excitation (donor excitation), traces corresponding to donor (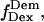 green) and acceptor trough FRET (
green) and acceptor trough FRET (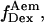 red) emissions are obtained (top). The resulting
red) emissions are obtained (top). The resulting 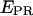 trace, presented at the bottom, shows an interconversion between a high and a low
trace, presented at the bottom, shows an interconversion between a high and a low 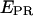 state. (Right) msALEX-DI allows one to excite the acceptor directly and monitor its emitted fluorescence (
state. (Right) msALEX-DI allows one to excite the acceptor directly and monitor its emitted fluorescence (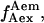 black). In this case, it is clearly shown that acceptor blinking (cycling between active and inactive states of the acceptor) is responsible for the observed anticorrelated behavior of the
black). In this case, it is clearly shown that acceptor blinking (cycling between active and inactive states of the acceptor) is responsible for the observed anticorrelated behavior of the 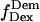 and
and 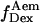 traces. Removal of the time points where acceptor is inactive generates a new
traces. Removal of the time points where acceptor is inactive generates a new 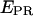 trace (bottom) where only time points with active acceptor are retained.
trace (bottom) where only time points with active acceptor are retained.
To account for cross talks between channels (20), 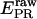 and
and 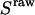 were corrected, by accounting for the donor emission into the acceptor channel, and for direct excitation of the acceptor at the donor-excitation wavelength, leading to the corrected ratios
were corrected, by accounting for the donor emission into the acceptor channel, and for direct excitation of the acceptor at the donor-excitation wavelength, leading to the corrected ratios 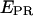 and S, respectively, (see Materials and Methods; Eqs. 4 and 5, respectively). The S-value allows sorting of D-A complexes from D-only and A-only species even in the absence of FRET. Indeed, S for D-only species is high, ≈1 (because
and S, respectively, (see Materials and Methods; Eqs. 4 and 5, respectively). The S-value allows sorting of D-A complexes from D-only and A-only species even in the absence of FRET. Indeed, S for D-only species is high, ≈1 (because 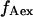 ≈ 0), and S for A-only species is low, ≈0 (because
≈ 0), and S for A-only species is low, ≈0 (because 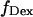 ≈ 0). D-A complexes characterized by any RD-A distance typically assume an S-value between 0.2 and 0.8.
≈ 0). D-A complexes characterized by any RD-A distance typically assume an S-value between 0.2 and 0.8.
msALEX-DI: identification and elimination of compositional heterogeneity
When represented in a two-dimensional 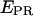 -S histogram, D-only, A-only, and D-A complexes are sorted into three clusters (Fig. 3 b, left). Fig. 3 b (right) shows an example of a mixture containing a transcription complex with
-S histogram, D-only, A-only, and D-A complexes are sorted into three clusters (Fig. 3 b, left). Fig. 3 b (right) shows an example of a mixture containing a transcription complex with 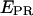 ≈ 0.1 (a D-A species), free DNA (an A-only species), and free RNAPσ70 (D-only species); all (
≈ 0.1 (a D-A species), free DNA (an A-only species), and free RNAPσ70 (D-only species); all (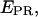 S) couples, for all time points of the single-molecule time traces, are represented. The three subpopulations are clearly resolved on the histogram, as done with similar measurements in solution (7). By selecting molecules having fDex and/or fAex above a certain threshold (typically >1000 counts), the D-only, A-only, or D-A species can be selected. For example, applying a fDex threshold on the data in Fig. 3 b removes the A-only peak (Fig. 3 c, left), and subsequently, applying a similar fAex threshold removes the D-only peak, leaving solely D-A species for further analysis (Fig. 3 c, right). For this data set, assuming that the D-A species in Fig. 3 b have an S-value comprised between 0.2 and 0.8, the described protocol removes 99 and 96% of the D-only and A-only populations, respectively, whereas 97% of the D-A complexes are kept for further analysis. We note that the
S) couples, for all time points of the single-molecule time traces, are represented. The three subpopulations are clearly resolved on the histogram, as done with similar measurements in solution (7). By selecting molecules having fDex and/or fAex above a certain threshold (typically >1000 counts), the D-only, A-only, or D-A species can be selected. For example, applying a fDex threshold on the data in Fig. 3 b removes the A-only peak (Fig. 3 c, left), and subsequently, applying a similar fAex threshold removes the D-only peak, leaving solely D-A species for further analysis (Fig. 3 c, right). For this data set, assuming that the D-A species in Fig. 3 b have an S-value comprised between 0.2 and 0.8, the described protocol removes 99 and 96% of the D-only and A-only populations, respectively, whereas 97% of the D-A complexes are kept for further analysis. We note that the 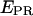 distribution is much broader for A-only species, because acceptors are not appreciably excited upon donor excitation, and thus all the terms in Eq. 9 are close to zero.
distribution is much broader for A-only species, because acceptors are not appreciably excited upon donor excitation, and thus all the terms in Eq. 9 are close to zero.
As seen in the collapse of the two-dimensional histogram on the 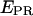 axis (Fig. 3 c, top), the D-only peak usually observed in single-molecule FRET experiments can be removed from the
axis (Fig. 3 c, top), the D-only peak usually observed in single-molecule FRET experiments can be removed from the 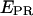 histograms, without affecting the low-FRET population. The ability to remove the D-only peak extends the range of distance measurement by single-molecule FRET, because it allows monitoring of D-A species characterized by large donor-acceptor distances that correspond to
histograms, without affecting the low-FRET population. The ability to remove the D-only peak extends the range of distance measurement by single-molecule FRET, because it allows monitoring of D-A species characterized by large donor-acceptor distances that correspond to 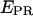 values close or equal to zero (a range of E-values with minimal uncertainties for FRET-based distance measurements (26)). Thus, it becomes possible to detect association/dissociation events between molecules (one of them being immobilized), without requirement for close proximity between donor and acceptor.
values close or equal to zero (a range of E-values with minimal uncertainties for FRET-based distance measurements (26)). Thus, it becomes possible to detect association/dissociation events between molecules (one of them being immobilized), without requirement for close proximity between donor and acceptor.
msALEX-DI: identification and elimination of photophysical heterogeneity
Usually, single-molecule FRET experiments using D-only excitation cannot distinguish between low- 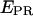 states arising from a large donor-acceptor distance, and low- EPR states arising from inactive (e.g., nonabsorbing and nonemitting) states of the acceptor. This latter photophysical phenomenon has been observed with various acceptors, dictating greater care in the interpretation of single-molecule FRET data at low
states arising from a large donor-acceptor distance, and low- EPR states arising from inactive (e.g., nonabsorbing and nonemitting) states of the acceptor. This latter photophysical phenomenon has been observed with various acceptors, dictating greater care in the interpretation of single-molecule FRET data at low 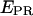 (27–29). msALEX-DI determines unambiguously the nature of low-
(27–29). msALEX-DI determines unambiguously the nature of low-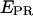 states by probing directly the acceptor photophysical state. This becomes clear after examining a time trace representative of a D-A molecule that populates both high-E and low-E states (Fig. 3 d, left), where traces corresponding to the donor and acceptor emission upon donor excitation (
states by probing directly the acceptor photophysical state. This becomes clear after examining a time trace representative of a D-A molecule that populates both high-E and low-E states (Fig. 3 d, left), where traces corresponding to the donor and acceptor emission upon donor excitation (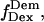 green line, and
green line, and 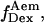 red line) are presented, along with the corresponding
red line) are presented, along with the corresponding 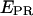 trace. The time trace shows numerous interconversions between two states: a high-
trace. The time trace shows numerous interconversions between two states: a high- 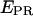 state (
state (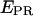 = 0.72 ± 0.15) and a low-
= 0.72 ± 0.15) and a low- 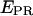 state (
state (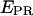 = 0.03 ± 0.09). The high-
= 0.03 ± 0.09). The high- 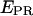 value is due to close D-A proximity in the transcription complex under study. The low-
value is due to close D-A proximity in the transcription complex under study. The low- 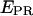 state is either due to a conformational change that increases the D-A distance, or due to a photophysical event, e.g., conversion of the acceptor from an active to an inactive state. A direct probing of the acceptor photophysical state, using msALEX-DI, supports the latter hypothesis, as seen by superimposing the acceptor emission upon acceptor excitation (i.e.,
state is either due to a conformational change that increases the D-A distance, or due to a photophysical event, e.g., conversion of the acceptor from an active to an inactive state. A direct probing of the acceptor photophysical state, using msALEX-DI, supports the latter hypothesis, as seen by superimposing the acceptor emission upon acceptor excitation (i.e., 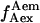 ) with the traces obtained upon donor excitation (Fig. 3 d, right); clearly, the events leading to the low-
) with the traces obtained upon donor excitation (Fig. 3 d, right); clearly, the events leading to the low- 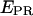 value correspond to an inactive state of the acceptor. Thus, by filtering the time trace using a threshold for the
value correspond to an inactive state of the acceptor. Thus, by filtering the time trace using a threshold for the 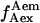 value (typically
value (typically 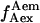 > 1000 counts), we generate automatically a new time trace, devoid of any points where the acceptor blinks (Fig. 3 d, right). The ability of msALEX-DI to remove photophysically induced FRET changes from single-molecule time traces is critical for accurate interpretation of low-
> 1000 counts), we generate automatically a new time trace, devoid of any points where the acceptor blinks (Fig. 3 d, right). The ability of msALEX-DI to remove photophysically induced FRET changes from single-molecule time traces is critical for accurate interpretation of low- 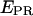 values, and for dynamical studies of systems that populate states of various FRET values, even when one of these states involves an interprobe distance out of the FRET range (RD-A > 1.5 R0).
values, and for dynamical studies of systems that populate states of various FRET values, even when one of these states involves an interprobe distance out of the FRET range (RD-A > 1.5 R0).
Promoter escape: detection
A prerequisite for performing experiments with single immobilized transcription complexes is the retention of significant transcriptional activity after surface immobilization; we test the transcriptional activity by showing that the majority of immobilized RNA polymerase molecules undergoes the transition from initiation to early elongation (from RPitc,≤2 to RDe,11). Immobilized RPitc,≤2 complexes show a single D-A population (e.g., Fig. 3 c, right panel), characterized by 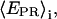 the average of
the average of 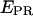 values for each single molecule
values for each single molecule 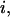 taking into account only the time points where the donor and acceptor probes are active.
taking into account only the time points where the donor and acceptor probes are active. 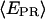 histograms for RPitc,≤2, Cy5 + 20 (Fig. 4, top), show a distribution centered around
histograms for RPitc,≤2, Cy5 + 20 (Fig. 4, top), show a distribution centered around 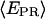 = 0.29. Upon addition of ATP, GTP, and UTP (80 μM) to form the first stable elongation complex (RDe,11), a second D-A subpopulation appears at higher
= 0.29. Upon addition of ATP, GTP, and UTP (80 μM) to form the first stable elongation complex (RDe,11), a second D-A subpopulation appears at higher 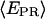 (Fig. 4, bottom). The new subpopulation represents transcription complexes that have translocated downstream to form the first stable elongation complex, whereas the subpopulation with
(Fig. 4, bottom). The new subpopulation represents transcription complexes that have translocated downstream to form the first stable elongation complex, whereas the subpopulation with 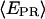 identical to that of the open complex represent inactive open complexes.
identical to that of the open complex represent inactive open complexes.
FIGURE 4.

Promoter escape leading edge FRET (Cy5, +20); 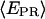 distributions, calculated by averaging
distributions, calculated by averaging 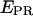 values for each single complex, taking into account only the time points where donor and acceptor probes are active. Transition from initiation (top) to elongation (bottom) results in the appearance of a new population at
values for each single complex, taking into account only the time points where donor and acceptor probes are active. Transition from initiation (top) to elongation (bottom) results in the appearance of a new population at 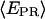 ≈ 0.61. Inactive complexes are seen as an immobile population at
≈ 0.61. Inactive complexes are seen as an immobile population at 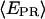 ≈ 0.3. Translocation activity is 72%.
≈ 0.3. Translocation activity is 72%.
Fitting of the data by two Gaussian functions allows us to quantify the relative fractions of active and inactive transcription complexes. The assignment of “activity” ratio is based on the ability of a transcription complex to translocate downstream and enter elongation (and, as a consequence, yield species with high LE-FRET compared to the open complex); the activity is expressed as the fraction of complexes present in the forward translocated population (see Materials and Methods; Eq. 1). In all cases, the translocational activity was >65%, consistent with the activity observed in ensemble measurements (64%) (9). The high activity clearly demonstrates that our immobilization conditions do not perturb the transcription system under study.
Promoter escape: retention of σ70
To estimate the retention of σ70 upon transition from initiation to elongation, we counted A-only molecules (i.e., DNA alone or DNA-RNAP complex) and D-A molecules (i.e., DNA-RNAP-σ70 complexes), in the context of RPitc,≤2 and RDe,11 (see Materials and Methods; (7)). In all cases, σ70 retention was >80%, consistent with earlier ensemble studies (8–10) and a recent single-molecule study (7). In these previous investigations, it has not been possible to establish unequivocally that the σ70 molecule present in RDe,11 complex was the same σ70 molecule that had been present in RPitc,≤2 (with translocation of that σ70 molecule, as opposed to dissociation of that σ70 molecule and rebinding of a different σ70 molecule, as suggested (30,31)). In this work, it is possible unequivocally to establish that the σ70 molecule present in RDe,11 complex is the same σ70 molecule that had been present in RPo. Indeed, the transition from initiation to elongation is triggered under conditions where σ70 rebinding can be excluded: first, immobilization of RPitc,≤2 is performed at very low concentration (<100 pM), followed by washes that remove unbound complexes; second, in the event of σ70 dissociation, any free σ70 would be present in the solution at concentrations several orders of magnitude below its Kd for the elongation complex (≈2 μM; (32)), and thus will not rebind to the elongation complex. Thus, we conclude that, under our conditions in vitro, the single σ70 molecule associated to the initiation complex remains associated and translocates with the initial elongation complex.
Abortive initiation: detection
For detection of abortive initiation, different NTPs subsets were added to immobilized RPitc,≤2 complexes to generate RPitc,≤4, RPitc,≤7 (initial transcribing complexes “locked” in abortive initiation), and RDe,11 (the first stable elongation complex) (Fig. 5). Data were analyzed by fitting the RPitc,≤2 histogram to a single Gaussian, followed by fitting the RPitc,≤4 and RPitc,≤7 histograms with a two-Gaussian function; in the second fit, one Gaussian function was constrained to have the mean and width values for RPitc,≤2, and an amplitude equal to the fraction of open complexes that fail to enter elongation (calculated using the RDe,11 data). In this calculation, we assume that the fraction of molecules that do not escape from the promoter are also not engaged in abortive initiation. This assumption is supported by the observation of time traces for individual molecules (see below). From this analysis, and visual inspection of the distributions, we show that successive NTP additions during abortive initiation and promoter escape shift the mean of the 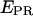 distribution toward higher values (Fig. 5 e). This means that, during abortive initiation, the RNAP leading edge translocates forward relative to the downstream DNA, consistent with DNA footprinting data (33,34). We note that this analysis can only detect relative motion of one macromolecule versus the other; it does not settle the question of whether the mobile element during abortive initiation is mainly RNAP or DNA.
distribution toward higher values (Fig. 5 e). This means that, during abortive initiation, the RNAP leading edge translocates forward relative to the downstream DNA, consistent with DNA footprinting data (33,34). We note that this analysis can only detect relative motion of one macromolecule versus the other; it does not settle the question of whether the mobile element during abortive initiation is mainly RNAP or DNA.
FIGURE 5.

Abortive initiation (a–d). (Left) Schematic diagram depicting the heterogeneity and dynamic behavior of the RPitc,≤2, RPitc,≤4, and RPitc,≤7 complexes engaged in abortive cycling (double-headed black arrows), as opposed to the static nature of the RDe,11. (Center and right) 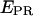 histograms (for all time points where donor and acceptor probes are active) of D-A complexes for (a) RPitc,≤2, (b) RPitc,≤4, (c) RPitc,≤7, and (d) RDe,11, for LE-FRET experiments (Cy5, +20, and +25). Data were analyzed by fitting the RPitc,≤2 histogram to a single Gaussian, followed by fitting the RPitc,≤4 and RPitc,≤7 histograms with a two-Gaussian function; in the second fit, one Gaussian function was constrained to have the mean and width values for RPitc,≤2, and an amplitude equal to the fraction of complexes that fail to enter elongation (see text). The recovered values for the center and width of the
histograms (for all time points where donor and acceptor probes are active) of D-A complexes for (a) RPitc,≤2, (b) RPitc,≤4, (c) RPitc,≤7, and (d) RDe,11, for LE-FRET experiments (Cy5, +20, and +25). Data were analyzed by fitting the RPitc,≤2 histogram to a single Gaussian, followed by fitting the RPitc,≤4 and RPitc,≤7 histograms with a two-Gaussian function; in the second fit, one Gaussian function was constrained to have the mean and width values for RPitc,≤2, and an amplitude equal to the fraction of complexes that fail to enter elongation (see text). The recovered values for the center and width of the 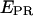 distribution for the active molecules are: for Cy5, +20 (center (width)), RPitc,≤2, 0.38 (0.45); RPitc,≤4, 0.51 (0.43); RPitc,≤7, 0.58 (0.45); and RDe,11, 0.68 (0.31); for Cy5, +25 (center (width)), RPitc,≤2, 0.11 (0.30); RPitc,≤4, 0.24 (0.31); RPitc,≤7, 0.29 (0.31); and RDe,11, 0.51 (0.31). The vertical dotted line represents the mean value of the
distribution for the active molecules are: for Cy5, +20 (center (width)), RPitc,≤2, 0.38 (0.45); RPitc,≤4, 0.51 (0.43); RPitc,≤7, 0.58 (0.45); and RDe,11, 0.68 (0.31); for Cy5, +25 (center (width)), RPitc,≤2, 0.11 (0.30); RPitc,≤4, 0.24 (0.31); RPitc,≤7, 0.29 (0.31); and RDe,11, 0.51 (0.31). The vertical dotted line represents the mean value of the 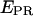 distribution for RPitc,≤2. (e) Recovered values for the
distribution for RPitc,≤2. (e) Recovered values for the 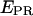 and D-A distances for the active molecules, as a function of the RNA product length (▵, Cy5, +20; ▪, Cy5, +25). For both constructs, transition from RPitc,≤2 to RPitc,≤4, to RPitc,≤7 to RDe,11 leads to an increase in the
and D-A distances for the active molecules, as a function of the RNA product length (▵, Cy5, +20; ▪, Cy5, +25). For both constructs, transition from RPitc,≤2 to RPitc,≤4, to RPitc,≤7 to RDe,11 leads to an increase in the 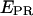 value (left), consistent with a decrease in distance between the leading edge of the enzyme and the downstream DNA (right).
value (left), consistent with a decrease in distance between the leading edge of the enzyme and the downstream DNA (right).
Abortive initiation: identification of the rate-limiting step
In addition to 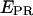 distributions, msALEX-DI has the unique capability to generate time traces of
distributions, msALEX-DI has the unique capability to generate time traces of 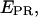 i.e.,
i.e., 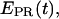 for individual complexes, free from complication due to compositional and photophysical heterogeneity. Representative
for individual complexes, free from complication due to compositional and photophysical heterogeneity. Representative 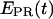 traces for RPitc,≤2, RPitc,≤7, and RDe,11 with Cy5 at position +25 are presented in Fig. 6. In Fig. 6 a, RPitc,≤2 complexes typically show a narrow distribution of
traces for RPitc,≤2, RPitc,≤7, and RDe,11 with Cy5 at position +25 are presented in Fig. 6. In Fig. 6 a, RPitc,≤2 complexes typically show a narrow distribution of 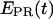 values (range 0.0–0.3), with
values (range 0.0–0.3), with 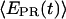 ∼ 0.1–0.2. This indicates that the leading edge of the enzyme is relatively immobile relative to the downstream DNA in this initial complex. For RPitc,≤7 complexes (Fig. 6 b),
∼ 0.1–0.2. This indicates that the leading edge of the enzyme is relatively immobile relative to the downstream DNA in this initial complex. For RPitc,≤7 complexes (Fig. 6 b), 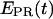 values are more widely distributed (range 0.0–0.8), with the majority of values in the 0.35–0.60 range; the distribution of points increases mean
values are more widely distributed (range 0.0–0.8), with the majority of values in the 0.35–0.60 range; the distribution of points increases mean 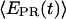 to ∼0.30–0.45. Thus, at this saturating concentration of NTPs, the RNAP leading edge spends most of the time in a forward translocated state relative to downstream DNA, and not in the open complex state where RNA synthesis has not yet started (for which
to ∼0.30–0.45. Thus, at this saturating concentration of NTPs, the RNAP leading edge spends most of the time in a forward translocated state relative to downstream DNA, and not in the open complex state where RNA synthesis has not yet started (for which 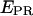 ≈ 0.1). Finally, for RDe,11 complexes, a more narrow
≈ 0.1). Finally, for RDe,11 complexes, a more narrow 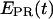 distribution is observed (
distribution is observed (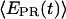 ∼ 0.5–0.6), demonstrating, as expected, that promoter escape has occurred, a stable elongation state has been reached, and that the RNAP does not translocate backward anymore. Similar results for all complexes were obtained with Cy5 at position +20 (data not shown). We note that the higher apparent noise of
∼ 0.5–0.6), demonstrating, as expected, that promoter escape has occurred, a stable elongation state has been reached, and that the RNAP does not translocate backward anymore. Similar results for all complexes were obtained with Cy5 at position +20 (data not shown). We note that the higher apparent noise of 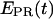 for RPitc,≤7 in Fig. 6 b is possibly due to cycles of forward and reverse active-center translocation, currently unresolved with the available temporal resolution (400 ms per frame). We also note the presence of a significant fraction of inactive complexes on the time traces. These complexes, observed in the context of RPitc,≤7 and RDe,11, are characterized by a narrow distribution of
for RPitc,≤7 in Fig. 6 b is possibly due to cycles of forward and reverse active-center translocation, currently unresolved with the available temporal resolution (400 ms per frame). We also note the presence of a significant fraction of inactive complexes on the time traces. These complexes, observed in the context of RPitc,≤7 and RDe,11, are characterized by a narrow distribution of 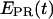 values around 0.1 (data not shown), similar to traces typically obtained with RPitc,≤2 (Fig. 6 a). From the presence of these complexes in the context of RPitc,≤7, we infer that the majority of inactive molecules that do not escape from the promoter are also not engaged in abortive initiation.
values around 0.1 (data not shown), similar to traces typically obtained with RPitc,≤2 (Fig. 6 a). From the presence of these complexes in the context of RPitc,≤7, we infer that the majority of inactive molecules that do not escape from the promoter are also not engaged in abortive initiation.
FIGURE 6.

Single-molecule time traces 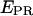 traces are plotted as a function of time, for different representative complexes (Cy5, +25). Only time points with active donors and acceptors are shown. The average
traces are plotted as a function of time, for different representative complexes (Cy5, +25). Only time points with active donors and acceptors are shown. The average 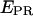 values obtained for RPitc,≤2 and RDe,11 (0.11 and 0.5, respectively) are represented by two horizontal lines to guide the eye. (a) RPitc,≤2, static distribution with
values obtained for RPitc,≤2 and RDe,11 (0.11 and 0.5, respectively) are represented by two horizontal lines to guide the eye. (a) RPitc,≤2, static distribution with 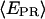 ∼ 0.11 (see Fig. 5 a). (b) RPitc,≤7, scattered distribution with a majority of time points distributed around a relatively high
∼ 0.11 (see Fig. 5 a). (b) RPitc,≤7, scattered distribution with a majority of time points distributed around a relatively high 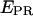 (t) value (distribution centered around
(t) value (distribution centered around 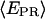 = 0.3 for the active molecules (see Fig. 5 c)), consistent with the forward translocation of the leading edge of RNAP relative to downstream DNA during abortive initiation. (c) RDe,11, static distribution with
= 0.3 for the active molecules (see Fig. 5 c)), consistent with the forward translocation of the leading edge of RNAP relative to downstream DNA during abortive initiation. (c) RDe,11, static distribution with 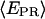 ∼ 0.5 (see Fig. 5 d).
∼ 0.5 (see Fig. 5 d).
The analysis of the 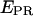 histograms for initial transcribing complexes indicates that the width of the
histograms for initial transcribing complexes indicates that the width of the 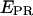 distribution for the active molecules does not change between RPitc,≤2, RPitc,≤4, and RPitc,≤7 (Fig. 5, a–c). This indication, and the displacement of the mean of the distribution toward higher
distribution for the active molecules does not change between RPitc,≤2, RPitc,≤4, and RPitc,≤7 (Fig. 5, a–c). This indication, and the displacement of the mean of the distribution toward higher 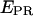 values (Fig. 5 e) also suggest that, at saturating or near-saturating NTP concentrations (80 μM), the active complexes engaged in abortive initiation spend most of the time in states with forward translocation of the RNAP leading edge relative to downstream DNA. In addition, preliminary experiments conducted at nonsaturating NTP concentration (5 μM) have shown lower
values (Fig. 5 e) also suggest that, at saturating or near-saturating NTP concentrations (80 μM), the active complexes engaged in abortive initiation spend most of the time in states with forward translocation of the RNAP leading edge relative to downstream DNA. In addition, preliminary experiments conducted at nonsaturating NTP concentration (5 μM) have shown lower 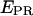 values for the mean of the distribution of initial transcribing complexes, reflecting the fact that in this case the RNAP spends more time incorporating NTPs in states without, or with less, forward translocation of the RNAP leading edge.
values for the mean of the distribution of initial transcribing complexes, reflecting the fact that in this case the RNAP spends more time incorporating NTPs in states without, or with less, forward translocation of the RNAP leading edge.
From the time traces, the 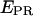 distributions, and the observed NTP-concentration dependence, we infer that, at saturating NTP concentration, the RNAP leading edge spends most of the time in a forward translocated state relative to downstream DNA. Thus, we infer that abortive-product synthesis and RNAP-active-center forward translocation must be fast relative to abortive-product dissociation and RNAP-active-center reverse translocation. We conclude that the abortive-product dissociation and RNAP-active-center reverse translocation are the rate-limiting steps of the abortive cycling process. At the achieved time resolution, however, the release of one abortive product, and the next cycle of abortive-product synthesis have not been resolved.
distributions, and the observed NTP-concentration dependence, we infer that, at saturating NTP concentration, the RNAP leading edge spends most of the time in a forward translocated state relative to downstream DNA. Thus, we infer that abortive-product synthesis and RNAP-active-center forward translocation must be fast relative to abortive-product dissociation and RNAP-active-center reverse translocation. We conclude that the abortive-product dissociation and RNAP-active-center reverse translocation are the rate-limiting steps of the abortive cycling process. At the achieved time resolution, however, the release of one abortive product, and the next cycle of abortive-product synthesis have not been resolved.
Abortive initiation and promoter escape: measurement of distances within active complexes
We previously have demonstrated on single diffusing molecules that the use of alternating-laser excitation allows recovery of accurate FRET efficiencies E and thus calculation of accurate D-A distances (20). Here, we use the same approach on immobilized molecules to convert the proximity ratio, 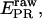 into the FRET efficiency, E. This was achieved by using the unique ability of msALEX-DI to sort D-only, A-only, and D-A species on a
into the FRET efficiency, E. This was achieved by using the unique ability of msALEX-DI to sort D-only, A-only, and D-A species on a 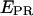 -S histogram, and to extract cross-talk terms and detection correction factors needed to perform this conversion, as shown in Fig. 7 and described in the Materials and Methods section. For RPitc,≤2 and RDe,11, and Cy5 at position +25, the recovered E-values and the corresponding D-A distances are presented in Table 1, together with the distances obtained using ALEX in solution (7,20), and ensemble measurements (9). The distances obtained for the immobilized complexes are in excellent agreement (within 5 Å) with those obtained from previous measurements. This agreement validates the accurate-FRET calculations using msALEX-DI, and thus offers a robust method for measuring accurate FRET on single immobilized molecules.
-S histogram, and to extract cross-talk terms and detection correction factors needed to perform this conversion, as shown in Fig. 7 and described in the Materials and Methods section. For RPitc,≤2 and RDe,11, and Cy5 at position +25, the recovered E-values and the corresponding D-A distances are presented in Table 1, together with the distances obtained using ALEX in solution (7,20), and ensemble measurements (9). The distances obtained for the immobilized complexes are in excellent agreement (within 5 Å) with those obtained from previous measurements. This agreement validates the accurate-FRET calculations using msALEX-DI, and thus offers a robust method for measuring accurate FRET on single immobilized molecules.
TABLE 1.
FRET efficiencies (E) and calculated distances (RD-A) for the RPitc,2 and RDe,11 complexes, with Cy5 at position +25; comparison with previous measurements
In addition, on Fig. 5 e (right) are presented the RD-A distances recovered for RPitc,≤2, RPitc,≤4, RPitc,≤7, and RDe,11, with Cy5 at position +20 and +25. As expected for a LE-FRET experiment, RD-A is shorter for Cy5 at +20 than for Cy5 at position +25, and its value decreases for each NTP addition. We note that the recovered distances for RPitc,≤2 and RDe,11 represent a reliable estimate of the interdye distance due to the relatively static nature of these complexes. However, the RD-A value recovered for RPitc,≤4 and RPitc,≤7 involves a mixture of different complexes, each with a different RD-A. In this case, the recovered distances should be considered as a qualitative measurement of different distances, even if our data show that the most forward translocated state is likely the most populated.
CONCLUSION
msALEX-DI
In this work, we describe a new methodology based on alternating-laser excitation of single immobilized molecules and its applications to the study of transcription initiation by RNA polymerase. The msALEX-DI methodology provides a general platform for studying association/dissociation events and conformational changes within single immobilized biomolecular complexes; the methodology is fully compatible with epifluorescence microscopy. However, by using a prism-based total internal reflection microscopy a better signal/background ratio is achieved, mainly because the excitation light does not propagate toward the detector and the autofluorescence from the optics and sample is reduced (35). As compared to the standard single-laser excitation microscopy of single immobilized molecules, we note the following advantages:
The ability to separate complexes with low
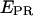 from D-only complexes. This ability permits monitoring of association events that form a complex, even when the donor-acceptor distance in the resulting complex exceeds considerably the dynamic range of FRET.
from D-only complexes. This ability permits monitoring of association events that form a complex, even when the donor-acceptor distance in the resulting complex exceeds considerably the dynamic range of FRET.The ability to identify and remove inactive states of the acceptor from
 time traces, extending the lower observable
time traces, extending the lower observable 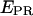 limit to zero, and thus increasing the distance scale where conformational changes can be studied (e.g., when
limit to zero, and thus increasing the distance scale where conformational changes can be studied (e.g., when 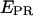 equals zero in one of the conformational states, due to a long donor-acceptor distance). Inactive states of the acceptor are frequently observed in single-molecule FRET experiments, especially for carbocyanine fluorophores (e.g., Cy5 or Alexa 647); in fact, these dyes can even act as efficient reversible single-molecule optical switches, whose fluorescent state after apparent photobleaching can be restored upon irradiation at shorter wavelengths (36,37).
equals zero in one of the conformational states, due to a long donor-acceptor distance). Inactive states of the acceptor are frequently observed in single-molecule FRET experiments, especially for carbocyanine fluorophores (e.g., Cy5 or Alexa 647); in fact, these dyes can even act as efficient reversible single-molecule optical switches, whose fluorescent state after apparent photobleaching can be restored upon irradiation at shorter wavelengths (36,37).The ability to calculate accurate FRET efficiencies and corresponding distances within single immobilized transcription complexes, after performing corrections as described before for single diffusing complexes (20). We note that the presence of fast conformational changes (in the fluorescence timescale (0.1–10 ns)) of the segments labeled with the FRET probes could bias these distance measurements. Although it is not possible to rule out these kinds of fluctuation with the msALEX-DI technique, additional experiments in solution can be performed using, for example, nsALEX (16), which allows us to perform alternating-laser excitation measurements with two interlaced pulsed lasers. At the nanosecond timescale, conformational dynamics resulting in different D-A distances could be detected, because they would appear as a multiexponential decay of the donor lifetime.
Promoter escape
msALEX-DI has allowed us to study immobilized transcription complexes, from transcription initiation to early transcription elongation. Our studies were aided by an immobilization strategy that allowed >65% of the complexes to undergo the transition from initiation to elongation; this high level of translocational activity matches the activity observed in ensemble-FRET studies in solution (9).
Our promoter-escape studies settle the important mechanistic question of whether the single molecule of σ70 present in transcription elongation is the same molecule present in transcription initiation (as opposed to a molecule of σ70 that binds to a σ70-free elongation complex formed after promoter escape). This is due to two facts: first, all free σ70 is removed during immobilization of the complexes, and second, any free σ70 resulting from release during promoter escape would result in exceedingly small σ70 concentrations in the reaction solution (≪10 pM, many orders of magnitude lower than the Kd of 2 μM for the interaction of σ70 for the elongation complex (31)). Our observations of high σ70 retention in elongation in the absence of free σ70 in solution clearly establish that the observed σ70 retention reflects retention of σ70 upon transition to elongation and translocation of σ70 with RNAP. We note that this property of σ70 is fully compatible with its property to bind σ70-free elongation complexes formed at later stages during elongation (31). Finally, our results establish that the presence of σ70 in the transcription complex is fully compatible with the process of promoter escape, and raise the possibility that σ70 may be an active participant in this process.
Abortive initiation
The high activity of the immobilized transcription complexes also enabled us to study abortive initiation, the mode of initial RNA synthesis during which RNAP synthesizes and releases short RNA products. The mechanism of abortive initiation is still unsolved, mainly due to its asynchronous nature that presents a challenge for conventional experimental methods, such as detection of abortive RNA products and chemical footprinting (33,34). Our single-molecule approach overcame the problem of synchronization by the first real-time observations of single transcription complexes involved in abortive initiation. Thus, we have been able to show that abortive initiation involves the forward translocation of the RNAP leading edge relative to downstream DNA. Moreover, our results establish that, during the iterative abortive synthesis at saturating NTP concentrations, the transcription complex spends the majority of the time in states with forward translocation of the RNAP leading edge relative to downstream DNA. This implies that the abortive-product release and RNAP-active-center reverse translocation are the rate-limiting steps, and represents the first available information regarding kinetics of individual reaction in abortive initiation on a multisubunit RNA polymerase. Interestingly, similar observations have been made on a single-subunit RNAP (T7 RNA polymerase) using exonuclease and KMnO4 footprinting (38), and kinetics analysis of RNA synthesis (39).
The fact that the RNAP spends most of its time in states with forward translocation of the RNAP leading edge relative to downstream DNA should permit structural analysis of those states. By incorporating pairs of donor and acceptor probes at various positions on RNAP, on DNA, or on RNAP and DNA, and measuring the distances in complexes engaged in iterative abortive synthesis, it should be possible to distinguish among the three models proposed for the mechanism of abortive synthesis: “RNAP inchworming” (which predicts conformational changes within RNAP during abortive initiation), “DNA scrunching” (which predicts DNA compaction during abortive initiation), and “transient excursions” (which predicts changes in distance between the trailing edge of RNAP and DNA) (A. Kapanidis, E. Margeat, S. Weiss, and R. H. Ebright, unpublished data).
Prospect
The methodologies presented in this article will allow real-time, single-molecule observations of the transitions between various states of the transcription complex throughout transcription, such as the transition from the close to the open complex, or the observation of the promoter escape. Higher temporal resolution, which is necessary for monitoring some of these transitions, can be obtained by combining the specific immobilization scheme presented here and confocal detection of single complexes (using avalanche photodiodes, which afford a better signal/noise ratio and time resolution than CCD cameras), or through use of faster and more sensitive cameras. The generality of ALEX-based methods, on immobilized or freely diffusing molecules (16), and their extension to three-color excitation and detection (N. Lee, A. Kapanidis, and S. Weiss, unpublished data) will pave the way for new experiments that monitor the kinetic coordination of multiple transitions within single macromolecular complexes and help unlock the mechanisms of transcription.
Acknowledgments
We thank Sören Doose, Sam Ho, Nam-Ki Lee, and Xavier Michalet for their advice and assistance on data acquisition and analysis, Taekjip Ha and Michelle Nahas for advice on immobilization chemistry and the prism-based TIR, Ekaterine Kortkhonjia for Ro measurement, and Fabien Pinaud and Nathalie Marcotte for discussion and suggestions.
This work was funded by U.S. Department of Energy grants 02ER63339 and 04ER63938, National Institutes of Health grant GM069709-01A1 (S.W.), National Institutes of Health grant GM41376 (R.H.E.), and a Howard Hughes Medical Institute Investigatorship (R.H.E).
Emmanuel Margeat's present address is Centre de Biochimie Structurale, CNRS UMR 5048, INSERM UMR 554, Université Montpellier I, 29 rue de Navacelles, 34090 Montpellier cedex, France.
Achillefs N. Kapanidis's present address is Dept. of Physics, Clarendon Laboratory, University of Oxford, Parks Rd., Oxford, OX1 3PU, UK.
Philip Tinnefeld's present address is Applied Laser Physics & Laser Spectroscopy, Physics Faculty, University of Bielefeld, Germany.
References
- 1.Record, M. T. Jr., W. Reznikoff, M. Craig, K. McQuade, and P. Schlax. 1996. Escherichia coli and Salmonella. F. C. Neidhart, editor. ASM Press, Washington, DC.
- 2.deHaseth, P. L., M. L. Zupancic, and M. T. Record, Jr. 1998. RNA polymerase-promoter interactions: the comings and goings of RNA polymerase. J. Bacteriol. 180:3019–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsu, L. M. 2002. Promoter clearance and escape in prokaryotes. Biochim. Biophys. Acta. 1577:191–207. [DOI] [PubMed] [Google Scholar]
- 4.Murakami, K. S., and S. A. Darst. 2003. Bacterial RNA polymerases: the wholo story. Curr. Opin. Struct. Biol. 13:31–39. [DOI] [PubMed] [Google Scholar]
- 5.Young, B. A., T. M. Gruber, and C. A. Gross. 2002. Views of transcription initiation. Cell. 109:417–420. [DOI] [PubMed] [Google Scholar]
- 6.Lilley, D. M., and T. J. Wilson. 2000. Fluorescence resonance energy transfer as a structural tool for nucleic acids. Curr. Opin. Chem. Biol. 4:507–517. [DOI] [PubMed] [Google Scholar]
- 7.Kapanidis, A. N., E. Margeat, T. Laurence, S. Doose, S. O. Ho, J. Mukhopadhyay, E. Kortkhonjia, R. Ebright, and S. Weiss. 2005. Retention of transcription initiation factor σ70 in transcription elongation complexes: single molecule analysis. Mol. Cell. 20:347–356. [DOI] [PubMed] [Google Scholar]
- 8.Mukhopadhyay, J., V. Mekler, E. Kortkhonjia, A. N. Kapanidis, Y. W. Ebright, and R. H. Ebright. 2003. Fluorescence resonance energy transfer (FRET) in analysis of transcription-complex structure and function. Methods Enzymol. 371:144–159. [DOI] [PubMed] [Google Scholar]
- 9.Mukhopadhyay, J., A. N. Kapanidis, V. Mekler, E. Kortkhonjia, Y. W. Ebright, and R. H. Ebright. 2001. Translocation of sigma(70) with RNA polymerase during transcription: fluorescence resonance energy transfer assay for movement relative to DNA. Cell. 106:453–463. [DOI] [PubMed] [Google Scholar]
- 10.Nickels, B. E., J. Mukhopadhyay, S. J. Garrity, R. H. Ebright, and A. Hochschild. 2004. The sigma(70) subunit of RNA polymerase mediates a promoter-proximal pause at the lac promoter. Nat. Struct. Mol. Biol. 11:544–550. [DOI] [PubMed] [Google Scholar]
- 11.Travers, A. A., and R. R. Burgess. 1969. Cyclic re-use of the RNA polymerase sigma factor. Nature. 222:537–540. [DOI] [PubMed] [Google Scholar]
- 12.Hansen, U. M., and W. R. McClure. 1980. Role of the sigma subunit of Escherichia coli RNA polymerase in initiation. II. Release of sigma from ternary complexes. J. Biol. Chem. 255:9564–9570. [PubMed] [Google Scholar]
- 13.Straney, D. C., and D. M. Crothers. 1985. Intermediates in transcription initiation from the E. coli lac UV5 promoter. Cell. 43:449–459. [DOI] [PubMed] [Google Scholar]
- 14.Krummel, B., and M. J. Chamberlin. 1989. RNA chain initiation by Escherichia coli RNA polymerase. Structural transitions of the enzyme in early ternary complexes. Biochemistry. 28:7829–7842. [DOI] [PubMed] [Google Scholar]
- 15.Metzger, W., P. Schickor, T. Meier, W. Werel, and H. Heumann. 1993. Nucleation of RNA chain formation by Escherichia coli DNA-dependent RNA polymerase. J. Mol. Biol. 232:35–49. [DOI] [PubMed] [Google Scholar]
- 16.Kapanidis, A. N., T. A. Laurence, N. K. Lee, E. Margeat, X. Kong, and S. Weiss. 2005. Alternating-laser excitation of single molecules. Acc. Chem. Res. 37:523–533. [DOI] [PubMed] [Google Scholar]
- 17.Kapanidis, A., N.-K. Lee, T. Laurence, S. Doose, E. Margeat, and S. Weiss. 2004. Fluorescence-aided molecule sorting. Analysis of structure and interactions by alternating laser excitation of single molecules. Proc. Natl. Acad. Sci. USA. 101:8936–8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yildiz, A., J. N. Forkey, S. A. McKinney, T. Ha, Y. E. Goldman, and P. R. Selvin. 2003. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 300:2061–2065. [DOI] [PubMed] [Google Scholar]
- 19.Kinosita, K., H. Itoh, S. Ishiwata, K. Hirano, T. Nishizaka, and T. Hayakawa. 1991. Dual-view microscopy with a single camera: real-time imaging of molecular orientations and calcium. J. Cell Biol. 115:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee, N. K., A. N. Kapanidis, Y. Wang, X. Michalet, J. Mukhopadhyay, R. H. Ebright, and S. Weiss. 2005. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys. J. 88:2939–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mekler, V., E. Kortkhonjia, J. Mukhopadhyay, J. Knight, A. Revyakin, A. N. Kapanidis, W. Niu, Y. W. Ebright, R. Levy, and R. H. Ebright. 2002. Structural organization of bacterial RNA polymerase holoenzyme and the RNA polymerase-promoter open complex. Cell. 108:599–614. [DOI] [PubMed] [Google Scholar]
- 22.Clegg, R. M. 1992. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 211:353–388. [DOI] [PubMed] [Google Scholar]
- 23.Ha, T., I. Rasnik, W. Cheng, H. P. Babcock, G. H. Gauss, T. M. Lohman, and S. Chu. 2002. Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature. 419:638–641. [DOI] [PubMed] [Google Scholar]
- 24.Zhuang, X., L. E. Bartley, H. P. Babcock, R. Russell, T. Ha, D. Herschlag, and S. Chu. 2000. A single-molecule study of RNA catalysis and folding. Science. 288:2048–2051. [DOI] [PubMed] [Google Scholar]
- 25.Axelrod, D. 2001. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2:764–774. [DOI] [PubMed] [Google Scholar]
- 26.Wu, P., and L. Brand. 1992. Orientation factor in steady-state and time-resolved resonance energy transfer measurements. Biochemistry. 31:7939–7947. [DOI] [PubMed] [Google Scholar]
- 27.Ha, T., A. Y. Ting, J. Liang, A. A. Deniz, D. S. Chemla, P. G. Schultz, and S. Weiss. 1999. Temporal fluctuations of fluorescence resonance energy transfer between two dyes conjugated to a single protein. Chem. Phys. 247:107–118. [Google Scholar]
- 28.Rhoades, E., M. Cohen, B. Schuler, and G. Haran. 2004. Two-state folding observed in individual protein molecules. J. Am. Chem. Soc. 126:14686–14687. [DOI] [PubMed] [Google Scholar]
- 29.Blanchard, S. C., R. L. Gonzalez, H. D. Kim, S. Chu, and J. D. Puglisi. 2004. tRNA selection and kinetic proofreading in translation. Nat. Struct. Mol. Biol. 11:1008–1014. [DOI] [PubMed] [Google Scholar]
- 30.Brodolin, K., N. Zenkin, A. Mustaev, D. Mamaeva, and H. Heumann. 2004. The σ70 subunit of RNA polymerase induces lacUV5 promoter-proximal pausing of transcription. Nat. Struct. Mol. Biol. 11:551–557. [DOI] [PubMed] [Google Scholar]
- 31.Mooney, R. A., and R. Landick. 2003. Tethering sigma70 to RNA polymerase reveals high in vivo activity of sigma factors and sigma70-dependent pausing at promoter-distal locations. Genes Dev. 17:2839–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gill, S. C., S. E. Weitzel, and P. H. von Hippel. 1991. Escherichia coli sigma 70 and NusA proteins. I. Binding interactions with core RNA polymerase in solution and within the transcription complex. J. Mol. Biol. 220:307–324. [DOI] [PubMed] [Google Scholar]
- 33.Carpousis, A. J., and J. D. Gralla. 1985. Interaction of RNA polymerase with lacUV5 promoter DNA during mRNA initiation and elongation. Footprinting, methylation, and rifampicin-sensitivity changes accompanying transcription initiation. J. Mol. Biol. 183:165–177. [DOI] [PubMed] [Google Scholar]
- 34.Carpousis, A. J., and J. D. Gralla. 1980. Cycling of ribonucleic acid polymerase to produce oligonucleotides during initiation in vitro at the lac UV5 promoter. Biochemistry. 19:3245–3253. [DOI] [PubMed] [Google Scholar]
- 35.Ha, T. 2001. Single-molecule fluorescence resonance energy transfer. Methods. 25:78–86. [DOI] [PubMed] [Google Scholar]
- 36.Heilemann, M., E. Margeat, R. Kasper, M. Sauer, and P. Tinnefeld. 2005. Carbocyanine dyes as efficient reversible single-molecule optical switch. J. Am. Chem. Soc. 127:3801–3806. [DOI] [PubMed] [Google Scholar]
- 37.Bates, M., T. R. Blosser, and X. Zhuang. 2005. Short-range spectroscopic ruler based on a single-molecule optical switch. Phys. Rev. Lett. 94:108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brieba, L. G., and R. Sousa. 2001. T7 promoter release mediated by DNA scrunching. EMBO J. 20:6826–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia, Y., and S. S. Patel. 1997. Kinetic mechanism of transcription initiation by bacteriophage T7 RNA polymerase. Biochemistry. 36:4223–4232. [DOI] [PubMed] [Google Scholar]