

Abstract
We use molecular dynamics (MD) simulations to understand the structure and stability of various paranemic crossover (PX) DNA molecules, synthesized recently by Seeman and co-workers at New York University. These studies include all atoms of the PX structures with an explicit description of solvent and ions. The average dynamics structures over the last 1 ns of the 3-ns simulation preserve the Watson-Crick hydrogen bonding as well as the helical structure. The root mean-square deviation in coordinates with respect to the MD averaged structure converges to 2–3 Å for PX55, PX65, and PX85, but for PX75 and PX95 the root mean-square deviation in coordinates exhibits large fluctuations, indicating an intrinsic instability. The PX structures are structurally more rigid compared to the canonical B-DNA without crossover. We have developed a strain energy analysis method based on the nearest-neighbor interaction and computed the strain energy for the PX molecules compared to the B-DNA molecules of the same length and sequence. PX65 has the lowest calculated strain energy (∼−0.77 kcal/mol/bp), and the strain increases dramatically for PX75, PX85, and PX95. PX55 has the highest strain energy (∼1.85 kcal/mol/bp) making it unstable, which is in accordance with the experimental results. We find that PX65 has helical twist and other helical structural parameters close to the values for normal B-DNA of similar length and sequence. Vibrational mode analysis shows that compared to other PX motifs, PX65 has the smallest population of the low-frequency modes that are dominant contributors for the conformational entropy of the PX DNA structures. All these results indicate that PX65 is structurally more stable compared to other PX motifs, in agreement with experiments. These results should aid in designing optimized DNA structures for use in nanoscale components and devices.
INTRODUCTION
A major goal in biotechnology is to create self-assembling nanostructures that utilize the informational and signal transduction capabilities of proteins and nucleic acids to make useful nanoscale devices (1–6). DNA-based nanomechanical devices can be used for performing computations (7–9) and mechanical work (translation and rotation) (10,11), and as sensors detecting specific molecules (12,13). The progress over the last decade in atomic force microscopy and scanning tunneling microscopy manipulation and in designing submicron templates and self-assembling systems based on DNA provides evidence that DNA nanostructures will produce useful nanoscale devices (14–16). However, practical design and manufacture of nanoscale machines and devices requires overcoming numerous formidable hurdles in synthesis, processing, characterization, design, optimization, and fabrication of the nanocomponents. Each of these areas presents experimentalists with significant challenges because the properties of nanoscale systems differ significantly from macroscopic and molecular systems and it is difficult to manipulate and characterize structures at the nanoscale. We believe that theory and simulation can help with critical decisions in the design and interpretation of these experiments, and illustrate some of the approaches and conclusions here.
The Seeman Laboratory at New York University has made critical advances toward practical DNA nanotechnology (2,3). The branched motifs for DNA provide components for the self-assembly of 2D and 3D arrays at the nanoscale, some of which have already been made (3). Here DNA “crossover points” provide a conceptual basis for making rigid DNA motifs. These crossover points connect two double helices by connecting either strand from one double helix to either strand of the second double helix. Such crossover points connect the two flexible double helices into one rigid structure. Rigid DNA crossover units such as the DAO- and DAE-motif double-crossover (DX) molecules are critical to the construction of nanomechanical devices (17,18). The nomenclature was introduced by Seeman: D stands for double, A for antiparallel, O for the odd number of half-turns between crossovers, and E for the even number of half-turns between crossovers.
Recently, Yan et al. synthesized a new DNA motif, paranemic-crossover (PX) DNA, and its one variant, JX2 DNA, that provided the basis for a robust sequence-dependent nanomechanical device (10). JX2 is a topoisomer of PX65 without the two middle crossover points. Since the operation of this device is sequence-dependent, one can imagine an array of such devices organized so that each device would respond individually to a specific set of signals.
Potential crossover points in PX structures occur at each point where either strand from one double helix comes together with that of another (Fig. 1). Various PX nanostructures shown in Fig. 1 have been built with a varying number of nucleotides in the major and minor grooves (19). For example, PX65 contains six nucleotides in the major groove and five in the minor groove, creating a helical duplex with eleven nucleotides per helical turn. Structures that have already been synthesized in solution include PX55, PX65, PX75, PX85, and PX95 (19). These PX structures consist of four individual strands specifically designed to complement in exactly one way. The W and N notations in the center of the molecule in Fig. 1 indicate the wide- and narrow-groove juxtapositions of the two helices.
FIGURE 1.

Basepair sequences used inthe generation of PX55, PX65, PX75 PX85, and PX95.
The experimental techniques for making such nanostructures can be time-consuming and difficult to validate. Thus, atomistic simulations to predict the structural properties of nanostructures before experiments would be particularly valuable. In this article, we demonstrate how molecular dynamics (MD) simulations of structural properties of the PX DNA lead to the improved understanding of the stability of various DNA-based nanostructures. Here we include explicit water and salt to predict the thermodynamic stability and structural properties of the PX nanostructures. This is the first simulation of such large DNA-based nanostructures in explicit water at a realistic timescale.
Nondenaturing gel electrophoresis shows that these molecules are stable, with structures similar to their B-DNA forms (19). The goal of this study is to determine the relative stability of PX nanostructures and to elucidate various structural factors underlying their stability. In particular, we are concerned with how the stability of these molecules depends on the length, base sequence, and ratio of the number of basepairs between the major and minor grooves, and how the presence of crossover points and their location affect the stability and structure of these molecules. Seeman and co-workers have used intuition and empirical methods to suggest the best sequences and spacing between the major and minor grooves to build these molecules. These approaches have been successful, but we believe that the design principles concerning the optimum basepair sequence and ratios of the number of basepairs between major and minor grooves could be improved by examining the various structures using atomistic MD simulations. We expect that this will help suggest the best PX motifs to be used in various nanoscale applications.
Our studies indicate that the PX65 structure is the most stable structure among the five PX structures considered, with helicoidal properties close to those of normal B-DNA. We also find that increasing the length of the PX structure increases the writhing factor of the entire double helix. This feature should be taken into account in designing 2D arrays using PX structures.
The details of building PX nanostructures and the simulation methods are presented in Methods. The results from the MD simulation on the five PX structures are presented, analyzed, and discussed in Results and Discussion. Finally, the summary of the main results and the conclusions are given in Summary and Conclusions.
METHODS
Building atomic-level PX nanostructures
The basepair sequences used for building the PX molecules are shown in Fig. 1. Each PX structure has four independent strands and is structurally similar to parallel DX molecules (20), except for the crossovers in the PX structure. In the notation PXMN (for example, PX65), the first integer, M = 6, indicates the number of basepairs in the major groove; the second integer, N = 5, indicates the number in the minor groove. Thus, as shown in Fig. 2, PX65 has two green strands and two red strands that intertwine with each other with six crossover points. The five cases considered here (PX55, PX65 PX75, PX85, and PX95) all have five nucleotides in the minor groove, and from five to nine nucleotides in the major groove. Seeman and co-workers have used sequence symmetry minimization to model PX molecules. They have found empirically that the best spacing for the minor groove is five nucleotide pairs, but the major groove can contain six to nine nucleotide pairs (19).
FIGURE 2.

Generation of PX DNA by reciprocal exchange. This illustrates the consequences of performing a crossover at every possible juxtaposition in the same-polarity case. The result is the remarkable PX structure, drawn with green and red strands that are related to each other by a dyad axis vertical in the page. This is a paranemic joining of two backbone structures, and it is very stable. This figure is adapted from Fig. 6 of Seeman. (16).
The construction of these five PX DNA motifs was done using the nucleic acid builder program Namot2 (version 2.2.) (21). The procedure for constructing these structures is as follows:
Building the DNA double helices: We first constructed two regular B-DNA structures and accommodated a different number of basepairs per helical turn by adjusting the twist angle of a selected number of basepairs. Table 1 shows the twist angles used for building the various PX structures. We assigned the same twist angle for all the basepairs in the helical half turn. The helical rise value of 3.4 Å was used to build the PX structures.
Building the crossover points: The two double helices thus built in Namot2 were oriented so that the 5′ and 3′ ends of the double helices were parallel to the y axis and the individual helices rotated so that the desired crossover points were at the closest distance to each other (rotation angles shown in Table 2). To find this point we wrote a computer program that starts with the first crossover point and rotates the first helix in 1° increments to find the rotation leading to the shortest distance between these crossover points. Once found, the first helix is rotated by the prescribed value and held steady while the second helix is rotated and the shortest distance between the crossover points is determined. The second helix is rotated 180° more than the first helix so that the helices are arranged as shown in Fig. 2. The crossovers were then created using the “nick” and “link” commands in Namot2. These structures were saved in the Protein Database file format.
TABLE 1.
Helical twist used in building different PX molecules
| PX structure | Twist angle (°) | Basepairs per turn |
|---|---|---|
| PX55 | 36.0 | 10 |
| PX65 | 30.0 | 11 |
| PX75 | 27.7 | 12 |
| PX85 | 22.5 | 13 |
| PX95 | 20.0 | 14 |
TABLE 2.
Rotation angles used in building different PX starting structures
| Rotation (about z axis) angles (°)
|
||
|---|---|---|
| PX structure | Helix 1 | Helix 2 |
| PX55 | 75 | 255 |
| PX65 | 60 | 240 |
| PX75 | 45 | 225 |
| PX85 | 30 | 210 |
| PX95 | 15 | 195 |
Simulation details for the PX structures
All simulations reported in this article were performed using the AMBER7 software package (22) with the all-atom AMBER95 force field (FF) (23). The AMBER95 FF has already been validated for performing MD simulations of B-DNA in explicit water with salt, starting from the crystal structure (24–27). These validation studies found that the root mean-square deviation in coordinates (CRMSD) from the crystal structure for a dodecamer structure is typically < 4.0 Å. Simulations have also been performed in solution phase (28–34) where canonical B-DNA form was preserved in nanosecond-long unrestrained MD simulations using the AMBER95 FF.
The electrostatic interactions were calculated with the particle mesh Ewald method (35,36) using a cubic B-spline interpolation of order 4 and a 10−4 tolerance set for the direct-space sum cutoff. A real-space cutoff of 9 Å was used for both the electrostatic and van der Waals interactions with a nonbond list update frequency of 10.
Using the LEAP module in AMBER, the PX nanostructures were immersed in a water box using the TIP3P (37) model for water. The box dimensions were chosen to ensure a 10-Å solvation shell around the DNA structure. In addition, some waters were replaced by Na+ counterions to neutralize the negative charge on the phosphate groups of the backbone of the PX structures. This procedure resulted in solvated structures containing between 33,000 atoms (for PX55) and 42,000 atoms (for PX95). The details of the simulation conditions are given in Table 3. The solvated structures were then subjected to 1000 steps of steepest-descent minimization of the potential energy, followed by 2000 steps of conjugate-gradient minimization. During this minimization, the PX DNA nanostructures were fixed in their starting conformations using harmonic constraints with a force constant of 500 kcal/mol/Å2. This allowed the water molecules to reorganize to eliminate bad contacts with the PX structures.
TABLE 3.
Details of the MD simulation conditions for various PX molecules
| Molecules | Number of base-pairs | Number of atoms in DNA | Number of water molecules | Number of Na(+) ions | Initial box dimension of the solvated PX DNA |
|---|---|---|---|---|---|
| PX55 | 70 | 4432 | 9211 | 140 | 45 Å × 66 Å × 149 Å |
| PX65 | 76 | 4813 | 10360 | 152 | 46 Å × 67 Å × 159 Å |
| PX75 | 86 | 5455 | 12272 | 172 | 47 Å × 68 Å × 176 Å |
| PX85 | 92 | 5833 | 11331 | 184 | 45 Å × 64 Å × 186 Å |
| PX95 | 98 | 6215 | 12138 | 196 | 45 Å × 65 Å × 196 Å |
The minimized structures were then subjected to 40 ps of MD, using a 2-fs time step for integration. During the MD, PX DNA nanostructures were fixed in their starting conformations using harmonic constraints with a force constant of 20 kcal/mol/Å2 and the system was gradually heated from 0 to 300 K. This allows for slow relaxation of the built PX structures. In addition, SHAKE constraints (38) using a geometrical tolerance of 5 × 10−4 Å were imposed on all covalent bonds involving hydrogen atoms. This is needed to prevent dynamics in NH and OH bonds from disrupting hydrogen bonds. Subsequently, MD was performed under constant pressure-constant temperature conditions, with temperature regulation achieved using the Berendsen weak coupling method (39) (0.5-ps time constant for heat bath coupling and 0.2 ps pressure relaxation time). This was followed by another 5000 steps of conjugate-gradient minimization while decreasing the force constant of the harmonic restraints from 20 kcal/mol/Å2 to zero in steps of 5 kcal/mol/Å2.
We then carried out 100 ps of unconstrained constant pressure-constant temperature MD to equilibrate the system at 300 K. Finally, for analysis of structures and properties, we carried out 3 ns of constant volume-constant temperature MD using a heat bath coupling time constant of 1 ps.
Methods used for calculating properties of the PX nanostructures
Flexibility of the PX nanostructures
To obtain the solution structure of each PX nanostructure equilibrated in salt and water, we averaged the coordinates of each MD snapshot from 2 to 3 ns at every 1-ps time interval. This averaging was done only for the last 1 ns to ensure that the structure had converged. This average structure represents the “solution structure” of the PX nanostructure.
To obtain a measure of the flexibility of these structures, we calculated the CRMSD of all atoms from this average solution structure at each time step. This was done at every 1-ps time interval for the whole 3-ns MD trajectory. This CRMSD is a measure of the overall flexibility of the PX structures in solution. It shows the fluctuation in the overall structure compared to the average solution structure during the MD simulation. We also calculated the CRMSD for each basepair from the minimized starting canonical structure using the time average over the last 200 ps for each basepair. This CRMSD from the minimized starting structure shows the flexibility of various regions of the PX structure in solution.
Thermodynamic stability of the PX nanostructures: strain energy and the vibrational density of states
The calculation of strain energy in the PX structure largely depends on the reference state used for the calculation. Although experimental measurement of stability using the melting temperature is straightforward, the conformation(s) of the molten state is not known. To determine the strain in the PX molecules, it is necessary to define a reference state and this reference state should be an unstrained conformation and should be transferable to calculate strain energy in any DNA nanostructure.
To this end, the reference energy for each of the DNA systems is obtained using the theory of nearest-neighbor interactions (40), in which each basepair is assumed to only interact with its nearest neighbor. From this, enthalpies of 10 unique basepair sequences (doublet energies) are determined as follows:
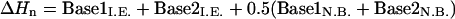 |
(1) |
where I.E. = internal energy of the base pair (bonds, angles, torsions, and inversions); and N.B. = nonbonded energy of the base pair (electrostatics, hydrogen bonds, and van der Waals).
To obtain the reference nearest-neighbor energies, 12 double helices of random sequences were created and subjected to the standard DNA simulation protocol. Analysis was performed on these double helices, extracting the individual components of energy for each nearest-neighbor pair. To obtain the energy of each basepair we first partition the potential energy into a sum over atoms. This is done by assigning half the energy for every two-body interaction to each of the two atoms, all the energy for each three-body interaction and each four-body inversion term to the central atom, and half the energy for every four-body dihedral (torsion) interaction to each of the two central atoms. Then we collect these atomic energies together for each base of the DNA. Thus, each nearest-neighbor pair self-consistently contains the interactions of that pair with the rest of the system. It also includes the solvation effects, as the interaction energy term for each of the bases includes the contribution from water as well as counterions. Thermal and conformational fluctuations also enter into the calculated energy as it was averaged over the canonical ensemble of structures. The effective nearest-neighbor energy was calculated as shown above for each pair, and the reference energy of that pair was determined by the arithmetic mean, as shown in Table 4. The doublet energy obtained this way correctly captures the sequence dependence and correlates in a reasonable way with the nucleotide energy obtained by Santa Lucia and co-workers (40–42), which is widely used for calculating thermodynamics for designed basepair sequence and determining optimal DNA length and sequence for various applications. Fig. 3 shows the correlation of the doublet energy obtained from our simulation with that obtained by Santa Lucia.
TABLE 4.
Reference energies for all 10 uniquely defined nearest-neighbor pairs
| Nearest neighbor | AVG (kcal/mol) | STDEV | No. points | Santa Lucia (kcal/mol) |
|---|---|---|---|---|
| C-C | −86.199 | 3.683 | 120 | −11.1 |
| C-G | −79.877 | 4.24 | 133 | −10.1 |
| A-C | −79.483 | 3.198 | 103 | −8.4 |
| G-G | −78.231 | 3.908 | 123 | −8.6 |
| A-G | −74.436 | 3.521 | 113 | −7.4 |
| A-A | −72.789 | 3.594 | 32 | −7.7 |
| C-T | −71.678 | 3.504 | 111 | −6.7 |
| G-T | −67.92 | 3.169 | 103 | −6.3 |
| A-T | −64.803 | 3.664 | 84 | −6.5 |
| T-T | −58.423 | 2.885 | 32 | −6.1 |
FIGURE 3.

Comparison of the base energy from the nearest-neighbor analysis with those from Santa Lucia (40). We find reasonable correlation with the experimental data. Note that there is a lot of noise even in their experimental data.
To validate the use of the calculated reference energy for designing DNA sequences we built additional DNA double helices of varying sequences, lengths, and topology, as shown in Table 5. The total energy, determined as a sum of the nearest-neighbor energies, was calculated and compared to the “predicted energy”—the energy computed by simply counting the number of nearest neighbors and assigning them their energy as given in Table 4. As shown in Table 5, the predicted energy is accurate to within 1% of the total energy, even for a system of topology totally different from that used to calculate the energy of the reference state.
TABLE 5.
Comparison of total energy obtained from simulation with predicted energy calculated from reference energies in Table 4 using the nearest-neighbor analysis
| Basepairs | % GC | Topology | Total energy (kcal/mol) | Predicted energy (kcal/mol) | Difference (kcal/mol) | % Difference |
|---|---|---|---|---|---|---|
| 70 | 42.85 | B-DNA | −27436.04 | −27115.8 | −320.27 | 1.17 |
| 86 | 44.18 | A-DNA | −33251.41 | −33387.9 | 136.51 | 0.41 |
| 92 | 39.13 | B-DNA | −31920.4 | −32112.5 | 192.08 | 0.6 |
| 98 | 48.97 | Z-DNA | −33057.76 | −33039 | −18.8 | 0.06 |
Thus, the strain energy for each PX nanostructure was calculated by determining the energy of every nearest-neighbor basepair in the PX molecule and subtracting the reference energy of the basepair from Table 4. This gives strain energy for each pair, which is then summed to obtain the total strain energy for the PX molecule.
This strain energy represents the energy cost for making a crossover structure and does not include the dependence of the strain energy on the length or sequence of the PX structures. The average strain energy is calculated by averaging over 200 snapshots uniformly distributed over the last 200–400 ps of the 3-ns MD run.
We also calculated vibrational density of states of PX nanostructures from the MD simulations as follows (43). The velocity autocorrelation function C(t), defined as the mass weighted sum of the atom velocity autocorrelation functions, was calculated using
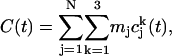 |
(2) |
where 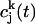 is the velocity autocorrelation of atom j in the k direction,
is the velocity autocorrelation of atom j in the k direction,
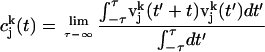 |
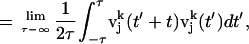 |
where 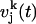 is the velocity of the atom j in the k direction at time t. The atomic spectral density
is the velocity of the atom j in the k direction at time t. The atomic spectral density 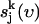 is the Fourier transform of
is the Fourier transform of 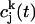 , which is given by
, which is given by
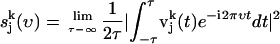 |
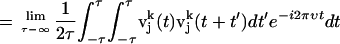 |
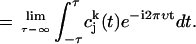 |
(3) |
From this we determine the vibrational density of states (power spectrum) as
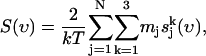 |
(4) |
where mj is the mass of atom j.
The entropy (S) can be calculated from S(υ) as in Lin et al. (44):
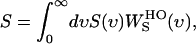 |
(5) |
where 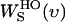 is the weighting function of entropy for a harmonic oscillator and is given by
is the weighting function of entropy for a harmonic oscillator and is given by
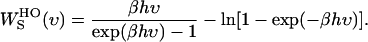 |
(6) |
Calculation of the double helical structural parameters
The double helical structure and the variations in the structure for a PX structure can be described by such structural parameters as rise, twist, roll, tilt, and slide. However, the only properties of the B-DNA helix that have a significant effect on the overall macroscopic shape of DNA are the twist and the roll (45). These helical properties were extracted from the equilibrated PX MD structures using the Curve 5.1 software package (46,47). Since these parameters are calculated for double helices, we partition the PX nanostructures into two double helices just for the analysis of helical structure parameters. The parameters for each PX structure were calculated by averaging the structural parameters for the snapshots of MD simulations over the last 200–400 ps over the 3-ns-long simulations.
RESULTS AND DISCUSSION
Differences in flexibility of the PX structures
Previous MD simulations have been reported on the crystal structure of B-DNA to validate the AMBER FF (48), using explicit salt and water and the particle mesh Ewald method for calculating the nonbond electrostatic interactions (24–27). These simulations have also been performed in solution. The simulations on crystalline B-DNA lead to an overall calculated CRMSD for all atoms of 1.0–1.5 Å (24–27). This validates the accuracy of the FF. For the solution phase, there are no reliable experimental structures with which to compare the simulations, which generally lead to RMSD differences of 3.6–4.2 Å from the crystal (26,27).
We carried out MD simulations for 2.5–3 ns in explicit salt and water for each of the five PX nanostructures (PX55, PX65, PX75, PX85, and PX95) at 300 K. In each case, we define an average MD structure by averaging the coordinates for various snapshots for the last 1 ns at intervals of 1 ps. This structure represents the time-averaged solution structure of the PX nanostructures (that one would compare to an NMR structure). These averaged structures for various PX structures are shown in Fig. 4, a and b.
FIGURE 4.

Averaged MD structures for various PX molecules: (a) front view, and (b) side views. For clarity, water molecules and counterions are not shown.
The base stacking and Watson-Crick hydrogen bonding are well maintained in the solution structure for all the PX molecules. Also, apart from PX65 and PX85, all the molecules undergo substantial writhing as seen from the side view of the PX structures shown in Fig. 4 b. This feature could make them unsuitable for creating 2D arrays of nanostructures. Further quantitative insight into the basepairing and other structural features is obtained from the calculated helical parameters shown in the next section.
To obtain some idea about the flexibility as well as relative stability of these structures, Fig. 5 a shows the CRMSD of the instantaneous PX snapshots from the time-averaged solution structure as a function of time. This CRMSD was calculated for the entire 3-ns MD simulation and it represents the flexibility in a PX structure in solution. We see that PX55 and PX65 have the least fluctuations in solution, with an average CRMSD of 2.0–2.5 Å over the final 1 ns. The other three cases, PX75, PX85, and PX95, have larger fluctuations of 2.5–3.0 Å, revealing a larger flexibility in solution. The CRMSD with respect to the initial minimized canonical structure is shown in Fig. 5 b for PX65 and PX95. To compare this to cases without crossovers, we also show the CRMSD for one double helix of each PX structure (namely 65_s1 and 95_s1). The CRMSD from the initial canonical structure remains within 3–4.5 Å for PX65 over the MD simulation, but it goes up to 7–8 Å for PX95. The CRMSD for PX55, PX75, and PX85 also goes up to 7–8 Å. The fluctuations in the PX structures are smaller than for the noncrossover form, indicating the increased rigidity of the crossover structure. For example the PX65 structure has an average CRMSD of 1.3 Å with respect to the average MD structure (average over the last 1 ns of the 3-ns runs) compared to 3.5 Å CRMSD of 65_s1 with respect to the average MD structure.
FIGURE 5.

(a) Variation of the CRMSD of all atoms of various snapshots from the MD simulation run with respect to the average MD structure for different PX molecules for the last 1 ns. (b) Root mean-square deviation (RMSD) with respect to the starting minimized canonical structures.
Fig. 6 shows the calculated average CRMSD (over the last 200 ps of the MD simulation time) from the time-averaged structure for each nucleotide in each of the five PX molecules studied here. The vertical dotted lines represent the location of crossover points in each PX structure. The CRMSD per nucleotide is typically <1 Å, except for PX95, where it is between 1 and 1.5 Å. The CRMSD at the crossover points in the middle of the strand is high for PX55, whereas PX95 has a large CRMSD at the end of the strands. The CRMSDs of the whole PX molecules, shown in Fig. 5, indicates a larger fluctuation, which implies that fluctuations are larger for the backbone than for the bases.
FIGURE 6.

CRMSD for individual bases for PX55, PX65, PX75, PX85, and PX95 from the starting MD structure. The data have been averaged for the last 200 ps of the MD run. The vertical line corresponds to the crossover point.
All PX structures lead to CRMSD of 2.0 to −3.0 Å, which are smaller than the values of 3.6 to 4.2 Å obtained for normal B-DNA. Thus the PX crossover molecules are structurally more rigid than their double helix counter parts. The rigidity of the PX structures is also exemplified through the vibrational density of state analysis (see Vibrational mode analysis of the PX structures).
The large deviation of PX95 structure from the canonical B-DNA form makes it unsuitable for forming larger nanostructures or a planar 2D array.
Comparison of the helicoidal parameters for the PX nanostructures
The helicoidal structural parameters such as roll, rise, tilt and twist collectively describe the overall backbone structure of each PX DNA nanostructure. These parameters were calculated for each basepair, averaged over the last 400 ps of the 3-ns MD run and as described in Calculation of the double helical structural parameters.
Published data on the solution simulations for B-DNA give helical twists of 30–32°, which are lower than those observed in crystal structures (35–36°) (28,29). This compares well to the 30–32° twist angle obtained in our calculations.
Fig. 7, a and b, show the rise, tilt, roll, and twist calculated for every basepair for the PX65 and PX95 structures. The helical twist angle for the two double helices of the PX65 fluctuates around 31° and the base tilt angle fluctuates around 0.31° (which is essentially zero). These values are close to the values obtained from simulation of the two separated double helices of PX65 (30° for twist and 0.22° for tilt) (49). Thus the helical parameters for PX65 are close to those of the B-DNA double helix and, hence, PX65 could be a very stable structure like a B-DNA. On the other hand, for the PX55, PX75, PX85, and PX95 structures, the helical twist and tilt angles show large fluctuations about the corresponding values in B-DNA. These fluctuations are especially large at the crossover points.
FIGURE 7.

Average rise, tilt, roll, and twist for (a) PX65 and (b) PX95. Solid line is for helix1 and broken line is for helix2. The vertical lines correspond to the crossover points. The data has been averaged over the last 200 ps of the 3-ns MD simulation. The horizontal solid lines gives the upper bound and lower bound for the corresponding quantities expexted for the helices in their B-DNA form (non-crossover form) during the MD simulation.
In regular B-DNA, high twist angle is linked to a high phase angle for pseudorotation and negative roll (45). Hence, the spikes in the helical twist angles are expected to arise from the change in direction of the backbone at the crossover points, which produces negative roll and pseudorotation.
Table 6 gives a detailed comparison of the various helicoidal parameters for all PX structures. We find that the average values of twist angle for both helices in PX85 and PX95 are consistently lower than in regular B-DNA. Fig. 8 also shows the instantaneous snapshots of individual PX molecules including the global helical axis for each of the double helix. The global helical axis is drawn using Curve 5.1. The shape of the global helix shows the changes in the overall structure. We observe large writhing in the global axis of PX85 and PX95 since these structures with low twist have a tendency to unwind (45).
TABLE 6.
Helicoidal parameters for the PX molecules
| PX55
|
PX65
|
PX75
|
||||
|---|---|---|---|---|---|---|
| Parameter | Helix1 | Helix2 | Helix1 | Helix2 | Helix1 | Helix2 |
| Shift (Å) | 0.04 (0.7) | −0.03 (0.6) | −0.02 (0.7) | 0.00 (0.8) | 0.05 (1.0) | 0.01 (0.4) |
| Slide (Å) | −0.08 (0.6) | −0.08 (0.4) | −0.05 (0.8) | −0.07 (0.7) | −0.1 (0.6) | −0.09 (0.5) |
| Rise (Å) | 3.50 (0.4) | 3.46 (0.4) | 3.53 (0.6) | 3.57 (0.4) | 3.5 (0.4) | 3.4 (0.3) |
| Tilt (°) | −.58 (5.3) | −0.30 (3.6) | −0.25 (5.0) | 0.64 (4.3) | −2.8 (6.2) | −0.04 (3.4) |
| Roll (°) | 3.6 (12.1) | 3.99 (7.7) | 2.08 (9.8) | 3.34 (11.9) | 4.3 (12.1) | 3.9 (9.7) |
| Twist (°) | 33.5 (7.3) | 32.8 (5.9) | 32.10 (5.3) | 31.7 (7.4) | 30.7 (7.8) | 32.39 (6.9) |
For calculation purposes each PX molecules was considered as split into its two double helix. The data were averaged over the last 400 ps of the 3 ns MD runs. The standard deviation is shown in parenthesis.
FIGURE 8.

Instantaneous snapshots of PX55, PX65, PX75, PX85, and PX95 molecules after 2.5- to 3-ns MD runs with the global helical axis of both the double helix. To calculate the global helical axis we split the crossover molecules into two separate double helix domains. The global helical axes were drawn with CURVES.
Fig. 9, a and b, show the variation of the widths of the major and minor grooves in each of the two double helices for PX65 and PX95. Table 7 summarizes the average major- and minor-groove widths for all the PX structures. The major and minor grooves show a steady increase in width going from PX55 to PX95 structure. The average major groove width for a B-DNA is ∼11.7 Å, which can widen to 15 Å on binding a protein or drug (50). The minor-groove width in B-DNA is 5.7 Å. The PX55 structure shows an average width of 12.9 Å for the major groove and 5.5 Å for the minor groove, and this is close to the values for B-DNA. These values increase for PX65 (13.9 Å for the major groove and 6.4 Å for the minor groove), eventually going up to 15–17 Å for PX95. The instantaneous major-groove width deviates significantly from the average value for each nucleotide, especially at the crossover points.
FIGURE 9.

Average major groove and minor groove widths for (a) PX65 and (b) PX95. The vertical lines correspond to the crossover points. The data have been averagred over the last 200 ps of the 3-ns MD run.
TABLE 7.
Average major groove and minor groove widths for all PX molecules
| Molecules | Major groove width (Å) | Minor groove width (Å) | |
|---|---|---|---|
| PX55 | Helix1 | 12.5 ± 2.1 | 5.48 ± 2.0 |
| Helix2 | 13.32 ± 1.8 | 5.44 ± 1.8 | |
| PX65 | Helix2 | 13.93 ± 2.1 | 6.43 ± 2.0 |
| Helix2 | 13.93 ± 2.0 | 6.27 ± 2.4 | |
| PX75 | Helix1 | 13.48 ± 2.8 | 6.35 ± 2.1 |
| Helix2 | 13.68 ± 2.2 | 5.63 ± 1.6 | |
| PX85 | Helix1 | 16.52 ± 3.9 | 5.49 ± 3.0 |
| Helix2 | 15.1 ± 3.4 | 5.55 ± 2.5 | |
| PX95 | Helix1 | 16.83 ± 3.7 | 5.78 ± 3.2 |
| Helix2 | 14.7 ± 3.6 | 6.88 ± 3.2 | |
The data have been averaged over the last 200 ps of the 2.5-ns MD runs.
An interesting feature in Fig. 9 a is that the major-groove width exhibits alternating widening and narrowing between successive crossover points for PX65. Although this feature is not present for PX75, PX85, and PX95, there are enhanced variations in the groove width along the backbone for these structures. It is well known that the groove width is sequence-dependent, as supported by several simulations on DNA (51–57). Thus we cannot be certain whether the observed narrowing and widening of the groove width arises from differing base sequences between the crossover points or from the presence of the crossover points.
Other structural parameters such as shear, stagger, buckle, propeller twist, and opening, all describing the structure and stacking of the basepairs in the DNA, have been calculated using the Curve 5.1 software package (46,47). Since they do not provide any further insight into the structure of these DNA motifs we have not presented them here.
Macroscopic structural properties of PX nanostructures
The macroscopic structural features such as writhing, overall bending, and the solvent-accessible surface area of the PX structures have been calculated. We have also analyzed the vibrational modes of the PX structures to understand the relationship between the low-frequency modes and the structural stability. These properties throw light on the nature of the PX nanostructures.
Writhing in longer PX DNA nanostructures
Fig. 10 shows the variation of “strand shortening” for various PX structures averaged over the last 200 ps of the 3-ns MD simulation runs. Strand shortening is calculated as follows: the Curve algorithm outputs the vectorial direction of each local helical axis segment U and its reference point P. The path length between successive helical axis reference points can be calculated as
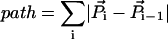 |
(7) |
and the end-to-end distance of the DNA fragment can be calculated as
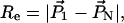 |
(8) |
where 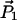 and
and 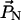 are the reference points for the two end-helical axes corresponding to two terminal nucleotides. The difference between the sum of all the path lengths and the end-to-end distance is a measure of the strand shortening. The strand shortening also indicates the overall flexibility of the DNA. Fig. 10 shows that the end-to-end distance decreases or the strand shortens more as the number of basepairs increases in the PX structures, indicating that strand shortening is highest for PX95. This shows that there is substantial distortion (e.g., writhing and/or bending) in the overall PX95 structure. This effect could be due to the total length or to the 95-crossover motif that places 14 basepairs in one helical turn.
are the reference points for the two end-helical axes corresponding to two terminal nucleotides. The difference between the sum of all the path lengths and the end-to-end distance is a measure of the strand shortening. The strand shortening also indicates the overall flexibility of the DNA. Fig. 10 shows that the end-to-end distance decreases or the strand shortens more as the number of basepairs increases in the PX structures, indicating that strand shortening is highest for PX95. This shows that there is substantial distortion (e.g., writhing and/or bending) in the overall PX95 structure. This effect could be due to the total length or to the 95-crossover motif that places 14 basepairs in one helical turn.
FIGURE 10.

Strand shortening, as defined in text, for various PX molecules. The data have been averaged over the last 200 ps of the MD run.
The bending effect is further confirmed by calculating the overall bending angle of each double helix in every PX structure. The bending angle is calculated as the angle between the successive 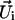 vector and is defined as
vector and is defined as
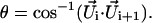 |
(9) |
Fig. 11 a shows the bending angle variation for every basepair calculated for various PX nanostructures averaged over the MD simulations. The writhing of the PX95 structure is further confirmed by the large bending angle seen for PX95 compared to PX65. We also give the comparison for PX65 and PX95 along with their double helical counterparts in Fig. 11 b (again for 65_s1 and 95_s1, which have the same lengths as PX65 and PX95, respectively). This shows clearly the significant bending of the helical axis for PX95 compared to PX65. Note that although the bending decreases for PX65 compared to its noncrossover double helical form (65_s1), it increases for PX95 compared to the noncrossover double helical form (95_s1). This suggests some intrinsic instability arising out of the specific sequence for the PX95 structure. This also can be attributed to the large number of basepairs (nine) in the major groove in PX95 compared to six basepairs in PX65. This leads to the writhing in the structure, which in turn leads to large bending. The bend is also evident from Fig. 4, which shows the snapshots from MD simulations for each PX structure.
FIGURE 11.

Bending angle between every ith and i + 5th base for helix1 of each PX structure.
Combining the effect of strand shortening with the bending, we infer that PX95 shows a larger writhing in its solution structure compared to the PX65 structure. The effect of writhing is likely to be an important structural feature in designing nanostructures. For example, because of the writhing, PX95 may not be a good choice for constructing a 2D array using PX nanostructures. Using the average solution structures from the MD run, Fig. 4 b gives a comparison between the side views of the solution structures of PX95 and the PX65. Clearly PX95 is more bent than PX65. This is an example of a structural feature deduced from theory and can be used as a design parameter for DNA-based nanotechnology.
We also calculated global helical bending for each of the two helices using the algorithm developed by Strahs and Schlick (58). This method computes the DNA curvature by summing the projected components of local base-pair-step tilt and roll angles after adjusting the helical twist. Our analysis for the global angles is based on the values of local tilt and roll angles for each basepair step computed by the Curves program (59). Bends in the helical axis defined by a negative roll angle indicate bending toward the minor groove, whereas bends defined by a positive roll angle correspond to bending toward the major groove (58). Fig. 12 plots the global bending for each PX molecule.
FIGURE 12.

Global bend angle calculated for each double helix of each PX structure. The error bars indicate the fluctuations that occur in the MD simulation.
The curvature of the double helix axis is similar for both helices in the PX55 and PX65 molecules. On the other hand, the curvature of the two helical axes shows a bending angle differing by 10–20° for the PX75, PX85, and PX95 structures, indicating the effect of writhing of the helical axis for these three structures. Also, for PX75 and PX95, the two helices bend in different directions, as is evident from the opposite sign of the roll angle for the two helices (see Table 8 for details).
TABLE 8.
Average global bending, global roll and tilt angle.
| Molecules | Global bend (degrees) | Global tilt (degrees) | Global roll (degrees) | |
|---|---|---|---|---|
| PX55 | Helix1 | 38.42 (13.7) | 34.86 (13.6) | −12.32 (10.6) |
| Helix2 | 39.49 (8.9) | 0.4 (11.4) | −37.57 (9.9) | |
| PX65 | Helix1 | 39.01 (13.1) | −15.06 (13.4) | −33.68 (12.3) |
| Helix2 | 36.68 (12.3) | 2.41 (12.2) | −34.28 (12.9) | |
| PX75 | Helix1 | 44.87 (10.0) | −5.94 (11.4) | −42.89 (10.41) |
| Helix2 | 27.05 (12.1) | 23.81 (14.3) | 4.72 (9.2) | |
| PX85 | Helix1 | 42.1 (12.5) | −12.4 (14.7) | −37.2 (12.9) |
| Helix2 | 26.73 (10.7) | 10.37 (13.3) | −20.84 (10.5) | |
| PX95
|
Helix1 | 39.83 (12.7) | −26.1 (20.6) | −19.05 (16.8) |
| Helix2 | 106.6 (60.1) | 48.68 (48.9) | 62.48 (79.4) | |
The data were averaged over the last 200 ps of the 2.5-ns long MD runs. The standard deviations are shown in brackets.
Relative stability and calculation of free energies of formation
To study the relative stability of the different PX molecules, we calculated the strain energy for each nucleotide. The average strain energy was calculated as the total strain energy from Eq. 1, described in the Methods section Thermodynamic stability of the PX nanostructures, divided by the total number of basepairs in each PX structure. The strain energy is the energy cost for making a crossover compared to the double helix B-DNA. The calculated strain energy per basepair calculated for different PX structures is plotted in Fig. 13. The strain energy is highest for PX55 and decreases to a minimum for PX65. Subsequently, the strain energy increases going from PX75 to PX95. The difference in strain energies between PX55 and PX65 is ∼3 kcal/mol/bp. The stability of the PX65 structure is compounded and could be attributed to basepair sequence, the length of the PX65 nanostructure, or the number of crossover points in the structure. We are currently examining the effect of each of these factors on the stability of PX nanostructures.
FIGURE 13.

Strain energy for various PX structures. The solid line serves as a guide to the eye only. Clearly, PX65 is the most stable molecule with the least strain energy, in agreement with the experimental observation.
The original set of experiments by Seeman and co-workers (New York University, private communication, 2003) showed that PX55 and PX95 were not formed as monomers in solution, but instead were dimers or multimers. Experimentally, PX95 was formed with a certain amount of dimer. The simulations showed that PX95 is less stable than PX65 but certainly comparable in strain energy to PX85. Hence we proposed to the Seeman Laboratory that PX95 should be a thermodynamically feasible structure. Subsequent experiments by Seeman and co-workers showed that at lower concentrations of the single strands PX95 was indeed formed as a monomer (19). Thus, a prediction made from simulation results guided the experiments leading to synthesis of PX95. Thus, optimization of nanodevice parameters using theory and simulations before synthesis would enable faster progress in nanotechnology.
Vibrational-mode analysis of the PX structures
Analysis of the low-frequency vibrational modes and their differences for various PX structures provides a measure of the relative stiffness of these polymers. To understand the structural stability in terms of flexibility, we calculated the distribution of vibrational modes of each PX structure using analysis of the velocity autocorrelation function, as described in the Methods section Thermodynamic stability of the PX nanostructures. This vibrational spectrum is shown in Fig. 14 for each of the PX structures and for normal B-DNA. In the high-frequency range the spectrum is quite similar for all the PX molecules. However, for low frequencies (∼10 cm−1), the density of states increases with the increase in the length of the PX structure, as expected. Integrating the power spectrum leads to the integrated density of states shown in Fig. 15 for all the PX molecules. This is compared with the integrated density of states for a small decamer (1BD1) and for the 95S1 double helices of PX95.
FIGURE 14.

Power spectrum for various PX molecules. For comparison, we also show the spectrum from 95S1, which is a B-DNA with the same length and sequence as that of one of the double helix of PX95.
FIGURE 15.

Integrated density of state S(ν) as a function of ν (cm−1) for various PX molecules.
The population of low-frequency modes provides a direct measurement of the rigidity of the PX molecules. Compared to 1BD1 or 95S1 (normal B-DNA with the same sequence and length as PX95), we see that all PX molecules have significantly lower density of states at low frequency. This indicates that the crossover structure enhances the rigidity of all PX molecules relative to B-DNA. Since PX55 and PX65 have the smallest population of low-frequency modes, they are the stiffest, whereas PX75, with the highest population, is the most flexible. These results are consistent with the recent experimental findings from the Seeman group (60) on the DX class of crossover molecules, which show that they are twice as stiff as linear DNA molecules.
The low-frequency modes dominate the differential conformational entropic contributions, which are obtained by integrating the continuous vibrational density of states (using the weighting function discussed in Thermodynamic stability of the PX nanostructures). These results, summarized in Table 9, show that the entropy per basepair (TS at room temperature) for PX65 is lower (∼36 kcal/mol/bp) compared to other PX motifs (∼44 kcal/mol/bp). This indicates that PX65 is the most rigid of the PX motifs studied here.
TABLE 9.
Entropy of the various PX structures calculated from the vibrational density of states
| PX | Entropy (kcal/mol/bp) |
|---|---|
| PX55 | 44.72 |
| PX65 | 36.0 |
| PX75 | 44.9 |
| PX85 | 44.54 |
| PX95 | 45.25 |
We also calculated the difference in the integrated power spectrum between various PX structures at each frequency spanning from 10 cm−1 (low frequency) up to 1400 cm−1. Since we used the SHAKE algorithm to constrain the high-frequency XH bond vibrations, these modes do not show up in the MD calculations and hence are omitted from this analysis. Fig. 16 compares the integrated density of states for each PX structure with the corresponding B-DNA at each frequency (wave number). The observed decrease in the population of the low-frequency modes for the PX structures compared to the B-DNA structure is compensated by an increase in the population of the mid-range frequencies from 600 cm−1 to 1400 cm−1. We note that the stability of the PX65 structure comes through a decrease in the low-frequency modes corresponding to large-scale domain motion in the nanostructure while increasing the number of high-frequency modes to distribute the thermal energy. The decrease in the low-frequency modes for PX95 is less than that for PX65, which could help explain the relative flexibility and hence instability of PX95 structures compared to PX65 structures.
FIGURE 16.

The difference in integrated density of states as a function of frequency. The integrated density of states of 1BD1 (normal B-DNA) has been subtracted from the integrated density of states of the PX molecules. This shows the effect of crossovers on the integrated density of states.
SUMMARY AND CONCLUSIONS
We have developed an automated procedure for building the crossover PX-DNA nanostructures and for performing long-timescale MD simulations on these nanostructures. Long-timescale MD simulations provide critical information on the structural features and relative stability of the various DNA motifs that are the building blocks for DNA-based nanostructures. We have also developed a consistent and universal reference state energy analysis using nearest-neighbor interactions. This reference state energy is transferable to the strain energy analysis of any crossover DNA structure and, for that matter, any DNA structure, and hence enables the relative stability analysis of PX molecules of varying length and sequence. Our reference energy calculated based on the nearest-neighbor interactions allows us to get a predicted energy accurate within 1% of the total energy, even for a system of different topology, thus validating the nearest-neighbor approximation. The calculated strain energy correlates very well with the experimental results. In accordance with the experimental results, we find that PX65 is the most stable molecule, with as little strain as −0.77 kcal/mol/bp, with the strain increasing for PX75, PX85, and PX95 up to 4 kcal/mol/bp. PX55 has the highest strain energy (∼1.84 kcal/mol/bp), indicating that this molecule may not form in accordance with the experimental results. Thus the strain energy analysis developed here can be used to calculate the stability of various crossover molecules of varying length, sequence, and number of crossover points, which in turn will help experimentalists to optimize the DNA motifs for building nanoscale devices before synthesis. Another interesting result of this study is that the PX-DNA motifs with seven, eight, and even nine base pairs (for PX75, PX85, and PX95, respectively) within a major groove can be stable molecules. The large numbers of basepairs are accommodated in the major groove via writhing of the DNA backbone instead of by distortion or twist only. Thus we find that the writhing factor is high in PX95 compared to PX65. This potentially important observation from theory is an important design factor to be considered for making 2D arrays of the PX structures.
The CRMSD values calculated from the average MD structure show that, PX65 fluctuates less than PX75, PX85, and PX95, suggesting increased stability for PX65. PX55 also has low fluctuations but the localized strain at the crossover points makes it unstable, which may explain the inability to form this structure.
The integrated density of states shows a decrease in population of the low-frequency modes for the crossover PX structures compared to the normal B-DNA structure without crossover points. This decrease in low-frequency modes makes the PX structures more rigid due to the presence of the crossover points.
These studies validate that atomistic theory can provide guidance and interpretations of experiments, making it valuable for progress in DNA-based nanotechnology. Also, the large amounts of structural data obtained from these atomistic simulations are currently being used for developing mesoscale force fields for the DNA-based nanostructures. The mesoscale force fields will be used for microsecond-scale simulations of real DNA-based nanodevices.
Acknowledgments
We thank Prof. Nadrian Seeman (New York University) and Prof. Erik Winfree (California Institute of Technology) for useful discussions on this subject. We also thank Mr. Gene Carter for collaborating with us in making changes to his Namot2 code to facilitate building of PX crossover points. We also thank Dr. Shiang-Tai Lin for help with the vibrational density of states analysis. P.K.M. thanks Supercomputer Research and Education Centre, Indian Institute of Science, Bangalore, for the computational facility where some of the computations were carried out.
This research was supported by the National Science Foundation (NIRT-CTS-0103002). The use of the Materials and Process Simulation Center facilities in these studies was funded by grants from ARO (Defense University Research Instrumentation Program), Office of Naval Research (Defense University Research Instrumentation Program), National Science Foundation (Major Research Instrumentation), and International Business Machines (Shared University Research). The Materials and Process Simulation Center is also supported by grants from the National Institutes of Health, the National Science Foundation, the Department of Energy, the Office of Naval Research, Chevron-Texaco, General Motors, Seiko Epson, Asahi Kasei, and Beckman Institute.
References
- 1.Seeman, N. C. 1998. DNA nanotechnology: novel DNA constructions. Annu. Rev. Biophys. Biomol. Struct. 27:225–248. [DOI] [PubMed] [Google Scholar]
- 2.Seeman, N. C. 2003. DNA in a material world. Nature. 421:427–431. [DOI] [PubMed] [Google Scholar]
- 3.Seeman, N. C. 2003. Biochemistry and structural DNA nanotechnology: an evolving symbiotic relationship. Biochemistry. 42:7259–7269. [DOI] [PubMed] [Google Scholar]
- 4.Sarikaya, M., C. Tamerler, A. K. Y. Jen, K. Schulten, and F. Baneyx. 2003. Molecular biomimetics: nanotechnology through biology. Nat. Mater. 2:577–585. [DOI] [PubMed] [Google Scholar]
- 5.Reif, J. H. 2002. Molecular assembly and computation: from theory to experimental demonstrations.In Automata, Languages and Programming. 1–21.
- 6.Niemeyer, C. M. 2002. Nanotechnology: tools for the biomolecular engineer. Science. 297:62–63. [DOI] [PubMed] [Google Scholar]
- 7.Winfree, E. 1995. On the computational power of DNA annealing and ligation. In DNA Based Computers: Proceedings of a. DIMACS Workshop, April 4, Princeton University. R. J. Lipton and B. E. Baum, editors. American Mathematical Society, Providence, RI. 199–221.
- 8.Mao, C. D., T. H. LaBean, J. H. Reif, and N. C. Seeman. 2000. Logical computation using algorithmic self-assembly of DNA triple-crossover molecules. Nature. 407:493–496. [DOI] [PubMed] [Google Scholar]
- 9.Liu, Q. H., L. M. Wang, A. G. Frutos, A. E. Condon, R. M. Corn, and L. M. Smith. 2000. DNA computing on surfaces. Nature. 403:175–179. [DOI] [PubMed] [Google Scholar]
- 10.Yan, H., X. P. Zhang, Z. Y. Shen, and N. C. Seeman. 2002. A robust DNA mechanical device controlled by hybridization topology. Nature. 415:62–65. [DOI] [PubMed] [Google Scholar]
- 11.Alberti, P., and J. L. Mergny. 2003. DNA duplex-quadruplex exchange as the basis for a nanomolecular machine. Proc. Natl. Acad. Sci. USA. 100:1569–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang, J. 2000. From DNA biosensors to gene chips. Nucleic Acids Res. 28:3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albers, J., T. Grunwald, E. Nebling, G. Piechotta, and R. Hintsche. 2003. Electrical biochip technology—a tool for microarrays and continuous monitoring. Anal. Bioanal. Chem. 377:521–527. [DOI] [PubMed] [Google Scholar]
- 14.Seeman, N. C., and A. M. Belcher. 2002. Emulating biology: Building nanostructures from the bottom up. Proc. Natl. Acad. Sci. USA. 99:6451–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao, C. D., W. Q. Sun, and N. C. Seeman. 1999. Designed two-dimensional DNA Holliday junction arrays visualized by atomic force microscopy. J. Am. Chem. Soc. 121:5437–5443. [Google Scholar]
- 16.Seeman, N. C. 2001. DNA nicks and nodes and nanotechnology. Nano Lett. 1:22–26. [Google Scholar]
- 17.Fu, T. J., and N. C. Seeman. 1993. DNA double-crossover molecules. Biochemistry. 32:3211–3220. [DOI] [PubMed] [Google Scholar]
- 18.Li, X. J., X. P. Yang, J. Qi, and N. C. Seeman. 1996. Antiparallel DNA double crossover molecules as components for nanoconstruction. J. Am. Chem. Soc. 118:6131–6140. [Google Scholar]
- 19.Shen, Z. Y., H. Yan, T. Wang, and N. C. Seeman. 2004. Paranemic crossover DNA: a generalized Holliday structure with applications in nanotechnology. J. Am. Chem. Soc. 126:1666–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LaBean, T. H., H. Yan, J. Kopatsch, F. R. Liu, E. Winfree, J. H. Reif, and N. C. Seeman. 2000. Construction, analysis, ligation, and self-assembly of DNA triple crossover complexes. J. Am. Chem. Soc. 122:1848–1860. [Google Scholar]
- 21.Tung, C. S., and E. S. Carter. 1994. Nucleic acid modeling tool (NAMOT): An interactive graphic tool for modeling nucleic acid structures. Comput. Appl. Biosci.. 10:427–433. [DOI] [PubMed] [Google Scholar]
- 22.Case, D. A., D. A. Pearlman, J. W. Caldwell, T. E. Cheatham, J. Wang, W. S. Ross, C. Simmerling, T. Darden, K. M. Merz, R. V. Stanton, A. Cheng, J. J. Vincent, M. Crowley, V. Tsui, H. Gohlke, R. Radmer, Y. Duan, J. Pitera, I. Massova, G. L Seibel, U. C. Singh, P. Weiner, and P. A. Kollman. 1999. AMBER7 Users' Manual. 7th ed. University of California, San Francisco.
- 23.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- 24.Lee, H., T. Darden, and L. Pedersen. 1995. Accurate crystal molecular-dynamics simulations using particle-mesh-Ewald: RNA dinucleotides—ApU and GpC. Chem. Phys. Lett. 243:229–235. [Google Scholar]
- 25.York, D. M., W. T. Yang, H. Lee, T. Darden, and L. G. Pedersen. 1995. Toward the accurate modeling of DNA: the importance of long-range electrostatics. J. Am. Chem. Soc. 117:5001–5002. [Google Scholar]
- 26.Miaskiewicz, K., J. Miller, M. Cooney, and R. Osman. 1996. Computational simulations of DNA distortions by a cis,syn-cyclobutane thymine dimer lesion. J. Am. Chem. Soc. 118:9156–9163. [Google Scholar]
- 27.Bevan, D. R., L. P. Li, L. G. Pedersen, and T. A. Darden. 2000. Molecular dynamics simulations of the d(CCAACGTTGG)(2) decamer: influence of the crystal environment. Biophys. J. 78:668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheatham, T. E., and P. A. Kollman. 1996. Observation of the A-DNA to B-DNA transition during unrestrained molecular dynamics in aqueous solution. J. Mol. Biol. 259:434–444. [DOI] [PubMed] [Google Scholar]
- 29.Cheatham, T. E., and P. A. Kollman. 1997. Molecular dynamics simulations highlight structural differences among DNA:DNA, RNA:RNA, and DNA:RNA hybrid duplexes. J. Am. Chem. Soc. 119:4805–4825. [Google Scholar]
- 30.Cheatham, T. E., P. Cieplak, and P. A. Kollman. 1999. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 16:845–862. [DOI] [PubMed] [Google Scholar]
- 31.Cheatham, T. E., J. Srinivasan, D. A. Case, and P. A. Kollman. 1998. Molecular dynamics and continuum solvent studies of the stability of polyG-polyC and polyA-polyT DNA duplexes in solution. J. Biomol. Struct. Dyn. 16:265–280. [DOI] [PubMed] [Google Scholar]
- 32.Srinivasan, J., T. E. Cheatham, P. Cieplak, P. A. Kollman, and D. A. Case. 1998. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate: DNA helices. J. Am. Chem. Soc. 120:9401–9409. [Google Scholar]
- 33.Feig, M., and B. M. Pettitt. 1998. Structural equilibrium of DNA represented with different force fields. Biophys. J. 75:134–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Louise-May, S., P. Auffinger, and E. Westhof. 1996. Calculations of nucleic acid conformations. Curr. Opin. Struct. Biol. 6:289–298. [DOI] [PubMed] [Google Scholar]
- 35.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 36.Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 37.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 38.Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical-integration of Cartesian equations of motion of a system with constraints: molecular-dynamics of N-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- 39.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 40.Santa Lucia, J. 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA. 95:1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santa Lucia, J., H. T. Allawi, and A. Seneviratne. 1996. Improved nearest-neighbor parameters for predicting DNA duplex stability. Biochemistry. 35:3555–3562. [DOI] [PubMed] [Google Scholar]
- 42.Xia, T. B., J. Santa Lucia, M. E. Burkard, R. Kierzek, S. J. Schroeder, X. Q. Jiao, C. Cox, and D. H. Turner. 1998. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick basepairs. Biochemistry. 37:14719–14735. [DOI] [PubMed] [Google Scholar]
- 43.Berens, P. H., D. H. J. Mackay, G. M. White, and K. R. Wilson. 1983. Thermodynamics and quantum corrections from molecular-dynamics for liquid water. J. Chem. Phys. 79:2375–2389. [Google Scholar]
- 44.Lin, S. T., M. Blanco, and W. A. Goddard. 2003. The two-phase model for calculating thermodynamic properties of liquids from molecular dynamics: validation for the phase diagram of Lennard-Jones fluids. J. Chem. Phys. 119:11792–11805. [Google Scholar]
- 45.Gorin, A. A., V. B. Zhurkin, and W. K. Olson. 1995. B-DNA twisting correlates with base-pair morphology. J. Mol. Biol. 247:34–48. [DOI] [PubMed] [Google Scholar]
- 46.Lavery, R., and H. Sklenar. 1989. Defining the structure of irregular nucleic acids: conventions and principles. J. Biomol. Struct. Dyn. 6:655–667. [DOI] [PubMed] [Google Scholar]
- 47.Lavery, R., and H. Sklenar. 1989. The definition of generalized helicodal parameters and of axis curvature for irregular nucleic acids. J. Biomol. Struct. Dyn. 6:63–91. [DOI] [PubMed] [Google Scholar]
- 48.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- 49.Maiti, P. K., T. A. Pascal, N. Vaidehi, and W. A. Goddard. 2004. The stability of Seeman JX DNA topoisomers of paranemic crossover (PX) molecules as a function of crossover number. Nucleic Acids Res. 32:6047–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schumacher, M. A., M. C. Miller, S. Grkovic, M. H. Brown, R. A. Skurray, and R. G. Brennan. 2002. Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR. EMBO J. 21:1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shafer, G. E., M. A. Price, and T. D. Tullius. 1989. Use of the hydroxyl radical and gel-electrophoresis to study DNA-structure. Electrophoresis. 10:397–404. [DOI] [PubMed] [Google Scholar]
- 52.Boutonnet, N., X. W. Hui, and K. Zakrzewska. 1993. Looking into the grooves of DNA. Biopolymers. 33:479–490. [DOI] [PubMed] [Google Scholar]
- 53.Young, M. A., B. Jayaram, and D. L. Beveridge. 1998. Local dielectric environment of B-DNA in solution: results from a 14 ns molecular dynamics trajectory. J. Phys. Chem. B. 102:7666–7669. [Google Scholar]
- 54.Sprous, D., M. A. Young, and D. L. Beveridge. 1999. Molecular dynamics studies of axis bending in d(G(5)- (GA(4)T(4)C)(2)-C-5) and d(G(5)-(GT(4)A(4)C)(2)-C-5): effects of sequence polarity on DNA curvature. J. Mol. Biol. 285:1623–1632. [DOI] [PubMed] [Google Scholar]
- 55.Young, M. A., and D. L. Beveridge. 1998. Molecular dynamics simulations of an oligonucleotide duplex with adenine tracts phased by a full helix turn. J. Mol. Biol. 281:675–687. [DOI] [PubMed] [Google Scholar]
- 56.Sarma, R. H., M. H. Sarma, L. S. Dai, and K. Umemoto. 1997. GC rich DNA oligonucleotides with narrow minor groove width. FEBS Lett. 418:76–82. [DOI] [PubMed] [Google Scholar]
- 57.Nelson, H. C. M., J. T. Finch, B. F. Luisi, and A. Klug. 1987. The Structure of an Oligo(Da).Oligo(Dt) tract and its biological implications. Nature. 330:221–226. [DOI] [PubMed] [Google Scholar]
- 58.Strahs, D., and T. Schlick. 2000. A-tract bending: insights into experimental structures by computational models. J. Mol. Biol. 301:643–663. [DOI] [PubMed] [Google Scholar]
- 59.Lavery, R., and H. Sklenar. 1988. The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic-acids. J. Biomol. Struct. Dyn. 6:63–91. [DOI] [PubMed] [Google Scholar]
- 60.Sa-Ardyen, P., A. V. Vologodskii, and N. C. Seeman. 2003. The flexibility of DNA double crossover molecules. Biophys. J. 84:3829–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]