

Abstract
Increasing evidence suggests an important role for VPAC2-activated signal transduction pathways in maintaining a synchronized biological clock in the suprachiasmatic nucleus (SCN). Activation of the VPAC2 signaling pathway induces per1 gene expression in the SCN and phase-shifts the circadian clock. Mice without the VPAC2 receptor lack an overt, coherent circadian rhythm in clock gene expression, SCN neuron firing rate, and locomotor behavior. Using a systems approach, we have developed a kinetic model integrating VPAC2 signaling mediated by the cyclic AMP (cAMP)/protein kinase A (PKA) pathway and leading to induced circadian clock gene expression. We fit the model to experimental data from the literature for cAMP accumulation, PKA activation, cAMP-response element binding protein phosphorylation, and per1 induction. By linking the VPAC2 model to a published circadian clock model, we also simulated clock phase shifts induced by vasoactive intestinal polypeptide (VIP) and matched experimental data for the VIP response. The simulated phase response curve resembled the hamster response to a related neuropeptide, GRP1–27, and light. Simulations using pulses of VIP revealed that the system response is extraordinarily robust to input signal duration, a result with physiologically relevant consequences. Lastly, simulations using varied receptor levels matched literature experimental data from animals overexpressing VPAC2 receptors.
INTRODUCTION
The suprachiasmatic nucleus (SCN) is the site of the master biological clock in mammals, where many of the circadian oscillations throughout the body, including overt locomotor behavioral rhythmicity, are orchestrated (1). Synchronization of the SCN with daily light/dark cycles, food availability, and temperature variations, etc., is maintained by numerous environmental inputs. In addition, the SCN receives physiological feedback from peripheral tissues for fine-tuning the phase relationships between the various rhythms in the body. Furthermore, within the SCN itself, cells communicate with one another to maintain synchrony. Synchronization and entrainment in the SCN is in part accomplished through intracellular signal transduction pathways.
There is abundant evidence indicating that many neuropeptides modulate the SCN circadian clock oscillation (2,3). Vasoactive intestinal polypeptide (VIP), a versatile neuropeptide, plays important roles in the circadian clock system. Microinjection of VIP into the SCN region of Syrian hamsters during the early or late subjective night produces phase-shifts similar to those induced by light (3,4). VIP also induces mammalian per1 gene expression in SCN neurons (5,6) and phase-shifts the rat SCN clock in vitro (7,8). Two types of VIP receptors are present in the SCN: VPAC2 and PAC1 (9). Detailed reporter localization and immunohistochemical studies have demonstrated a high density of VPAC2 expression in SCN neurons and SCN VIP efferent target neurons (10,11). The VPAC2 receptor affinity for VIP is three orders of magnitude higher than that of PAC1 (12). In addition, alterations in VPAC2 expression profoundly disrupt the circadian system. For example, mice overexpressing VPAC2 exhibit a shorter free-running period (13), whereas mice deficient in this receptor lack circadian rhythms in both locomotor behavior and clock gene expression (14). Loss of VIP also disrupts locomotor behavior rhythms, abolishes circadian firing rhythms in approximately half of all SCN neurons, and disrupts synchrony between rhythmic neurons (15). These observations suggest that VPAC2 may contribute to autoregulation and/or synchronization within the SCN.
Although the signaling pathways mediating the actions of VIP are becoming established in other systems, they are less clearly delineated in the SCN circadian clock cells. VPAC2 is a G-protein-coupled receptor that has been clearly linked to the stimulating guanine nucleotide binding protein (Gs) and adenylyl cyclase (AC), with cyclic AMP (cAMP) implicated as a key second messenger in many tissues (12,16). In adenocarcinoma cells, VIP binding to VPAC2 has been shown to activate the cAMP/protein kinase A (PKA) signal transduction pathway (17), whereas in pinealocytes, VPAC2 activation leads to phosphorylation and activation of the transcription factor cAMP-response element binding protein (CREB), a PKA target (18). In pituitary tumor cells, activation of the phospholipase C/inositol-phosphate pathway downstream of VPAC2 also leads to CREB activation through mitogen-activated protein kinase pathways (19). Activated CREB in turn induces gene expression, for example, in human choriocarcinoma cells CREB induces per1 gene expression through CRE elements present in the per1 gene promoter (20). In SCN slices and dissociated SCN cells, VIP induces cAMP synthesis (21, 22). In ex vivo experiments, both PKA and mitogen-activated protein kinase have been shown to play roles in VIP-induced phase shifts in the spontaneous firing rhythms of SCN neurons (7). These results suggest that signaling downstream of VPAC2 in the SCN is similar to VPAC2 signaling in other systems.
Many signaling pathways in the SCN are gated by the circadian clock such that the functional consequences of signaling depend on the phase of the clock when the signaling is initiated (2). The functional consequences of VIP signaling through cAMP/PKA is also gated by the circadian clock in such a way that VIP, like light, only induces phase shifts and per1 expression during the circadian night (3,4,5,6,23). Where along the signaling pathway the gating occurs is not clear.
To understand the dynamics of VPAC2 receptor signaling, how it modulates circadian clock function, and how it is gated by the circadian clock, we have undertaken a computational modeling study of this system in this work. Specifically, we have focused on how the topology and interacting components of the VPAC2 signaling pathway contribute to the control of clock gene induction and how the circadian clock responsiveness to VIP stimulation is gated. We fit the model to a wide range of experimental data from the literature for cAMP accumulation, PKA activation, and CREB phosphorylation. We linked the VPAC2 model to a mammalian circadian clock model (24,25) and observed phase shifts similar to what has been observed in vivo and in vitro. Interestingly, our simulations reveal that the system response to short pulses of VIP is nearly equivalent to the response to step changes in VIP concentration, a result with physiologically relevant consequences. Results of simulation studies with varied receptor levels agree with published experimental results for mice overexpressing the VPAC2 receptor, providing additional validation of the model. Together, the modeling and simulation results of this study provide a framework for gaining increased understanding of the modulation of circadian clock properties by extracellular inputs.
MATERIALS AND METHODS
The VPAC2 model of this work is an extension of the cAMP/PKA signaling pathway framework described by Bhalla (26). Key reaction modules are shown in Fig. 1, A–H, describing the VIP-activated cAMP/PKA signal transduction that ultimately leads to gene induction in the nucleus. The model incorporates the following molecular processes originally modeled by Bhalla (26):
Ligand (L) binding to receptor (R) (Fig. 1 A). The receptors exist either by themselves or coupled with G-protein (R.GDP.Gs).
G-protein activation (Fig. 1 B). When ligand binds to the receptor coupled with G-protein (L.R.GDP.Gs), it induces guanine triphosphate (GTP)/guanine diphosphate (GDP) exchange and G-protein activation (GTP.Gα).
Adenylate cyclase (AC) activation (Fig. 1 C). Activated Gα binds AC, forms activated adenylate cyclase (Gas.AC), and produces cAMP.
PKA activation (Fig. 1 D). Four cAMP molecules bind sequentially to each PKA inactive heterotetramer (R2C2) and release two activated PKA catalytic subunits.
Phosphodiesterase activation (Fig. 1 E). Activated PKA activates cAMP phosphodiesterase (PDE), which converts cAMP to AMP.
PKA inhibition (Fig. 1 H). A cytoplasmic protein slowly inhibits PKA activity.
FIGURE 1.

(A-H) Schematic kinetic reactions of VIP-activated signaling process leading from receptor binding to activated gene transcription. Reversible reactions are represented by double arrows and enzymatic reactions are represented by enzyme species over the single arrow reactions. Ligand receptor complex internalization is represented by dotted line arrows. L, ligand (VIP); R, receptor; L.R, ligand receptor complex; R.GDP.Gs, receptor G-protein complex; L.R.GDP.Gs, ligand bound receptor G-protein complex; GTP.Gα, activated G-protein; Gβγ, G-protein β- and γ-subunit; AC, adenylyl cyclase; Gα.AC, stimulatory G-protein-activated AC; GDP.Gs, GDP-bound G-protein trimer; R2C2, protein kinase A catalytic unit/regulatory unit heterotetramer; cAMP.R2C2-cAMP4.R2C2, cAMP complexed with PKA tetramer; PDE, phosphodiesterase; PDE*, phosphorylated phosphodiesterase; CREB*, phosphorylated CREB. (I) Flow sheet of the overall VPAC2 signaling reaction network. Abbreviations are as above, except that PR2 indicates PKA regulatory subunit dimer and IPKA indicates PKA inhibitor. (J) Flow sheet for the Leloup and Goldbeter (24,25) mammalian circadian rhythm model with input from the VPAC2 model, adapted from Leloup and Goldbeter (24). Degradations are indicated by dotted arrows.
We extended the above framework to describe VPAC2 signaling by including the following processes:
Receptor internalization (Fig. 1,A and B). The receptor-ligand complex is internalized as described by Langlet et al. (27), providing another mechanism for the attenuation of signaling.
Basal cAMP formation. To maintain a nonzero steady-state level of cellular cAMP, a nonspecific low level of G-protein activity was added, maintaining cAMP at ∼0.15 nM (21,28).
Additional PKA dynamics (Fig. 1 H). PKA nuclear translocation was added to bridge cytoplasmic signaling with the nucleus. PKA nuclear inhibition and translocation back to the cytoplasm was added to terminate PKA nuclear action. PKA heterodimerization with regulatory units was added to replenish the inactive PKA pool.
CREB activation (Fig. 1 F). CREB phosphorylation by nuclear PKA was modeled, extending the intracellular signaling to gene regulation.
- Clock gene (per1) induction (Fig. 1 G). Per1 promoter binding by phosphorylated CREB and subsequent transcriptional activation was modeled, bridging the VPAC2 signaling pathway model to the core circadian clock. Transcriptional regulation of per1 by CREB was modeled using the framework employed by Leloup and Goldbeter (24,25) to model circadian regulation of per1:

(1)
where Mp is the per1 mRNA level, CREB* is the phosphorylated CREB level, CREB*0 is the level of phosphorylated CREB in the absence of VIP, vsp is the maximum per1 transcription rate, KCP is the phosphorylated CREB level for half-maximal transcription, n is the degree of cooperativity of activation of per1 transcription by CREB, vmp is the maximal saturating enzymatic degradation of per1 mRNA, Kmp is the per1 mRNA level for half-maximal degradation, and kdmp is the nonsaturating per1 mRNA degradation constant.
The overall VPAC2 signaling network is shown in flow diagram format in Fig. 1 I, where the complex feedback loops and intrinsically self-limiting nature of the pathway are apparent. Overall, the VPAC2 signaling pathway model consists of 36 ordinary differential equations (ODEs) and 66 parameters. It is a single-cell model where the cytoplasm and nucleus are assumed to be “well-stirred (but separate) compartments”, i.e., perfect diffusional access to all components within each compartment. All reaction rates were based on standard mass action kinetics, except for the Hill function and Michaelis-Menten terms in Eq. 1.
The model parameters were tuned as follows:
The initial values for the first 40 parameters were obtained from Bhalla (26); the remaining parameters were assigned coarse initial estimates.
Experimental data from the literature were collected for the VIP responses of several system components, including cAMP, PKA, and CREB phosphorylation.
We fit the experimental data for each system component individually, starting with cAMP (early in the signaling pathway), and finish with CREB phosphorylation (bridging the signaling pathway to the nucleus).
For each system component model, we performed a sensitivity analysis in which all of the model parameters were tested for how strongly they affected the system response. Values of the parameters that most strongly controlled the concentration profile of the system component were varied until the simulation predictions matched the experimental data closely.
Parameters that remain unchanged from the original values reported in Bhalla (26) were related to ligand binding, receptor/G-protein coupling, G-protein activation, AC activation, and PKA inhibition (specifically, k1, k2, k5, k6, k7, k9, k10, k11, k16, k18, and k29—see Supplementary Material). A complete list of all equations, parameters, and initial values is provided in the Supplementary Material.
As described above, sensitivity analysis played an important role in our parameter fitting procedure. We also employed sensitivity analysis to gain insight into the model behavior itself. We used the accumulated per1 mRNA concentration over the increasing phase of the induction as a system output and evaluated the extent to which it was affected by variations in the model parameters. More specifically, the following formula was used to calculate a scaled change in per1 output in response to a change in parameter value (S):
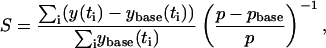 |
(2) |
where y(ti) is the level of per1 at time ti for the perturbed parameter value, ybase(ti) is the level of per1 in the unperturbed case, pbase is the initial parameter value, and p is the perturbed parameter value. The simulations were performed until t = 2000 s (VIP treatment at t = 0) and S value were calculated using 50 time points from t = 1000 s to t = 2000 s with a sampling time of 20 s. Parameters were varied individually, over a range from 0.5 to 2 times the nominal values. This local sensitivity analysis (29) provided insights into the biological processes most important in controlling the system output.
We also investigated the sensitivity of the model responses to the duration of the VIP input signal. As for the local sensitivity analysis described above, we used per1 mRNA concentration time course profile as the output, but instead of computing sensitivity coefficients, we simply compared the time courses for differing pulse durations. We considered VIP inputs at 100 nM concentration with pulse durations ranging from 1 to 30 min.
To simulate the circadian phase-shifting properties of VIP in the SCN, we linked the VPAC2 signaling pathway model to the short form of Leloup and Goldbeter (24,25) mammalian circadian clock model. The Leloup and Goldbeter (24,25) model describes the transcriptional feedback network consisting of the core clock proteins Per1, Cry, and Bmal1/Clock and their posttranslational regulation by reversible phosphorylation. It consists of 16 ODEs with 53 parameters and is shown schematically in Fig. 1 J. We linked the Leloup and Goldbeter (24,25) model to our VPAC2 signaling model through transcriptional regulation of per1, a gene induced downstream of VPAC2 that is also a core clock gene. Regulation of per1 transcription by VPAC2 signaling and the circadian clock is accomplished by two different mechanisms. In the first case, per1 induction is mediated by CREB (as described in the model above), whereas in the second it is mediated by nuclear Bmal1/Clk protein complexes. It has been shown that the two mechanisms are independent (to a degree) in that disruption of CREB binding sites has no impact on per1 regulation by Bmal1/Clk (20). This independence has led us to postulate two functional hypotheses about the mechanisms by which the circadian clock and VPAC2 signaling jointly regulate per1 induction, shown here in Eqs. 3 and 4:
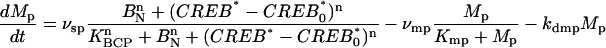 |
(3) |
 |
(4) |
where the symbols are as defined for Eq. 1 with the addition of BN representing the level of nuclear Bmal1/Clk complex. Equation 3 is a model for the case where CREB and Bmal1/Clk function independently but recruit the transcriptional apparatus by the same means. In this case, if the levels of Bmal1/Clk are high enough for transcriptional recruitment to be saturated, activation of CREB will not lead to further increase in transcriptional initiation (Fig. 2 A). The functional form is based on that employed by Ueda et al. (30). Equation 4 is a model for the case where CREB and Bmal1/Clk recruit the transcriptional apparatus through separate mechanisms, and thus activation of CREB may lead to either additive or synergistic transcriptional activation of per1 in connection with Bmal/Clk (Fig. 2 B).
FIGURE 2.

Schematic representation of the two mechanisms for the combined VIP signaling- and circadian clock-regulated per1 gene expression. (A) The mechanism represented by Eq. 3, where either VIP signaling-activated CREB or the circadian transcription factor Bmal1/Clk can activate the transcription machinery to reach its maximum rate. When both are present, it is effectively as if only one is binding to the transcriptional machinery at a time to reach the maximum rate of transcription. (B) The mechanism represented by Eq. 4, where both CREB and Bmal1/Clk can activate the gene transcription to their individual respective maxima. When both are present, they both bind to the transcriptional machinery and have an additive or synergistic effect on the rate of transcription. C, CREB; B/C, Bmal1/Clk; Pol II, RNA polymerase II. The arrow denotes the direction of per1 transcription.
The models were implemented in MATLAB (The MathWorks, Natick, MA) and simulations were performed using the MATLAB stiff ODE integrator ode15s (31).
RESULTS
Model development and validation
We expanded the framework developed by Bhalla (26) for the cAMP/PKA signaling pathway and used it to describe the VIP receptor VPAC2-activated signaling through the cAMP/PKA pathway to circadian clock gene per1 induction in the circadian pacemaker SCN cells. This model has incorporated PKA nuclear translocation upon activation, CREB phosphorylation by PKA, and the ensuing gene activation. To capture the dynamics of VPAC2 signaling with our model, we systematically tuned the model parameters to match experimental data from the literature. Specifically, we systematically varied the parameters until we converged on a set of values for which the model predictions optimally matched the experimental kinetic profiles for cAMP accumulation, PKA activation, and CREB phosphorylation simultaneously. The details for each model component are given below.
Fitting VIP-induced cAMP accumulation
As described above, VPAC2 is a GPCR coupled with Gs that activates the cAMP/PKA signaling pathways. To fit the model to literature data on VIP-induced cAMP accumulation, we performed a local sensitivity analysis to identify the most sensitive parameters for cAMP production, as described in the Materials and Methods section. The parameters most important for matching the cAMP profile were related to G-protein coupling to ligand-receptor complex (k3 and k4), ligand binding to receptor-G-protein complex (k7 and k8), G-protein activation (k10), cAMP production (k12, k13, and k14), cAMP hydrolysis (k35, k36, and k37), and GTP hydrolysis (k43) (data not shown). These parameters were then varied individually to fit experimental data. After fitting the model, we obtained kinetic profiles in cAMP accumulation in response to VIP treatment similar to those obtained with cultured GH3 cells (28), as shown in Fig. 3. The simulated cAMP accumulation shows a fast increasing phase, a peak in ∼5–10 min after VIP treatment, and then a slow decrease back to basal level (Fig. 3 A). A comparison of our simulation results to the cAMP increase in GH3 cells, experimentally measured at a single time point, is shown in Fig. 3 A. The simulated cAMP accumulation also showed dose dependence with an EC50 = 1.5 nM compared to EC50 = 2.2 nM as reported in GH3 cells (28) (Fig. 3 B). It is also interesting to note that the experimentally obtained dose response curve showed a decrease in cAMP level at 100 nM VIP concentration as compared to at 10 nM. However, our simulated cAMP kinetic profile showed a corresponding increase in cAMP peak level with increasing VIP concentration (Fig. 3 A). The discrepancies arise from the timing of the sampling: the experimental data were obtained 15 min after VIP treatment, which our simulations indicate (at least for higher VIP concentrations) was already beyond the time for the peak level of cAMP (Fig. 3 A). Because our simulation indicated a slightly different kinetic profile for different concentrations, and because of the discrepancies between our simulation and reported dose response curve, it would be desirable to measure cAMP increases across a time span and at different VIP concentrations to cover the whole dynamic range of cAMP concentration.
FIGURE 3.

VIP-induced cAMP accumulation. (A) Simulated cAMP accumulation at different VIP concentrations. VIP addition was at t = 0. Symbols represent experimentally observed cAMP concentration at 800 s in GH3 cells (see Mackenzie et al. (28)). Different symbols correspond to different VIP concentrations as denoted in the legend. (B) Simulated dose response curve (solid line) compared to experimental data (○) that obtained from GH3 cells (28), 15 min after VIP treatment.
Fitting VIP-induced PKA activation
One effect of elevated cAMP concentration in the cytosol is activation of PKA. In our simulation, PKA activation follows cAMP accumulation with a slight delay, with increasing cAMP accumulation resulting in increased PKA activation. The procedure for fitting the model to reported PKA activation profile is similar to the procedure described above for cAMP (see previous section). We matched our model predictions to experimental observations in cultured cells (17), where PKA activation peaked ∼5 min after VIP treatment and returned to baseline within 20 min (Fig. 4). The most important parameters for fitting the PKA activation profile are related to active PKA nuclear translocation (k45), nuclear active PKA inhibition (k49 and k50), PKA inhibitor nuclear translocation (k57 and k58), and cAMP hydrolysis from PKA regulatory units (k60). The association constant (Ka) value we obtained from the model fitting, 0.5 nM, is very close to the experimentally determined value of 0.4 nM (17). However, without quantitative kinetic data, the fitting had to be qualitative, and the simulated results are in arbitrary units. Nonetheless, the simulated PKA activation resembles experimentally observed PKA activation profiles quite well (17).
FIGURE 4.

Simulated PKA, phosphorylated CREB, and per1 mRNA concentrations after VIP administration at time 0. All concentrations are normalized to give respective peak levels of 1. Dotted line is simulated PKA concentrations, dashed line is simulated phosphorylated CREB concentration, and solid line is simulated per1 mRNA concentration. (♦) Per1 mRNA expression data following foskolin stimulation from Yagita and Okamura (32).
Fitting VIP-induced CREB phosphorylation
Activation of the cAMP/PKA signaling pathway leads to transcriptional activation via activation of the CREB. Using a procedure similar to the ones in the two previous sections, we matched our model predictions to data from one detailed study in the rat pinealocyte that considered phosphorylation of CREB after VIP treatment (18). In both the experimental study and our simulations, phospho-CREB reaches peak levels 30 min after VIP treatment and returns to baseline in ∼2 h (Fig. 4). The most important parameters controlling the CREB phosphorylation profile were related to CREB phosphorylation (k47 and k48) and dephosphorylation (k59). Similar to PKA activation, only qualitative fitting was possible and the simulated CREB activation profile is in arbitrary units.
VIP-induced per1 gene expression
VIP induces the clock genes per1 and per2 in the SCN at least partly through the cAMP/PKA signaling pathways (6). We included CREB-mediated gene activation in our model using the scheme employed by Leloup and Goldbeter (24,25) (Eq. 1) and including per1 induction after VIP treatment. Using the parameters for per1 mRNA accumulation from the Leloup and Goldbeter model, our simulations results show per1 transcripts peaking ∼100 min after VIP treatment, returning to basal level in ∼240 min (Fig. 4). These results resemble forskolin-induced per1 expression in rat-1 fibroblasts (32), an in vitro model of the mammalian circadian clock (33).
Model sensitivity analysis
To assess the sensitivity of per1 induction to parameter changes, we systematically varied each individual parameter by a factor of 0.5, 0.95, 1.05, or 2, and computed scaled percentage changes in per1 mRNA concentrations during the increasing phase of induction as described in the Materials and Methods section (Eq. 2). Of the 66 parameters, 16 gave changes >±25% under at least one of the changed conditions; 16 parameters gave changes <25% but >5% under at least one of the changed conditions; and 34 parameters either did not change the per1 expression under any of the changed conditions or the changes were <5%. The most sensitive parameters include, in order of importance, vsp (maximum rate for per1 mRNA synthesis), k49 (binding rate for nuclear inhibitor binding to nuclear PKA), k59 (CREB* dephosphorylation rate), k12 (rate of cAMP production by activated AC), k13 (rate of ATP binding to activated AC), k51 (rate for nuclear-inhibited PKA translocation to cytoplasm), k55 (rate of inhibited PKA to reform holoenzyme with PKA regulatory unit), k46 (rate for PKA binding to CREB), k50 (dissociation rate for nuclear inhibitor bound to PKA), k57 (rate of PKA inhibitor nuclear translocation), k10 (rate of G-protein activation), k52 (rate of backward translocation of inhibited PKA to nuclei), vmp (maximum rate for per1 mRNA degradation), kmp (Michaelis constant for per1 mRNA degradation), k14 (dissociation rate for ATP-bound AC), and k43 (rate of GTP hydrolysis) (Fig. 5 A). The model showed considerable sensitivity to both increases and decreases in vsp, vmp, kmp, k12, k13, k14, and k43, but was more sensitive to increases in k46 and k50 and to decreases in k49, k51, k55, and k59. Because vsp, vmp, and kmp are directly involved in the per1 mRNA synthesis and degradation, it is not surprising that the per1 mRNA time course was directly proportional to changes in these parameters. Similarly, k10, k12, k13, k14, and k43 are involved in AC activation and cAMP synthesis, and, not surprisingly, per1 mRNA accumulation was found to be very sensitive to changes in their values.
FIGURE 5.

Model sensitivity analysis. (A) Model sensitivity to parametric changes: parameters were varied individually in the range of 0.5, 0.95, 1.05, and 2 times the original value; the 16 most sensitive parameters, which gave rise to at least 30% changes in one of the four perturbations introduced, are shown. (B) Model sensitivity to perturbation signal duration: short pulses of VIP input were similarly effective as a step VIP input in inducing per1 transcription.
The parameters with moderate sensitivity include, in order of importance, k56 (rate of backward reaction for inactive PKA holoenzyme re-formation), k58 (rate of backward reaction for PKA inhibitor nuclear translocation), n (degree of transcription factor cooperativity), k35/36/37 (phosphodiesterase-catalyzed cAMP degradation), kAP (activation constant for per1 transcription), k3/4 (liganded receptor binding to G-protein), k41 (rate of basal G-protein activation), k19/20/21/22/23 (rate for cAMP binding to PKA holoenzyme), and k42 (rate of receptor internalization) (data not shown).
Per1 induction was not sensitive to the remaining parameters, including k1/2 (ligand binding to G-protein free receptors), k5/6 (receptor and G-protein coupling), k7/8 (ligand binding to G-protein-coupled receptors), k9 (trimeric G-protein formation), k11 (GTP hydrolysis), k15/16 (Gα and AC binding), k17/18 (rate for first cAMP molecule binding to PKA holoenzyme), k24–28 (rate for releasing PKA catalytic units), k29/30 (cytoplasmic PKA inhibition), k31–34 (phosphodiesterase phosphorylation and dephosphorylation), k38/39 (phosphorylated phosphodiesterase binding to cAMP), k44/45 (PKA nuclear translocation), k47 (PKA CREB dissociation), k48 (CREB phosphorylation), k53/54 (PKA tetramer re-formation), k60 (hydrolysis of cAMP bound to PKA regulatory units), and kdmp (basal per1 mRNA degradation) (data not shown).
Overall the sensitivity of the per1 mRNA time course was similar to what we observed earlier while tuning the parameters to fit experimental data to the predicted model responses of specific key signaling molecules, i.e., cAMP, PKA, CREB phosphorylation, and per1 induction. However, it was surprising to note that the overall model was not sensitive to the rate of PKA dissociation from CREB (k47) and the rate of CREB phosphorylation (k48). Phosphorylated CREB concentration is directly linked to per1 transcriptional activation. The fact that per1 transcriptional induction was not sensitive to CREB phosphorylation but sensitive to CREB dephosphorylation suggests that the balance of phosphorylated CREB is more dependent upon the rate of dephosphorylation.
To this point, all simulations have been performed with a step input in the VIP signal (i.e., VIP was added and the concentration held constant in the system throughout the simulation run). However, the bioactive effect of VIP in vivo only lasts a few minutes in biological fluids due to its rapid degradation and inactivation by enzymes, catalytic antibodies, and spontaneous hydrolysis (see Sethi et al. (34) for a review). To investigate the effects of transient VIP stimulation, we considered pulses in VIP concentration of 100 nM sustained for different durations. The results are shown in Fig. 5 B. A 1-min pulse of VIP gave rise to a peak level of per1 induction ∼20% of that obtained in response to a step signal; for a 2-min pulse, the resulting peak level is ∼50% of the step response and ∼80% for a 5-min pulse; the 10-min pulse response is almost indistinguishable from the step response (Fig. 5 B). These results suggest that a short pulse of VIP could be as effective as a step signal in inducing per1 gene expression in our model, reflecting the high sensitivity of the VPAC2 signaling pathway to short pulse signals in vivo, as well as the robustness of the system to pulse inputs of longer duration than 10 min. These results have physiologically important consequences, as discussed below.
Modeling VIP-induced phase shifts in the circadian clock
Injection of VIP into SCN has been shown to induce phase shifts in Syrian hamsters in vivo and in rat in vitro (3,4,8). We linked our VPAC2 signaling model to the Leloup and Goldbeter (24,25) circadian clock model to simulate the VIP-induced phase shift. Based on the observation that Bmal1/Clk- and CREB-mediated per1 promoter activation are at least partially independent of each other (20), we postulated two functional hypotheses about the mechanisms by which the circadian clock and VPAC2 signaling jointly regulate per1 induction (Materials and Methods, Eqs. 3 and 4). In both cases, VIP induced phase shifts of the circadian clock. The model represented by Eq. 3 generates phase shifts similar to experimentally measured locomotor behavior phase shifts induced by VIP injection into the SCN in vivo (3), though with a larger phase delay than the experimental observation (Fig. 6 A). In our simulation, VIP applied at late night (rising phase of per1 mRNA near nadir) induced a 1 h phase advance, which is nearly the same as in the experimental data (60 vs. 50 min) (Fig. 6 A). However, VIP applied at early night (lowering phase near per1 mRNA nadir) induced an ∼80 min phase delay, which was almost twice the mean phase delay observed in experiments (40 min.) (Fig. 6 B). The model represented by Eq. 4 generates phase shifts similar in magnitude to what has been observed in SCN neuron firing rhythm in vitro (7,8) (Fig. 6 B). VIP applied at late night induced a large 4 h phase advance (compared to an average 3 h phase advance observed in the SCN firing rhythm) (Fig. 6 B). VIP applied at early night induced a smaller 80 min phase delay, whereas VIP applied to SCN slices induced a 60 min phase delay in SCN firing rhythms (8) (Fig. 6 B).
FIGURE 6.

Simulated VIP-induced phase shift in SCN circadian clock using two different transcriptional regulation mechanisms. (A) Phase-shifting as predicted with combined transcriptional saturation represented by Eq. 3. (B) Phase-shifting as predicted with separate transcriptional saturation represented by Eq. 4.
To study the phase dependence of the VIP-induced phase shift, we simulated treatment with 100 nM VIP at each hour of the entire circadian cycle and generated phase response curves (Fig. 7 A). Lacking experimental VIP phase response curve data, we compared the phase response curve to that of another neuropeptide: gastrin-releasing peptide (GRP1–27) (4). GRP1–27 is a ligand for a different G-protein-coupled receptor that activates different signaling pathways (35,36). Nevertheless, it has been demonstrated that GRP1–27 and VIP induce similar phase shifts (4). Therefore, the GRP1–27 phase response curve is used currently as a reference for comparison. The simulated phase response curve generated using Eq. 3 closely resembles the phase response curves of light and GRP1–27. The simulated phase response curve generated using Eq. 4, however, shows VIP-induced phase shifts throughout the circadian cycle, with phase advances from late night to early day and phase delays from late day to early night (Fig. 7 A).
FIGURE 7.

Simulated VIP-induced phase response curve. (A) Phase response curve induced by a step increase in VIP. Open circles are data from GRP1–27-induced phase shift (see Piggins et al. (4)). (B) Phase response curve induced by a 5-min short pulse. Solid line is the phase response curve simulated with Eq. 3; dashed line is the phase response curve simulated with Eq. 4.
We showed earlier that a short pulse of VIP induced similar per1 transcriptional response as did the step signal (Fig. 5 B). To see if a short pulse of VIP will also induce appropriate phase shift throughout the circadian cycle, we simulated VIP phase response curves using a 5 min, 100 nM VIP pulse. The simulated phase response curve obtained from the model in Eq. 3 was almost identical to that for the step input in VIP (Fig. 7 B). The simulated phase response curve obtained from the model in Eq. 4 still followed the same general pattern as that resulting from the step input signal; however, phase advances were smaller, whereas the delay was about the same (Fig. 7 B). These results indicate that short pulse VIP signals were effective not only in generating per1 response from the VIP signaling model but also in phase-shifting the circadian clock model.
Transgenic mice overexpressing VPAC2 receptor showed faster re-entrainment to photoperiod phase-shifting as compared to wild-type animals (13). To investigate the modulation of the phase response curve by VPAC2 receptor overexpression or suppression, we simulated receptor overexpression and suppression in our linked model, using as the perturbation a step increase in the VIP signal (Fig. 8). With the model in Eq. 3, lowering the receptor level to half of the nominal level slightly reduced the peak phase shift for both phase advance and phase delay. However, doubling the receptor level showed a more pronounced phase advance than phase delay (Fig. 8 A). For the model in Eq. 4, the entire phase response curve was shifted either up or down in direct relation to an increase or decrease in the receptor concentration (Fig. 8 B). With reduced receptor concentration, the phase delay became larger and phase advance smaller, whereas with increased receptor concentrations, phase advance increased and phase delay decreased. For both models, the changes in receptor level led to changes in phase-shifting for whichever changes in phase advance were more pronounced. These observations are in accordance with the reported photoperiod phase-shifting (13), where animals overexpressing the VPAC2 receptors showed a significantly enhanced re-entrainment to a phase advancing photoperiod change. Based on the simulation, we would predict that a single pulse of light or VIP will induce larger phase advances in the transgenic mice overexpressing VPAC2 as compared to wild-type mice.
FIGURE 8.

Phase advance is more sensitive than phase delay to receptor level changes. (A) Simulated phase response curves using the model represented by Eq. 3. (B) Simulated phase response using the model represented by Eq. 4. Solid lines are simulated phase response curves with nominal receptor levels. Dotted lines are simulated phase response curves with doubled receptor levels. Dashed lines are simulated phase response curves with half amount of nominal receptor levels.
DISCUSSION
Circadian rhythms are generated by molecular oscillations in cells that are modulated by extracellular stimuli. The SCN, as the central oscillator, orchestrates coordinated rhythmic processes throughout the body and receives many stimuli originating from the environment and within the organism itself. How the SCN integrates these numerous stimuli into a consistent and precise molecular oscillation is not clear. In an initial effort toward understanding this process, we report in this work a mathematical model that describes VIP-activated VPAC2 receptor signaling and a coupling of the model to the circadian clock. We chose VIP for this in silico study because it has been suggested to be a paracrine/autocrine molecule contributing to the maintenance of sustained SCN oscillations, exerting its effect through the VPAC2 receptor (14,15).
Using a modeling framework for the cAMP/PKA signaling pathway developed by Bhalla (26), we built a kinetic model describing VIP-activated signaling in SCN cells that culminates in circadian clock gene induction. We tuned our model parameters to obtain good quantitative and qualitative fits to experimental data from the literature for VIP-induced cAMP accumulation (Fig. 3), PKA activation (Fig. 4), CREB phosphorylation (Fig. 4), and per1 gene induction (Fig. 4). It is worth noting that the different measurements we used to fit the model were from different cells. We recognize that different cells will potentially have different kinetics, and that SCN cell specific measurements are needed for better parameter estimates. Nonetheless, fitting the model to experimental measurements gives us a working initial model with which we can explore the VPAC2-mediated signaling dynamics and functionality in a biologically relevant context of the circadian clock. Importantly, linking the model to a circadian clock showed good agreement with experimentally observed phase shifts (Figs. 6 and 7).
Sensitivity analysis of the model allowed us to identify the 16 most sensitive model parameters for predicting per1 gene induction (Fig. 5 A): vsp, vmp, kmp, k10, k12, k13, k14, k43, k46, k49, k50, k51, k52, k55, k57, and k59. These were related to per1 mRNA synthesis, degradation, AC activation and deactivation, PKA inhibition, and CREB phosphorylation and dephosphorylation. It is entirely reasonable that the model prediction is sensitive to these parameters, given that they are involved in the critical steps of cAMP production, PKA activation, CREB activation, and mRNA synthesis. Surprisingly, per1 transcriptional induction in our model did not seem to be particularly sensitive to the rate of CREB phosphorylation in the range tested, rather showing sensitivity to the rate of CREB dephosphorylation—indicating that the rate of CREB dephosphorylation plays a more important role in maintaining the activated CREB concentration.
Since the half-life of VIP in biological fluid is only a few minutes (34), VIP signal duration is a critical issue in tissues. To test the sensitivity of the model to the VIP signal duration, we simulated the response to short pulses of VIP inputs. A 2-min VIP pulse was able to elicit a per1 induction up to ∼50% of the peak level of the step VIP response, whereas a 5-min pulse response achieved ∼80% of the step response peak level (Fig. 5 B). The sensitivity of the model to the short pulses, and the robustness of the response to variations in input duration, once a critical duration is exceeded—observations that have physiologically relevant consequences—seem to validate the biological relevance of the model. The robustness of the system response to pulse inputs with durations >∼10 min may provide a physiological advantage in that variability in VIP release (that may arise from intrinsic biological noise) does not lead to variability in the system response: noisy input signals are converted into precise output signals, provided that the input duration threshold (∼10 min) is exceeded. The fact that the system response to VIP stimulation is intrinsically limited also may provide a protective mechanism, preventing potentially cytotoxic overstimulation. Intrinsically limited responses to a single input also allow for the requirement of synergistic or combinatorial inputs to achieve sustained responses. Lastly, the similarity between system responses to pulse and step inputs indicates that experimental observations obtained using step inputs may accurately reflect what occurs under physiological conditions.
By linking our VPAC2 signaling model to the Leloup and Goldbeter (24,25) mammalian circadian clock model, we were able to simulate the phase-shifting effects of VIP on the SCN clock as evidenced in the per1 gene oscillation. Using two models for the combined circadian and VIP regulation of per1 gene expression, we matched VIP-induced phase shifts to experimentally observed phase shifts in locomotor behavior rhythm and SCN neuronal firing rhythm (Fig. 6). In the first case, the circadian clock regulated per1 gene expression, mediated by Bmal1/Clk, and the VIP-induced per1 gene expression, mediated by phosphorylated CREB, are independent but they recruit the transcription machinery in the same manner and thus can saturate per1 transcription individually (Fig. 2 A). When Bmal1/Clk regulation of per1 transcription is saturated, phosphorylated CREB can no longer induce transcription. Under such circumstances, a step increase in the VIP signal phase-shifted the circadian clock at early and late night as experimentally observed (4), although the simulated phase delays early in the night were greater than observed (Fig. 6 A). The simulated phase response curve mimics the light phase response curve and the GRP1–27 phase response curve (Fig. 7 A). Saturation of per1 transcription by Bmal1/Clk during the day limited the action of the VIP signaling. This effectively served as a mechanism for circadian clock gating on VIP induction of per1 gene expression and restricted phase shifts to the night phase (Fig. 7 A). The limitation of the model is that it cannot explain the large phase shift observed in vitro in SCN neuron firing rhythm (7).
In the second model, we described the circadian clock regulation of per1 gene expression as completely independent from VIP-induced per1 gene expression such that Bmal1/Clk recruits the transcriptional machinery separately from CREB. Thus CREB can induce per1 gene expression even when the Bmal1/Clk induced transcription is at its maximal rate (Fig. 2 B). The simulated phase shift matched the observed in vitro SCN neuron firing rhythm phase shift (Fig. 6 B); and the phase response curve in this case showed phase-shifting throughout the circadian cycle, thus necessitating an additional gating mechanism to match the experimentally observed phase response curve. However, this model was able to describe the large phase shift induced by VIP in SCN firing rhythm observed in vitro (7,8). With currently available data, it is not possible to determine whether either model or some appropriate combination of the two represents the mechanism of the integration of VIP signaling and circadian clock function more realistically. These different possibilities and their possible combined contributions to the VIP-induced phase shift warrant further experimental investigation.
It is interesting to note that with the limiting saturation rate of transcription as described in Eq. 3, the limitation serves as a gating mechanism for VIP-induced phase shift. Recently, Geier and colleagues (37) modeled light entrainment of the circadian oscillator based on the independence of light-induced transcription of per1 gene from Bmal1/Clk-driven transcription. In their model, a separate gating term was introduced to fit the light phase response curve. Our results indicate that with a limiting maximum transcription rate, clock-gated gene induction can be explained without an additional gating term. In terms of gene regulation, this makes sense: although various transcription factors can act independently to activate transcription, they do act to recruit transcriptional machinery to the same promoter; and the rate of transcription is limited to how fast the transcriptional machinery can be loaded onto the promoter and initiate transcription (of course, with the assumption that there is no interaction between the two sets of transcription factors). Therefore, if two separate transcription factors bind to the same promoter and they can function independently of each other, their combined transcriptional activation is still limited by the promoter capacity to initiate transcription.
We also tested if a short pulse of VIP is capable of phase-shifting the circadian clock. Our simulation using a 5-min pulse of VIP phase-shifted the circadian clock in a pattern similar to the simulated response to a sustained VIP step signal (Fig. 7 B)—an observation with physiological implications indicating that the model describes in vivo VIP signaling pathways reasonably well in the sense that the model response to short duration of VIP signals and the induced phase response are similar to behavioral responses.
Varying the quantity of the VPAC2 receptor in the model resulted in a corresponding variation in the magnitude of the phase shift induced by VIP. Increasing the VPAC2 receptor amount induced a larger phase advance using both coupling mechanism represented by Eqs. 3 and 4 (Fig. 8), whereas decreasing VPAC2 receptor amount reduced phase delay with Eq. 3 but increased phase delays with Eq. 4 (Fig. 8). However, changes in phase advances were more pronounced than changes in phase delays. These observations agree with the reported data from transgenic mice overexpressing VPAC2 (13). Our simulation also predicts that a single pulse of light or VIP will induce larger phase advances in the transgenic mice overexpressing VPAC2 as compared to wild-type mice. It would be interesting to see the prediction verified experimentally using transgenic animals.
In summary, we have presented the development of a kinetic model for the VIP-induced signal transduction process in SCN cells and compared our model simulation results to published data. These simulation results matched experimental data closely for cAMP accumulation, PKA activation, CREB phosphorylation, and per1 gene induction. When the model was linked to a circadian clock model, the predicted VIP-induced phase shifts closely resembled experimental observations for most of the circadian day. Also, simulations of varying amounts of the VPAC2 receptor predicted that overexpression of the receptor will cause a larger phase advance. However, we recognize that in reality, the VIP-induced phase shift is much more complicated than what we have described so far. For example, it is evident that the signaling pathways responsible for VIP signaling to the clock include both protein kinase A and phospholipase C pathways (6), and that mitogen-activated protein kinase signaling also contributes to the phase-shifting effect of VIP (7). Our next goal is to describe these signaling pathways and incorporate them into our model. We will take an approach of combined modeling and experimental validation to further our understanding of the role of VIP in maintaining the SCN circadian clock. Lastly, it is worth highlighting that other computational models for the circadian clock mechanism are available in the literature (38,39). It will be interesting to see if differences in the phase shift can be generated from simulations based on these other models.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health grant MH64459-01, Defense Advanced Research Projects Agency research contract BAA-01-26, and funding from Delaware Biotechnology Institute, University of Delaware.
References
- 1.Davidson, A. J., S. Yamazaki, and M. Menaker. 2003. SCN: ringmaster of circadian circus or conductor of the circadian orchestra. Novartis Found. Symp. 253:110–121. [PubMed] [Google Scholar]
- 2.Gillette, M. U., and J. W. Mitchell. 2002. Signaling in the suprachiasmatic nucleus: selectively responsive and integrative. Cell Tissue Res. 309:99–107. [DOI] [PubMed] [Google Scholar]
- 3.Albers, H. E., S. Y. Liou, E. G. Stopa, and R. T. Zoeller. 1991. Interaction of colocalized neuropeptides: functional significance in the circadian timing system. J. Neurosci. 11:846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piggins, H. D., M. C. Antle, and B. Rusak. 1995. Neuropeptides phase shift the mammalian circadian pacemaker. J. Neurosci. 15:5612–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cagampang, F. R., H. D. Higgins, W. J. Sheward, A. J. Harmar, and C. W. Coen. 1998. Circadian changes in PACAP type 1 (PAC1) receptor mRNA in the rat suprachiasmatic and supraoptic nuclei. Brain Res. 813:218–222. [DOI] [PubMed] [Google Scholar]
- 6.Nielsen, H. S., J. Hannibal, and F. Fahrenkrug. 2002. Vasoactive intestinal polypeptide induces per1 and per2 gene expression in the rat suprachiasmatic nucleus late at night. Eur. J. Neurosci. 15:570–574. [DOI] [PubMed] [Google Scholar]
- 7.Meyer-Spasche, A., and H. D. Piggins. 2004. Vasoactive intestinal polypeptide phase-advances the rat suprachiasmatic nuclei circadian pacemaker in vitro via protein kinase A and mitogen-activated protein kinase. Neurosci. Lett. 358:91–94. [DOI] [PubMed] [Google Scholar]
- 8.Reed, H. E., A. Meyer-Spasche, D. J. Cutler, C. W. Coen, and H. D. Higgins. 2001. Vasoactive intestinal polypeptide (VIP) phase-shifts the rat suprachiasmatic nucleus clock in vitro. Eur. J. Neurosci. 13:839–843. [DOI] [PubMed] [Google Scholar]
- 9.Kalamatinos, T., I. Kallo, H. D. Piggins, and C. W. Coen. 2004. Expression of VIP and/or PACAP receptor mRNA in peptide synthesizing cells within the suprachiasmatic nucleus of the rat and its efferent target sites. J. Comp. Neurol. 475:19–35. [DOI] [PubMed] [Google Scholar]
- 10.Kallo, I., T. Kalamatianos, N. Wiltshire, S. Shen, W. J. Sheward, A. J. Hamar, and C. W. Coen. 2004. Transgenic approach reveals expression of the VPAC2 receptor in phenotypically defined neurons in the mouse suprachiasmatic nucleus and its efferent target sites. Eur. J. Neurosci. 19:2201–2211. [DOI] [PubMed] [Google Scholar]
- 11.Usdin, T. B., T. I. Bonner, and E. Mezey. 1994. Two receptors for vasoactive intestinal polypeptide with similar specificity and complementary distributions. Endocrinology. 135:2662–2680. [DOI] [PubMed] [Google Scholar]
- 12.Laburthe, M., A. Corvineau, and J. C. Marie. 2002. VPAC receptors for VIP and PACAP. Receptors Channels. 8:137–153. [PubMed] [Google Scholar]
- 13.Shen, S., C. Spratt, W. J. Sherward, I. Kallo, K. West, C. F. Morrison, C. W. Coen, M. H. Marston, and A. J. Harmar. 2000. Overexpression of the human VPAC2 receptor in the suprachiasmatic nucleus alters the circadian phenotype of mice. Proc. Natl. Acad. Sci. USA. 97:11575–11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harmar, A. J., H. M. Marston, S. Shen, C. Spratt, K. M. West, W. J. Sheward, C. F. Morrison, J. R. Dorin, H. D. Piggins, J.-C. Reubi, J. S. Kelly, E. S. Maywood, and M. H. Hastings. 2002. The VPAC2 receptor is essential for circadian function in mouse suprachiasmatic nuclei. Cell. 109:497–508. [DOI] [PubMed] [Google Scholar]
- 15.Aton, S. J., C. S. Colwell, A. J. Harmar, J. Waschek, and E. D. Herzog. 2005. Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat. Neurosci. 8:476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCulloch, D. A., C. J. MacKenzie, M. S. Johnson, D. N. Robertson, P. J. Holland, E. Ronaldson, E. M. Lutz, and R. Mitchell. 2002. Additional signals from VPAC/PAC family receptors. Biochem. Soc. Trans. 30:441–446. [DOI] [PubMed] [Google Scholar]
- 17.Mangeat, P., J. Marvaldi, O. A. Ahmed, and G. Marchis-Mouren. 1981. Parallel activation of cyclic AMP phosphodiesterase and cyclic AMP-dependent protein kinase in two human gut adenocarcinoma cells (HT 29 and HRT 18) in culture, by vasoactive intestinal peptide (VIP) and other effectors activating the cyclic AMP system. Regul. Pept. 1:397–414. [DOI] [PubMed] [Google Scholar]
- 18.Schomerus, C., E. Maronde, E. Laedtke, and H. W. Korf. 1996. Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activating polypeptide (PACAP) induce phosphorylation of the transcription factor CREB in subpopulations of rat pinealocytes: immunocytochemical and immunochemical evidence. Cell Tissue Res. 286:305–313. [DOI] [PubMed] [Google Scholar]
- 19.Lin, J. H., Y. C. Jang, D. C. Wen, and F. F. Wang. 1996. Synergistic activation of cAMP and calcium on cAMP-response-element-mediated gene expression in GH3 pituitary tumor cells. Cell. Signal. 8:111–115. [DOI] [PubMed] [Google Scholar]
- 20.Travnickova-Bendova, Z., N. Cermakian, S. M. Reppert, and P. Sassone-Corsi. 2002. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc. Natl. Acad. Sci. USA. 99:7728–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rea, M. A. 1990. VIP-stimulated cyclic AMP accumulation in suprachiasmatic hypothalamus. Brain Res. Bull. 25:843–847. [DOI] [PubMed] [Google Scholar]
- 22.Vanecek, J., and K. Watanabe. 1998. Melatonin inhibits the increase of cyclic AMP in rat suprachiasmatic neurons induced by vasoactive intestinal peptide. Neurosci. Lett. 252:21–24. [DOI] [PubMed] [Google Scholar]
- 23.Shinohara, K., T. Funabashi, and F. Kimura. 1999. Temporal profiles of vasoactive intestinal polypeptide precursor mRNA and its receptor mRNA in the rat suprachiasmatic nucleus. Brain Res. Mol. Brain Res. 63:262–267. [DOI] [PubMed] [Google Scholar]
- 24.Leloup, J. C., and A. Goldbeter. 2003. Toward a detailed computational model for the mammalian circadian clock. Proc. Natl. Acad. Sci. USA. 100:7051–7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leloup, J. C., and A. Goldbeter. 2004. Modeling the mammalian circadian clock: Sensitivity analysis and multiplicity of oscillatory mechanisms. J. Theor. Biol. 230:541–562. [DOI] [PubMed] [Google Scholar]
- 26.Bhalla, U. S. 2002. Use of Kinetikit and GENESIS for modeling signaling pathways. Methods Enzymol. 345:3–23. [DOI] [PubMed] [Google Scholar]
- 27.Langlet, C., N. Gaspard, I. Nachtergael, P. Robberecht, and I. Langer. 2004. Comparative efficacy of VIP and analogs on activation and internalization of the recombinant VPAC2 receptor expressed in CHO cells. Peptides. 25:2079–2086. [DOI] [PubMed] [Google Scholar]
- 28.Mackenzie, C. J., E. M. Lutz, M. S. Johnson, D. N. Robertson, P. J. Holland, and R. Mitchell. 2001. Mechanisms of phospholipase C activation by the vasoactive intestinal polypeptide/pituitary adenylate cyclase activating polypeptide type 2 receptor. Endocrinology. 142:1209–1217. [DOI] [PubMed] [Google Scholar]
- 29.Saltelli, A., M. Ratto, S. Taratolla, and F. Campolongo. 2005. Sensitivity analysis for chemical models. Chem. Rev. 105:2811–2827. [DOI] [PubMed] [Google Scholar]
- 30.Ueda, H. R., M. Hagiwara, and H. Kitano. 2001. Robust oscillations within the interlocked feedback model of Drosophila circadian rhythm. J. Theor. Biol. 210:401–406. [DOI] [PubMed] [Google Scholar]
- 31.Shampine, L. F., and M. W. Reichelt. 1997. The MATLAB ODE suite. SIAM J. Sci. Comput. 18:1–22. [Google Scholar]
- 32.Yagita, K., and H. Okamura. 2000. Forskolin induces circadian gene expression of rPer1, Per2, and DBP in mammalian rat-1 fibroblasts. FEBS Lett. 465:79–82. [DOI] [PubMed] [Google Scholar]
- 33.Rosbash, M. 1998. Why the rat-1 fibroblast should replace the SCN as the in vitro model of choice. Cell. 93:917–919. [DOI] [PubMed] [Google Scholar]
- 34.Sethi, V., H. Onyuksel, and I. Rubinstein. 2005. Liposomal vasoactive intestinal peptide. Methods Enzymol. 391:377–395. [DOI] [PubMed] [Google Scholar]
- 35.Fitzgerald, L. R., I. J. Mannan, G. M. Dytko, H. L. Wu, and P. Nambi. 1999. Measurement of responses from Gi- Gs- or Gq-coupled receptors by a multiple response element/cAMP response element-directed reporter assay. Anal. Biochem. 275:54–61. [DOI] [PubMed] [Google Scholar]
- 36.Garcia, L. J., T. K. Pradhan, H. C. Weber, T. W. Moody, and R. T. Jensen. 1997. The gastrin-releasing peptide receptor is differentially coupled to adenylate cyclase and phospholipase C in different tissues. Biochim. Biophys. Acta. 1356:343–354. [DOI] [PubMed] [Google Scholar]
- 37.Geier, F., S. Becker-Weimann, A. Kramer, and H. Herzel. 2005. Entrainment in a model of the mammalian circadian oscillator. J. Biol. Rhythms. 20:83–93. [DOI] [PubMed] [Google Scholar]
- 38.Forgers, D. B., and C. S. Peskin. 2003. A detailed predictive model of the mammalian circadian clock. Proc. Natl. Acad. Sci. USA. 100:14806–14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurosawa, G., and Y. Iwasa. 2002. Saturation of enzyme kinetics in circadian clock models. J. Biol. Rhythms. 17:568–577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.