

Abstract
Fatty acid binding proteins (FABPs) carry fatty acids (FAs) and other lipids in the cellular environment, and are thus involved in processes such as FA uptake, transport, and oxidation. These proteins bind either one or two ligands in a binding site, which appears to be inaccessible from the bulk. Thus, the entry of the substrate necessitates a conformational change, whose nature is still unknown. A possible description of the ligand binding process is given by the portal hypothesis, which suggests that the FA enters the protein through a dynamic area known as the portal region. On the other hand, recent simulations of the adipocyte lipid binding protein (ALBP) suggested a different entry site (the alternative portal). In this article, we discuss molecular dynamics simulations of the apo-intestinal-FABP (I-FABP) in the presence of palmitate molecule(s) in the simulation box. The simulations were carried out to study whether the FA can enter the protein during the simulations (as in the ALBP) and where the ligand entry site is (the portal region, the alternative portal or a different domain). The analysis of the simulations revealed a clear difference between the ALBP and the I-FABP. In the latter case, the palmitate preferentially adsorbed to the portal region, which was more mobile than the rest of the protein. However, no ligand entry was observed in the multi-nanosecond-long simulations, in contrast to ALBP. These findings suggest that, although the main structural motif of the FABPs is common, the fine details of each individual protein structure grossly modulate its reactivity.
INTRODUCTION
Fatty acids (FAs) are essential metabolic and structural components of the cell. FAs are seldom present in the hydrophilic intracellular environment as free mobile species, since their affinity to the cellular membranes is much higher. For this reason, a multi-gene family of proteins named FA binding proteins (FABPs) evolved. These proteins carry the FAs through the aqueous cellular environment, and are involved in processes like FA uptake, transport, and oxidation (1,2).
Specific FABP genes are predominantly expressed in different tissues, and the proteins are named accordingly, e.g., intestinal-FABP (I-FABP), ileal lipid binding protein (ILBP), muscle-FABP (M-FABP), and adipocyte lipid binding protein (ALBP). Each individual FABP has its own sequence, but all share a common structure consisting of 10 antiparallel β-strands. Presumably, different FABPs have different metabolic roles, but the specific function of individual FABPs has not yet been fully elucidated (1).
FABPs bind either one or two ligands to a single binding site, which appears to be inaccessible from the bulk. Thus, the binding of the substrate necessitates a conformational change to permit its entry. The nature of this conformational change is unknown since, as seen in crystallographic structures of ALBP and I-FABP (3–12), there is almost no difference between the structures of apo- and holo-FABPs. A possible description of the ligand binding process is provided by the portal hypothesis, which suggests that the FA enters the protein through a dynamic area, made of α-helix II and the turns between β-strands βC-βD and βE-βF before binding in the ligand binding cavity (4). The dynamic area through which the entry occurs is termed the portal region.
The portal hypothesis is supported by a plethora of experimental studies, most of which were carried out using either the I-FABP or ALBP. Crystallographic studies of both proteins indicated that the lipid tail is located near the suggested entry site (4,8). NMR measurements of I-FABP indicated that the protein exhibits a pronounced backbone disorder at the portal region, thus suggesting that it is more mobile than the rest of the protein; hence this region may be involved in ligand insertion (13–16). To test whether mutations in the portal region influence FA binding, the dissociation constants (Kd) of a fluorescent fatty acid analog (1,8-ANS) from ALBP, mutated at the portal region (V32D, F57H, and the double mutant V32D/F57H), were measured and were found to be elevated by a factor of 2–12 when compared to the corresponding values of the wild-type protein (11). In a different study, the binding of various FAs to ALBP and I-FABP mutants was studied and the corresponding Kd values were measured (17). Most of the mutations in the portal region resulted in an increase of Kd: up to 70-fold for simple FA (palmitate, stearate, and oleate) or 250-fold in the extreme case of linolenate (maximal Kd changes were found in I-FABP R56A mutant). On the other hand, a 2–4-fold decrease of Kd was also observed in a few mutants (I-FABP, K27A, and K27F). In a study that followed, the kinetic rate constants kon for the binding of oleate and linoleate to I-FABP mutants were measured (18). kon values measured in the wild-type protein were in the order of 10−7 M−1 s−1. The values measured for the mutated proteins were decreased by up to 10-fold (in the D74A mutant). Other mutations showed a smaller decrease, or even a slight increase of kon (1.4-fold, in K27A). Overall, mutations that reduced kon values were generally associated with the portal region.
Computational methods such as molecular dynamics (MD) were used to study holo- and apo-ALBP, I-FABP, and M-FABP. These simulations allowed the comparison between different FABPs and different ligands. In a preliminary work, Zanotti et al. have simulated the exit of the ligand from the binding site of I-FABP (19). Their simulation indicated that the ligand might escape the binding cavity through the portal region. However, the atomic details of the process could not be resolved, since the temperature in their simulation was 1500 K. Due to the slow dissociation of the ligand from the protein at room temperature, simulations of the dissociation process are still impractical, but MD simulations of FABP were still proven useful in the past. Rich and Evans compared the properties of bound FAs in MD simulations of ALBP and found a few differences between various FAs (20). Woolf and Tycko have addressed similar questions by means of multiple MD simulations of holo-FABPs. They compared between different FABPs and ligands, and characterized the residues that were involved in ligand stabilization (21,22). A different approach was used by Likic, Prendergast, and co-workers. These authors carried out MD simulations for the study of the dynamics of the binding cavity (23) and internal water molecules in I-FABP (24,25). Their results supported the portal hypothesis, as the portal region was found to be more mobile than the rest of the protein, suggesting that ligand entry could occur from there (23). Furthermore, Likic and Prendergast found that water molecules could exchange between the protein interior and the solvent. The exchange sites were located in the portal region (25), which again suggests that molecules can enter or exit the protein through this area. In parallel, Bakowies and van Gunsteren studied the dynamics of holo- and apo-I-FABP. They analyzed the dynamics of the protein, the ligand, and the water molecules and reported that water exchange occurred between the protein interior and the bulk solvent, both in the portal region and on a different site that included the N-terminus and residues Gln-42, Gly-44, Met-84, Gly-86, and Asn-87 (26). Since the portal region was the main water entry and escape site, the simulations of Bakowies and van Gunsteren were in accord with the conjecture that the portal region is involved in ligand entry.
In contrast to simulations described above, where FABP were simulated either in the absence of FA or with bound FA, we have recently simulated the apo-ALBP in the presence of a FA on its surface. It was found that the FA adsorbed to the protein surface within 2 ns, and then penetrated ∼10 Å deep into the protein from a region antipodal to the portal region (27). This ligand entry site, which included residues Cys-1, Phe-4, Ile-42, Leu-66, and Leu-91, will be referred to here as the “alternative portal”. After those simulations, it was suggested that there may be more than a single region involved in ligand entry.
In this article, we discuss MD simulations of the apo I-FABP in the presence of palmitate molecule(s) in the simulation box. The simulations were run to study the dynamics of FA reaction with the I-FABP's surface: whether the FA can enter the protein during the simulations (as in the ALBP) and where the ligand entry site is (the portal region, the alternative portal, or a different domain). Seven independent simulations were run. The simulation setups (duration, number of palmitates in the simulation, etc.) are detailed in Table 1. In two of the simulations (termed MD_p and MD_i), a single palmitate was initially located ∼5 Å away from the protein surface. These locations (displayed in Fig. 1) were similar to those used in MD simulations of ALBP (27). Five more runs were executed to examine whether the palmitate adsorbed preferentially to specific regions on the protein surface. For this reason, five free ligands were randomly located in the simulation box before the start of each run. Performing the runs with several ligands is beneficial since better statistics are gained in a similar computational effort.
TABLE 1.
Details of the simulations
| Simulation name | Duration | Number of palmitates | Initial location of the palmitates |
|---|---|---|---|
| MD_i | 13 ns | 1 | Near the alternative portal* |
| MD_p | 11 ns | 1 | Near the portal region* |
| MD_1 | 8 ns | 5 | Random |
| MD_2 | 4 ns | 5 | Random |
| MD_3 | 4 ns | 5 | Random |
| MD_4 | 4 ns | 5 | Random |
| MD_5 | 4 ns | 5 | Random |
The starting configurations of simulations MD_i and MD_p were created by aligning the crystal structure of the I-FABP with the structures used for the simulations MD_I and MD_P in Friedman et al. (27), respectively.
FIGURE 1.

Configuration of the protein and the palmitate at the start of the simulations MD_p (A) and MD_i (B). The atoms that form the portal region are shown in green, whereas the atoms that form the alternative portal region are shown in red. The solvent and the salt ions, which are present in the simulation box, are not displayed.
The analysis of the simulations revealed a clear difference between the ALBP and the I-FABP. In the latter case, the palmitate preferentially adsorbed to the portal region, which was more mobile than the remainder of the protein. However, in contrast to a previous study of ALBP, where initial ligand entry occurred after 2ns of simulation time, no ligand entry was observed in the multi-nanosecond simulations of I-FABP. Our findings suggest that although the main structural motif of the FABPs is common, the fine details of each individual protein structure grossly modulate its reactivity.
METHODS
Molecular dynamics simulations
The MD simulations were performed using the GROMACS program, version 3.2.1 (28,29), with the GROMACS force field, which is a modified version of the GROMOS87 force field (30–34). The calculations were carried out using the structure of the apo-I-FABP (Protein Data Bank (pdb) code 1IFC), determined by Scapin and co-workers (6), that was downloaded from the Protein Data Bank (35). The lipid topology files, as needed in GROMACS, were prepared using the PRODRG server (36). The protein and ligand were embedded in a box containing SPC model water (37) that extended to at least 10 Å between the solutes and the edge of the box. Seven different simulations were run (as summarized in Table 1). In the first two (MD_i and MD_p), the protein was simulated in the presence of a single palmitate ion, located near its surface (see Fig. 1). The initial locations of the palmitates in these simulations were essentially the same as in previous simulations of the ALBP (27). Eleven Na+ and 10 Cl− ions were added to the system by replacing water molecules in random positions, making the whole system neutral. In the other five simulations, five palmitates were randomly put in the simulation cell before the simulations, and the numbers of Na+ and Cl− ions were 16 and 10, respectively.
Before each MD simulation, internal constraints were relaxed by energy minimization. After the minimization, MD equilibration run was performed under position restraints for 20 ps. Then, an unrestrained MD run was initiated. The first 100 ps of the run were treated as a further equilibration simulation, and the remainder were saved and used for the analysis. During the MD runs, the LINCS algorithm (38) was used to constrain the lengths of hydrogen-containing bonds; the waters were restrained using the SETTLE algorithm (39). The time step for the simulations was 2 fs. The simulations were run under NPT conditions, using Berendsen's coupling algorithm (40) to keep the temperature and the pressure constant (P = 1 bar, τP = 0.5 ps; T = 300 K; τT = 0.1 ps). Van der Waals forces were treated using a cutoff of 12 Å. Long-range electrostatic forces were treated using the particle-mesh Ewald method (41). The coordinates were saved every 0.5 ps.
Analysis and graphical presentation
The analysis of the simulations was performed by the tools available in the GROMACS package, aided by home-written software. When the electrostatic contributions to interaction energies are considered, we refer only to the short-range interactions (up to 10 Å). Long-range interactions between individual molecules cannot be extracted from the simulations due to the usage of particle-mesh Ewald (41) treatment for the calculation of long range electrostatics. It should be mentioned that, over the whole run, the long-range interactions contributed ∼10% of the total electrostatic energies.
The entropies of the palmitate molecules were calculated according to the quasi-harmonic assumption (42,43) by Eq. 1:
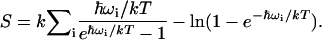 |
(1) |
k is the Boltzmann constant, T is the absolute temperature, ωi are the quasi-harmonic frequencies obtained from the diagonalized mass-weighed covariance matrix, and the summation is done on all eigenvalues. ωi is given by Eq. 2:
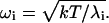 |
(2) |
λi are the eigenvalues of the diagonalized mass-weighted covariance matrix.
An approximation of the entropy loss upon FA adsorption could be achieved through an estimation of the reduction in the number of available trans/gauche states of the FA in its free and adsorbed form. However, due to the relatively short time in which the FA is found free in solution, such calculations were deemed as inaccurate. Therefore, quasi-harmonic analysis was our method of choice. It should be mentioned that a similar analysis was recently applied to study DNA binding to protein, in a system that involved many more degrees of freedom compared to our current study (44).
All protein figures were created using the VMD computer program (45).
RESULTS
Protein conformational stability
The protein conformational stability was examined by a calculation of the backbone root mean-square deviations (RMSD) relative to the protein crystal structure throughout the simulations. The results are summarized in Table 2 (second column). The maximal backbone RMSD values were between 0.156 and 0.202 nm, indicating that the protein did not undergo any significant conformational changes during the simulations.
TABLE 2.
Root mean-square deviations changes during the simulations*; all values are given in nanometers
| Simulation | Maximal backbone RMSD | Maximal heavy atom RMSD | Maximal heavy atom RMSD for the portal region† | Maximal heavy atom RMSD for the alternative portal region‡ | Residues with heavy atom RMSD > 0.3 nm§ |
|---|---|---|---|---|---|
| MD_i | 0.189 | 0.249 | 0.332 | 0.192 | Arg-10, Asn-24, Val-25, Lys-27, Lys-29, His-33, Lys-50, Tyr-70, Asp-74, Gly-75, Asn-87, Gly-110, Glu-131 |
| MD_p | 0.193 | 0.249 | 0.329 | 0.180 | Arg-10, Asn-24, Val-25, Val-26, Lys-27, Lys-29, His-33, Asp-74, Gly-75, Asn-87, Gly-110, Glu-131 |
| MD_1 | 0.156 | 0.261 | 0.361 | 0.220 | Arg-10, Val-26, Lys-27, Lys-29, Glu-43, Ala-73, Asp-74, Gly-75, Asn-87, Gly-110, Glu-131 |
| MD_2 | 0.177 | 0.229 | 0.270 | 0.163 | Lys-7, Asn-35, Asp-74, Asn-87, Glu-131 |
| MD_3 | 0.178 | 0.280 | 0.400 | 0.258 | Phe-2, Lys-7, Arg-10, Asn-24, Val-25, Val-26, Lys-27, Arg-28, Lys-29, Leu-30, Gly-31, His-33, Lys-50, Phe-55, Asp-74, Ile-108, Ser-109, Gly-110, Asn-111, Glu-120, Glu-131 |
| MD_4 | 0.170 | 0.234 | 0.279 | 0.257 | Lys-7, Arg-10, Lys-27, Lys-29, Asn-35, Lys-50, Arg-56, Ile-108, Ser-109, Gly-110, Glu-131 |
| MD_5 | 0.202 | 0.268 | 0.295 | 0.288 | Ala-1, Phe-2, Arg-10, Lys-27, Ala-73, Asp-97, Asn-98, Ile-108, Ser-109, Gly-110, Glu-131 |
RMSD values are calculated versus the crystal structure.
Residues 25–33, 53–56, and 72–75.
Residues 2, 40, 64, and 89.
Residues that are part of the portal region are shown in bold letters. Residues that are part of the alternative portal region are underlined.
Adsorption of the palmitate molecules on the protein surface
Simulations of I-FABP in the presence of a single palmitate
Before a reaction between the protein and the ligand can take place, the ligand should first come into contact with the protein surface. Therefore, the minimal distance between any of the protein atoms and the palmitate was calculated as a function of the simulation time for the simulations MD_p and MD_i, and the results are presented in Fig. 2.
FIGURE 2.

Minimal distance between any of the protein's and palmitate's atoms as a function of the simulation time during the simulations MD_p (A) and MD_i (B). The palmitate adsorbed to the protein surface after a few nanoseconds.
At the initial stages of the simulation MD_p, the palmitate molecule randomly diffused in the solution. After ∼2 ns, the FA approached the protein's surface and quickly adsorbed to it. Once the ligand was adsorbed, the minimal distance between the protein and the palmitate ranged between a contact distance of 0.17 nm and 0.4 nm (Fig. 2 A). In the simulation MD_i, the adsorption took ∼1 ns and was more gradual (Fig. 2 B), as the minimal distance between the protein and the palmitate gradually decreased from 0.52 nm to a range of 0.17–0.4nm. In both simulations, once the ligand came close to the protein surface, it did not lose contact with the protein during the rest of the simulation. Visual inspection of the dynamics revealed that the palmitate was indeed adsorbed at the protein surface as the simulations proceeded, i.e., the lipid carbons maintained their contact with the protein's surface.
In the simulation of MD_p, the adsorption started at the carboxylate headgroup, which was attracted to the protein's surface due to interactions with positively charged residues (Lys-27, Arg-56) on the portal region. A contact between the carbon atoms near the carboxylate headgroup and the protein surface was formed, and this initiated a gradual attachment of the rest of the palmitate carbons, one by one, to the protein's surface (for an animation file displaying the process, see the Supplementary Material). When the energy of interaction between the palmitate and the protein is calculated and decomposed into the electrostatic and Lennard-Jones (LJ) hydrophobic contributions (see Fig. 3), it can be seen that the LJ interactions become gradually more favorable, whereas the electrostatic contribution can either be favorable or slightly repulsive, depending on the dynamics of the palmitate headgroup and the positive residues on the protein surface. The electrostatic interactions play a significant role in the initial adsorption of the palmitate, as seen when the ligand starts to approach the protein (see the electrostatic interaction at t ∼ 2 ns in Fig. 3 A).
FIGURE 3.

Electrostatic (solid) and Lennard-Jones (shaded) contributions to the energy of interaction between the protein and palmitate(s) in the simulations MD_p (A) and MD_i (B). Interactions between the solutes and the solvent are not taken into account. The changes of total hydrophobic SASA for the protein and the palmitate are also depicted for the simulations MD_p and MD_i in frames C and D, respectively.
The hydrophobic nature of the FA adsorption is further demonstrated in the reduction of total solvent accessible surface area (SASA) of the protein and the palmitate, as displayed in Fig. 3 (frames C and D). After ligand adsorption, the reduction in the SASA parallels the strengthening of the LJ interactions between the protein and the FA (as seen when Fig. 3 A is compared to 3 C, and Fig. 3 B to 3 D). This reduction in the hydrophobic SASA implies that water molecules are excluded from the interfaces of the protein and the FA, which results in favorable entropic contribution due to the insertion of these molecules into the bulk solvent phase.
It should be noted that, in calculations of the interaction energies, such as displayed in Fig. 3, only the interactions between the solutes are taken into account. Since the electrostatic interactions are screened by the solvent, the actual interaction between the solutes will be less intense than that calculated by the interaction-energy treatment. The solvent screening effects cannot be computed from the simulations. These screening effects are smaller when the distance between the interacting species (e.g., the palmitate carboxylate and lysine amino-terminal) are small. Thus, one should be aware of the limitation of this analysis, but the conclusions made above, i.e., that the LJ contribution is larger, that the electrostatic interactions are favorable, and that electrostatic interactions play a significant role in the initial adsorption of the palmitate, still hold.
In the simulation MD_i (see Fig. 2 B), the adsorption started immediately as the simulation was initiated. The initial driving force was clearly hydrophobic. During the ligand adsorption, bursts of electrostatic interactions resulted from temporary interactions between the negative ligand and positive amino acids (Lys-94 and Lys-100). The LJ forces became slightly more favorable as the simulation proceeded, since the ligand accommodated itself near the protein.
On the average, the dominant term in both simulations is the LJ contribution. The electrostatic term is also favorable, but is weaker and more variable. It should also be mentioned that no global changes in the protein structure can be observed upon ligand adsorption. The ligand is more flexible and accommodates itself to the protein surface.
Simulations of I-FABP in the presence of five palmitate molecules
In the simulations MD_i and MD_p, the palmitate adsorbed to the protein surface in different locations. It is desirable to check whether there are other sites on the protein surface on which the ligand can adsorb, and if any of the sites is preferred. This was achieved by running five MD simulations of I-FABP (termed MD_1-MD_5) in which the I-FABP was simulated in the presence of five ligands (corresponding to a palmitate concentration of ∼30 mM) that were initially located in the bulk solution at random positions. The random assignment of 25 palmitates (five in each of the five simulations) in the simulation box prevented any bias due to the initial location of the FAs.
The number of palmitate molecules that formed contacts with the protein as a function of simulation time for the simulation MD_1 is presented in Fig. 4 A. A contact was defined when the distance between at least one pair of atoms was <0.3 nm. To reduce the fluctuations, the values presented in Fig. 4 A were averaged during 100 ps intervals. A single FA was already adsorbed at t = 0. The other ligands randomly diffused in the simulation box at first. Three palmitates approached the protein surface during the first nanosecond (see the peak, n = 4, in Fig. 4 A at t ≈ 1 ns), but two these ligands diffused away from the protein surface (as seen at t ≈ 2 ns, n ≈ 2). This indicates that the ligands fail to adsorb at the sites of contact, near their initial locations. After 3 ns, the number of palmitate molecules that remained at the protein's surface was stabilized at three. It should be mentioned that the palmitate ions neither aggregated into micelles nor folded around themselves during the simulations.
FIGURE 4.

(A) Number of palmitates, which form at least one contact with the protein surface as a function of the simulation time, calculated for the simulation MD_1. A contact is defined when the minimal distance between the palmitate and the protein is <0.3nm. The presented graph is an average over 100ps of simulation time. (B) The electrostatic (solid) and Lennard-Jones (shaded) contributions to the energy of interaction between the protein and palmitates in the simulations MD_1. (C) The total interaction energy per adsorbed palmitate as a function of simulation time (the presented graph is an average over 100 ps).
To analyze the energetics of the interaction between several ligands and the surface of I-FABP, the electrostatic and LJ terms of the energy of the interaction between the protein and the ligands during simulation MD_1 are presented in Fig. 4 B. The LJ interaction energy became more and more favorable during the simulation, whereas the electrostatic term was unstable. The electrostatic interactions are dictated by the transient interplay between the carboxylate headgroup of the palmitate and positive residues on the protein surface. These forces are opposed by the thermal motions of the protein and ligand, the interactions of both with the solvent (which result in screening of the electrostatic interaction energy), and the entropic tendency of the palmitate to keep at least its headgroup mobile rather than adsorbed to the protein's surface. As a result, the electrostatic stabilization is reversible in nature. Interactions between the salt ions in the solution and the palmitates do not play a significant role in the process, and no correlation was found between the palmitate-to-salt ions distances and the interaction energies presented in Fig. 4. It should be mentioned that our simulation time is significantly longer than the time needed for the equilibration of soluted ions with a protein (46).
The maxima in the interaction energies are correlated with the maxima in the average number of palmitate molecules that contact the protein (compare frames A and B in Fig. 4; for example, at t ≈ 1 ns). The total interaction energy (LJ plus Coulomb), divided by the number of palmitate molecules that contact the protein surface, is shown in Fig. 4 C. It can be seen that, after an equilibration period of ∼1 ns, the interaction energy per palmitate molecule is quite stable, and becomes slightly more favorable with time. This indicates that the adsorption of one ligand does not hinder the adsorption of others.
To save computer resources, simulations MD_2-MD_5 were run for 4 ns, not 8 ns as the simulation MD_1. In these simulations, the average number of palmitate molecules that formed contacts with the protein surface during the last 1 ns was 2–3, similar to the average number in MD_1.
The entropic contribution to ligand binding
The adsorption of the palmitate may limit its conformational freedom and therefore be entropically unfavorable. For this reason, the entropies of the free versus bound palmitates were examined during the simulations MD_i and MD_1, and the results are presented in Table 3. The total entropy of the ligand can be studied by assuming quasi-harmonic dynamics (42,43). The values correspond to the conformational freedom of the palmitate molecules during the simulations, and result mainly from vibrational contributions. These calculations were performed on three palmitate molecules, where we could clearly distinguish between periods when the ligands either adsorbed to the protein surface or freely diffused in the bulk solution. In all three cases, the entropy of the adsorbed ligand was significantly smaller than the entropy of the same molecule in its free state. We can estimate the contribution of the ligand binding entropy to a destabilization of the palmitate-protein complex by 10–30 kJ/mol (Table 3). Our calculations are supported by kinetic measurements of palmitate binding to I-FABP (47), where the change in TΔS upon ligand binding was −16.7 ± 8.35 kJ mol−1. This entropy change is mainly due to the reduction in the palmitate's mobility, as the binding of the relatively small ligand in the internal cavity is not expected to change the protein's entropy or the entropy of the solvent. The entropic interactions between the protein and the adsorbed palmitate are smaller than the favorable interaction energies (∼100–200 kJ mol−1, see Fig. 3), but are nonnegligible. This is in contrast to interactions between salt ions and the protein surface, where the unfavorable entropic interactions dominate over the electrostatic attraction (48). Although it is tempting to analyze the differences between TΔS values of different palmitate molecules (Table 3, right-most column) in terms of the ligand adsorption sites, the relatively small number of FAs for which we could calculate TΔS may render the differences between the individual molecules inconclusive. Apparently, differences between the entropies of palmitate molecules adsorbed at different sites on the protein surface will not play a dominant role in the preference of the substrate toward a specific site. This is due to the fact that the contribution of the interaction energies is larger than that of the entropy.
TABLE 3.
The entropy of specified ligands during simulations MD_p and MD_1
| Simulation | Time (ns) | State | Entropy(J mol−1 K−1) | TΔS* (kJ mol−1) |
|---|---|---|---|---|
| MD_p | 0.5–1.5 | Free | 1201.8 | −15.4 |
| MD_p | 2.5–11.0 | Adsorbed | 1150.3 | |
| MD_1† | 2.0–3.0 | Free | 1195.1 | −32.4 |
| MD_1† | 5.0–8.0 | Adsorbed | 1087.2 | |
| MD_1‡ | 1.5–3.0 | Free | 1218.1 | −10.9
|
| MD_1‡ | 4.0–8.0 | Adsorbed | 1181.7 |
TΔS = T (Sadsorbed minus Sfree).
There were five ligands in the simulation; the calculations were held on ligand No. 2.
There were five ligands in the simulation; the calculations were held on ligand No. 3.
The location of the adsorbed palmitate
It is of interest to examine the locations at which the palmitate adsorbed to the protein's surface. Accordingly, the residues that contacted the palmitate are listed in Table 4. A contact was defined when the FA and a certain residue shared a common solvation shell, i.e., d < 0.56 nm. To avoid counting residues that merely contacted the FA instantaneously, only residues that formed contacts with the protein for >1 ns of simulation time are listed.
TABLE 4.
The residues that contact the palmitates during the simulations*
| Simulation | Residues† |
|---|---|
| MD_i | Thr-79, Thr-81, Lys-92, Phe-93, Lys-94, Arg-95, Val-96, Asp-97, Asn-98, Gly-99, Lys-100, Glu-101, Leu-102, Ile-103, Thr-118, Tyr-119, Glu-120, Gly-121, Glu-123 |
| MD_p | Asn-24, Val-26, Lys-27, Lys-29, Lys-30, His-33, Asn-54, Phe-55, Arg-56,Asn-57 |
| MD_1 | Ala-1, Phe-2, Arg-10, Gly-22, Ile-23, Asn-24, Val-26, Lys-27, Lys-29, Leu-30, His-33, Gln-42, Gly-44, Asn-45, Phe-47, Phe-55, Arg-56, Asn-57, Ile-58, Leu-64, Asp-67, Phe-68, Ala-69, Ser-71, Leu-72, Ala-73, Asp-74, Gly-75, Glu-77, Thr-79, Gly-80, Met-84, Glu-5, Gly-86, Asn-87, Lys-94, Arg-95, Val-96, Asp-97, Asn-98, Gly-99, Ile-108, Gly-110 |
| MD_2 | - |
| MD_3 | Val-8, His-33, Asn-35, Lys-37, Ser-52, Ser-53, Asn-54, Phe-55, Asn-57, Asn-87, Ser-109, Gly-110, Asn-111, Lys-130 |
| MD_4 | Phe-2, Asp-3, Asn-21, Val-26, Lys-29, Leu-30, His-33, Asn-35, Gln-42, Gly-44, Asn-45, Asn-54, Phe-55, Arg-56, Leu-64, Gly-65, Val-66, Asp-67, Phe-68, Thr-79, Gly-80, Thr-81, Thr-83, Met-84, Gly-86, Asn-87, Lys-88, Lys-92, Phe-93, Lys-94, Glu-101, Leu-102 |
| MD_5 | Ala-1, Gly-22, Ile-23, Asn-24, Lys-27, Gln-42, Asn-45, Leu-64, Met-84, Glu-85, Gly-86, Asn-87 |
A contact was defined when the FA and a certain residue shared a common solvation shell, i.e., d < 0.56 nm.
Residues that are part of the portal region are shown in bold letters. Residues that are part of the alternative portal region are underlined.
The palmitates showed a strong tendency to adsorb to the portal region, as revealed by Table 4 and demonstrated in Fig. 5 A. As the portal region was also found to be more mobile than the rest of the protein in all simulations (see Table 2), it is very reasonable to assume that the adsorption recorded in this study delays the substrate molecule in the vicinity of the entry point. This increases the probability for ligand entry when the portal domain attains its open state.
FIGURE 5.

Structure of the protein with the palmitate adsorbed to its surface, for the simulations MD_p (A, at t = 10 ns) and MD_i (B, at t = 9 ns). The residues that form the portal region are shown in green. The residues that form the alternative portal region are shown in red. Please note that no ligand binding occurs near these residues. In the simulation MD_p, the lipid adsorbs to the portal region. The residues to which the palmitate adsorbs in the simulation MD_i are shown in pink. The palmitate is colored cyan, except for the carboxylate oxygens that are shown in red. It is visible near the portal region (green) in panel A or near the pink-colored region in panel B.
A different region, which had some tendency to adsorb FAs, is composed of the β-sheets βG, βH, and βI, and the turns that connect them (residues 77, 79–81, 83–87, and 92–103), located opposite to the portal region (Fig. 5 B). Residues Met-84, Gly-86, and Asn-87 were found to be involved in the water exit from and the entry into the protein interior in previous simulations of I-FABP, as carried out by Bakowies and van Gunsteren (26). This may indicate that ligand exchange reactions can initiate there. On the other hand, the residues of this region do not show high mobility, except for Asn-87, and it therefore seems that ligand entry is more likely to occur from the portal region.
A complete picture of all the residues that contact the protein is shown in Fig. 6. The protein surface is shown in gray, and the residues that bind the palmitate molecules are colored. As seen in the figure, no long-time ligand adsorption was found at the region that forms the scaffold of the clam-like structure of the protein. Examining the secondary structure of the residues to which the FAs adsorb reveals that the palmitates preferentially adsorbed to the coils that connect α-helices, the turns between adjacent β-sheets, and the edges of the helices and the sheets near the turns or coils. Due to their secondary structure and location, these regions are more mobile than the rest of the protein. Although no major conformational change of the protein could be detected in the simulations, the fact that the adsorption sites are more flexible suggests that the protein can adjust to the presence of the ligand at the local scale.
FIGURE 6.

Residues to which the palmitate molecules adsorb. The residues that contact the ligand are colored according to the structural elements (α-helices in purple, β-sheets in yellow, turns in cyan, and coils in white), and the rest of the protein is colored gray. The protein is viewed separately from both sides. It can be seen that the residues that contact the palmitates are located at or adjacent to loops or turns that connect secondary structures.
DISCUSSION
Fatty acid adsorption on the protein surface
The adsorption of the FA on the protein surface must precede its binding. In the reported MD simulations of I-FABP, in the presence of five randomly placed palmitate molecules, an average of ∼3 ligands were adsorbed to the protein surface. The portal region is one of two distinguishable adsorption sites (the other site involves residues 77, 79–81, 83–87, and 92–103). An average of 3.88 palmitate molecules adsorbed to each residue of the portal region (out of the 25 palmitate molecules that were simulated in the simulations MD_1-MD_5), compared to an average of 2.48 per any protein residue. This is another indication of the portal region's ability to attract the FA.
The adsorption process was dominated by hydrophobic interactions, which included both LJ contributions and reduction of the hydrophobic SASA (as demonstrated in Fig. 3). It should be stressed that the adsorption process necessitates some flexibility of the protein surface to which the FA is adsorbed. Indeed, the portal region accounts for 27.5% of the total hydrophobic SASA of the protein. Thus, its hydrophobicity, combined with its high mobility, account for its ability to adsorb FAs better than any other region on the protein surface. Apparently, the portal region of the I-FABP is designed to adsorb fatty acids.
The mechanism of ligand binding
MD simulations of the ALBP, where the apo-protein was simulated in the presence of a single palmitate that was initially located at different regions adjacent to the protein's surface, were recently reported (27). Those simulations indicated that the ligand could adsorb to the protein at more than one location. At a certain location, the ligand managed to penetrate the protein into a newly formed cavity some 10 Å deep, after several nanoseconds of simulation time. The entry site in ALBP (colored red in Figs. 1 and 5) was located near the N-terminal, at the junction between the loops connecting the β-strands; this site was termed the alternative portal region.
Based on the simulations of the ALBP, the ligand binding was suggested to proceed as described below. Initially, the ligand adsorbs to the protein surface. It then searches for a soft spot (i.e., a mobile region), through which it will be able to enter the protein. After finding this region, the ligand penetrates the protein surface, starting with its hydrophobic lipid tail. The next step will be protonation of the ligand, before its desolvation, which demands a large investment of energy (∼30 kJ mol−1). Ligand binding must be preceded by its protonation, since the insertion of the negative charged ligand into the low dielectric matrix of the protein is prohibitive; the insertion of the charged headgroup will demand an investment of >300 kJ mol−1 (for a complete discussion, see Friedman et al. (27)).
Since the ligand was adsorbed to the protein surface in simulations of I-FABP, and since the energetic cost involved with desolvation of charged palmitate is prohibitively high, we can assume that ligand binding to I-FABP occurs in a similar manner. As in the ALBP, the rate limiting step in I-FABP will be the desolvation of the carboxylate head of the FA anion, since the activation energies for palmitate binding are similar for both proteins (47).
The difference between I-FABP and ALBP
In simulations of ALBP, reported in Friedman et al. (27), five residues were involved in the ligand entry: Cys-1, Phe-4, Ile-42, Leu-66, and Leu-91. Four of these (all except for Cys-1) are conserved between ALBP from Mus musculus (pdb code 1LIB, (8)) and I-FABP from Rattus norvegicus (pdb code 1IFC, (6)). The alternative portal of ALBP was found to be the most mobile region, whereas the portal region exhibited the same mobility as the rest of the protein.
The situation is clearly different in I-FABP, where ligand adsorption occurred primarily at the portal region (Table 4). Moreover, the portal region had a higher mobility than the rest of the protein, whereas the mobility of the alternative portal did not reveal any significant trend (Table 2). The ligand did not adsorb at the alternative portal region, but at a different location (Fig. 5 B) when the FA was placed near the alternative portal region at the beginning of the simulation (simulation MD_i; see Fig. 1 B). Thus, there is no indication that ligand entry into the I-FABP can occur from the alternative portal region, in contrast to the ALBP.
Despite the structural similarity between I-FABP and ALBP, the proteins appear to have different roles: I-FABP is suggested to be involved in cellular fatty acid uptake (49–53), whereas ALBP is suggested to be involved in trafficking of fatty acids in response to lipolytic stimulation (54,55). The functional difference between the two proteins may be attributed to their interactions with other components in the cellular environment, as supported by the finding that the various FABPs are expressed predominantly at specific tissues. Each tissue contains other variants of proteins that interact with FABPs. Furthermore, there is a pronounced variation between the electrostatic potential surface around specific FABPs (56), which also suggests that they interact with different moieties. In that context, it should also be mentioned that the fine details of the structure of the ILBP render it much more flexible than other lipid binding proteins, allowing it to bind bile acids as well as fatty acids (57).
The conjecture that the ligand entry sites of ALBP and I-FABP are not the same indicates that separate regions on the FABP surface are free to interact with the cellular components. For example, if the ligand enters the protein from the portal region, the alternative portal region can be involved in protein-protein interactions and vice versa.
CONCLUDING REMARKS
A comparison of the protein substrate interactions, investigated by MD simulations, demonstrates that two members of the FABP family (I-FABP and ALBP) differ markedly in the mode of reaction with the substrate. I-FABP revealed enhanced reactivity at the portal domain, whereas ALBP preferred interactions at the alternative portal region. Thus, despite the structural similarity, the two proteins appear to have evolved to function by a different mechanism.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
Prof. Johan Åqvist (Dept. of Cell and Molecular Biology, Uppsala University, Uppsala, Sweden) is acknowledged for sharing the manuscript “Absolute and relative entropies from computer simulation with applications to ligand binding” (58) with us before its publication.
The authors acknowledge the use of computer resources belonging to the High Performance Computing Unit, a division of the Inter-University Computation Center in Israel, and to the Bioinformatics Unit at Tel Aviv University. R.F. acknowledges the Colton Foundation. This research is supported by the Israel Science Foundation (grant No. 427/01-1) and the United States-Israel Binational Science Foundation (grant No. 2002129).
References
- 1.Hertzel, A. V., and D. A. Bernlohr. 2000. The mammalian fatty acid-binding protein multigene family: Molecular and genetic insights into function. Trends Endocrinol. Metab. 11:175–180. [DOI] [PubMed] [Google Scholar]
- 2.Lucke, C., L. H. Gutierrez-Gonzales, and J. A. Hamilton. 2003. Intracellular lipid binding proteins: evolution, structure, and ligand binding. In Cellular proteins and their ligand fatty acids—emerging roles in gene expression, health and disease. A. K. Duttaroy and F. Spener, editors. Wiley, New York. 95–114.
- 3.Xu, Z., D. A. Bernlohr, and L. J. Banaszak. 1992. Crystal structure of recombinant murine adipocyte lipid-binding protein. Biochem. J. 31:3484–3492. [DOI] [PubMed] [Google Scholar]
- 4.Sacchettini, J. C., J. I. Gordon, and L. J. Banaszak. 1989. Crystal-structure of rat intestinal fatty-acid-binding protein—refinement and analysis of the Escherichia coli-derived protein with bound palmitate. J. Mol. Biol. 208:327–339. [DOI] [PubMed] [Google Scholar]
- 5.Sacchettini, J. C., G. Scapin, D. Gopaul, and J. I. Gordon. 1992. Refinement of the structure of Escherichia coli-derived rat intestinal fatty-acid binding-protein with bound oleate to 1.75-Å resolution—correlation with the structures of the apoprotein and the protein with bound palmitate. J. Biol. Chem. 267:23534–23545. [PubMed] [Google Scholar]
- 6.Scapin, G., J. I. Gordon, and J. C. Sacchettini. 1992. Refinement of the structure of recombinant rat intestinal fatty acid-binding apoprotein at 1.2- Å resolution. J. Biol. Chem. 267:4253–4269. [DOI] [PubMed] [Google Scholar]
- 7.Eads, J., J. C. Sacchettini, A. Kromminga, and J. I. Gordon. 1993. Escherichia coli-derived rat intestinal fatty-acid-binding protein with bound myristate at 1.5 Å resolution and I-Fabp(Arg106-Gln) with bound oleate at 1.74 Å resolution. J. Biol. Chem. 268:26375–26385. [PubMed] [Google Scholar]
- 8.Xu, Z., D. A. Bernlohr, and L. J. Banaszak. 1993. The adipocyte lipid-binding protein at 1.6- Å resolution. Crystal structures of the apoprotein and with bound saturated and unsaturated fatty acids. J. Biol. Chem. 268:7874–7884. [PubMed] [Google Scholar]
- 9.LaLonde, J. M., D. A. Bernlohr, and L. J. Banaszak. 1994. X-ray crystallographic structures of adipocyte lipid-binding protein complexed with palmitate and hexadecanesulfonic acid. Properties of cavity binding sites. Biochemistry. 33:4885–4895. [DOI] [PubMed] [Google Scholar]
- 10.LaLonde, J. M., M. A. Levenson, J. J. Roe, D. A. Bernlohr, and L. J. Banaszak. 1994. Adipocyte lipid-binding protein complexed with arachidonic acid. Titration calorimetry and X-ray crystallographic studies. J. Biol. Chem. 269:25339–25347. [DOI] [PubMed] [Google Scholar]
- 11.Ory, J., C. D. Kane, M. A. Simpson, L. J. Banaszak, and D. A. Bernlohr. 1997. Biochemical and crystallographic analyses of a portal mutant of the adipocyte lipid-binding protein. J. Biol. Chem. 272:9793–9801. [DOI] [PubMed] [Google Scholar]
- 12.Ropson, I. J., B. C. Yowler, P. M. Dalessio, L. Banaszak, and J. Thompson. 2000. Properties and crystal structure of a β-barrel folding mutant. Biophys. J. 78:1551–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hodsdon, M. E., and D. P. Cistola. 1997. Ligand binding alters the backbone mobility of intestinal fatty acid-binding protein as monitored by N-15 NMR relaxation and H-1 exchange. Biochemistry. 36:2278–2290. [DOI] [PubMed] [Google Scholar]
- 14.Hodsdon, M. E., and D. P. Cistola. 1997. Discrete backbone disorder in the nuclear magnetic resonance structure of apo intestinal fatty acid-binding protein: implications for the mechanism of ligand entry. Biochemistry. 36:1450–1460. [DOI] [PubMed] [Google Scholar]
- 15.Zhang, F. L., C. Lucke, L. J. Baier, J. C. Sacchettini, and J. A. Hamilton. 1997. Solution structure of human intestinal fatty acid binding protein: implications for ligand entry and exit. J. Biomol. NMR. 9:213–228. [DOI] [PubMed] [Google Scholar]
- 16.Zhang, F. L., C. Lucke, L. J. Baier, J. C. Sacchettini, and J. A. Hamilton. 2003. Solution structure of human intestinal fatty acid binding protein with a naturally occurring single amino acid substitution (A54T) that is associated with altered lipid metabolism. Biochemistry. 42:7339–7347. [DOI] [PubMed] [Google Scholar]
- 17.Richieri, G. V., P. J. Low, R. T. Ogata, and A. M. Kleinfeld. 1998. Thermodynamics of fatty acid binding to engineered mutants of the adipocyte and intestinal fatty acid-binding proteins. J. Biol. Chem. 273:7397–7405. [DOI] [PubMed] [Google Scholar]
- 18.Richieri, G. V., P. J. Low, R. T. Ogata, and A. M. Kleinfeld. 1999. Binding kinetics of engineered mutants provide insight about the pathway for entering and exiting the intestinal fatty acid binding protein. Biochemistry. 38:5888–5895. [DOI] [PubMed] [Google Scholar]
- 19.Zanotti, G., L. Feltre, and P. Spadon. 1994. A possible route for the release of fatty-acid from fatty-acid-binding protein. Biochem. J. 301:459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rich, M. R., and J. S. Evans. 1996. Molecular dynamics simulations of adipocyte lipid binding protein: effect of electrostatics and acyl chain saturation. Biochemistry. 35:1506–1515. [DOI] [PubMed] [Google Scholar]
- 21.Woolf, T. B. 1998. Simulations of fatty acid-binding proteins suggest sites important for function. I. Stearic acid. Biophys. J. 74:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woolf, T. B., and M. Tychko. 1998. Simulations of fatty acid-binding proteins. II. Sites for discrimination of monounsaturated ligands. Biophys. J. 74:694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Likic, V. A., and F. G. Prendergast. 1999. Structure and dynamics of the fatty acid binding cavity in apo rat intestinal fatty acid binding protein. Protein Sci. 8:1649–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Likic, V. A., N. Juranic, S. Macura, and F. G. Prendergast. 2000. A “structural” water molecule in the family of fatty acid binding proteins. Protein Sci. 9:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Likic, V. A., and F. G. Prendergast. 2001. Dynamics of internal water in fatty acid binding protein: computer simulations and comparison with experiments. Proteins. 43:65–72. [DOI] [PubMed] [Google Scholar]
- 26.Bakowies, D., and W. F. van Gunsteren. 2002. Simulations of apo and holo-fatty acid binding protein: structure and dynamics of protein, ligand and internal water. J. Mol. Biol. 315:713–736. [DOI] [PubMed] [Google Scholar]
- 27.Friedman, R., E. Nachliel, and M. Gutman. 2005. Molecular dynamics simulations of the adipocyte lipid binding protein reveal a novel entry site for the ligand. Biochemistry. 44:4275–4283. [DOI] [PubMed] [Google Scholar]
- 28.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- 29.van Der Spoel, D., E. Lindahl, B. Hess, A. R. van Buuren, E. Apol, P. J. Meulenhoff, D. P. Tieleman, A. L. T. M. Sijbers, A. K. Feenstra, R. van Drunen, and H. J. C. Berendsen. 2004. GROningen MAchine for Chemical Simulations. Version 3.2.1. Groningen, The Netherlands.
- 30.van Gunsteren, W. F., and H. J. C. Berendsen. 1987. Gromos-87 Manual. Biomos BV, Groningen, The Netherlands.
- 31.van Buuren, A. R., and H. J. C. Berendsen. 1993. Molecular dynamics simulation of the stability of a 22 residue alpha-helix in water and 30% trifluoroethanol. Biopolymers. 33:1159–1166. [DOI] [PubMed] [Google Scholar]
- 32.van Buuren, A. R., S. J. Marrink, and H. J. C. Berendsen. 1993. A molecular dynamics study of the decane/water interface. J. Phys. Chem. 97:9206–9212. [Google Scholar]
- 33.Mark, A. E., S. P. van Helden, P. E. Smith, L. H. M. Janssen, and W. F. van Gunsteren. 1994. Convergence properties of free energy calculations: alpha-cyclodextrin complexes as a case study. J. Am. Chem. Soc. 116:6293–6302. [Google Scholar]
- 34.Liu, H., F. Muller-Plathe, and W. F. van Gunsteren. 1995. A force field for liquid dimethyl sulfoxide and liquid properties of liquid dimethyl sulfoxide calculated using molecular dynamics simulation. J. Am. Chem. Soc. 117:4363–4366. [Google Scholar]
- 35.Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Aalten, D. M. F., R. Bywater, J. B. C. Findlay, M. Hendlich, R. W. Hooft, and G. Vriend. 1996. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided Mol. Des. 10:255–263. [DOI] [PubMed] [Google Scholar]
- 37.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. 1969. Interaction models for water in relation to protein hydration. Nature. 224:175–177.5343518 [Google Scholar]
- 38.Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- 39.Miyamoto, S., and P. A. Kollman. 1992. SETTLE: an analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comput. Chem. 13:952–962. [Google Scholar]
- 40.Berendsen, H. J. C., J. P. M. Postma, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 41.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 42.Levy, R. M., M. Karplus, J. Kushick, and D. Perahia. 1984. Evaluation of the configurational entropy for proteins—application to molecular-dynamics simulations of an alpha-helix. Macromolecules. 17:1370–1374. [Google Scholar]
- 43.Andricioaei, I., and M. Karplus. 2001. On the calculation of entropy from covariance matrices of the atomic fluctuations. J. Chem. Phys. 115:6289–6292. [Google Scholar]
- 44.Dixit, S. B., D. Q. Andrews, and D. L. Beveridge. 2005. Induced fit and the entropy of structural adaptation in the complexation of CAP and λ-repressor with cognate DNA sequences. Biophys. J. 88:3147–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 46.Pfeiffer, S., D. Fushman, and D. Cowburn. 1999. Impact of Cl− and Na+ ions on simulated structure and dynamics of betaARK1 PH domain. Proteins. 35:206–217. [PubMed] [Google Scholar]
- 47.Richieri, G. V., R. T. Ogata, and A. M. Kleinfeld. 1996. Kinetics of fatty acid interactions with fatty acid binding proteins from adipocyte, heart, and intestine. J. Biol. Chem. 271:11291–11300. [DOI] [PubMed] [Google Scholar]
- 48.Friedman, R., E. Nachliel, and M. Gutman. 2005. Protein surface dynamics: interactions with water and small solutes. J. Biol. Phys. 31:433–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prows, D. R., E. J. Murphy, and F. Schroeder. 1995. Intestinal and liver fatty-acid-binding proteins differentially affect fatty-acid uptake and esterification in L-cells. Lipids. 30:907–910. [DOI] [PubMed] [Google Scholar]
- 50.Baier, L. J., C. Bogardus, and J. C. Sacchettini. 1996. A polymorphism in the human intestinal fatty acid binding protein alters fatty acid transport across Caco-2 cells. J. Biol. Chem. 271:10892–10896. [DOI] [PubMed] [Google Scholar]
- 51.Prows, D. R., E. J. Murphy, D. Moncecchi, and F. Schroeder. 1996. Intestinal fatty acid-binding protein expression stimulates fibroblast fatty acid esterification. Chem. Phys. Lipids. 84:47–56. [DOI] [PubMed] [Google Scholar]
- 52.Atshaves, B. P., W. B. Foxworth, A. Frolov, J. B. Roths, A. B. Kier, B. K. Oetama, J. A. Piedrahita, and F. Schroeder. 1998. Cellular differentiation and I-FABP protein expression modulate fatty acid uptake and diffusion. Am. J. Physiol. 43:C633–C644. [DOI] [PubMed] [Google Scholar]
- 53.Shen, W. J., K. Sridhar, D. A. Bernlohr, and F. B. Kraemer. 1999. Interaction of rat hormone-sensitive lipase with adipocyte lipid-binding protein. Proc. Natl. Acad. Sci. USA. 96:5528–5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coe, N. R., M. A. Simpson, and D. A. Bernlohr. 1999. Targeted disruption of the adipocyte lipid-binding protein (aP2 protein) gene impairs fat cell lipolysis and increases cellular fatty acid levels. J. Lipid Res. 40:967–972. [PubMed] [Google Scholar]
- 55.Darimont, C., N. Gradoux, and A. De Pover. 1999. Epidermal growth factor regulates fatty acid uptake and metabolism in Caco-2 cells. Am. J. Physiol. 276:G606–G612. [DOI] [PubMed] [Google Scholar]
- 56.LiCata, V. J., and D. A. Bernlohr. 1998. Surface properties of adipocyte lipid-binding protein: response to lipid binding, and comparison with homologous proteins. Proteins. 33:577–589. [DOI] [PubMed] [Google Scholar]
- 57.Lucke, C., F. Zhang, H. Ruterjans, J. A. Hamilton, and J. C. Sacchettini. 1996. Flexibility is a likely determinant of binding specificity in the case of ileal lipid binding protein. Structure. 4:785–800. [DOI] [PubMed] [Google Scholar]
- 58.Carlsson, J., and J. Åqvist. 2005. Absolute and relative entropies from computer simulation with applications to ligand binding. J. Phys. Chem. B. 109:6448–6456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.