

Abstract
The bacterial K+ channel KcsA from Streptomyces lividans was analyzed by neutron and x-ray small-angle solution scattering. The C-terminally truncated version of KcsA, amenable to crystallographic studies, was compared with the full-length channel. Analyzing the scattering data in terms of radius of gyration reveals differences between both KcsA species of up to 13.2 Å. Equally, the real-space distance distribution identifies a 40 to 50 Å extension of full-length KcsA compared to its C-terminally truncated counterpart. We show that the x-ray and neutron scattering data are amenable for molecular shape reconstruction of full-length KcsA. The molecular envelopes calculated display an hourglass-shaped structure within the C-terminal intracellular domain. The C-terminus extends the membrane spanning region of KcsA by 54–70 Å, with a central constriction 10–30 Å wide. Solution scattering techniques were further employed to characterize the KcsA channel under acidic conditions favoring its open conformation. The full-length KcsA at pH 5.0 shows the characteristics of a dumbbell-shaped macromolecular structure, originating from dimerization of the tetrameric K+ channel. Since C-terminally truncated KcsA measured under the same low pH conditions remains tetrameric, oligomerization of full-length KcsA seems to proceed via structurally changed C-terminal domains. The determined maximum dimensions of the newly formed complex increase by 50–60%. Shape reconstruction of the pseudooctameric complex indicates the pH-induced conformational reorganization of the intracellular C-terminal domain.
INTRODUCTION
The structure of the membrane-spanning region of the tetrameric bacterial K+ channel KcsA, from Streptomyces lividans, has been solved to 3.2 and 2.0 Å resolution comprising amino acids 22–124 of the total 160 amino acids (1,2). The C-terminal domain of KcsA (amino acids 126–160) was removed proteolytically before crystallization and the 21 N-terminal amino acids are positionally disordered in the crystallographic analysis (1). However, primary sequence analysis suggests an α-helical organization of the N- and C-termini, which is supported by electron paramagnetic resonance spectroscopy (EPR) studies performed on the full-length KcsA channel (3). Accordingly KcsA forms a short amphipathic α-helix at its N-terminus and a helical bundle extending the membrane-spanning region past His-124.
This study investigates the suitability of solution x-ray and neutron scattering in characterizing the full-length KcsA K+ channel as a representative of a detergent-solubilized integral membrane protein. We systematically compare the C-terminally truncated version of KcsA with its full-length counterpart in terms of its structural parameters, such as the radii of gyration and maximum real-space dimensions. By using neutron as well as x-ray solution scattering data, we are able to estimate hydration effects observed in the scattering intensity curves as well as to distinguish between detergent micelle and protein scattering by contrast variation.
Characterizing the full-length architecture of KcsA is of particular interest, as this could provide valuable information on regulation of this ion-specific channel. In general, potassium channels consist of a pore-forming ion-conduction pathway endowed with additional N- or C-terminal domains involved in channel gating. To be of benefit for a cell, potassium channels have to display at least two properties: first, the ability to select and conduct K+ ions specifically, and second, the ability to control the processes underlying the opening and closing of the channel, known as gating. Activation (opening) of K+ channels may occur upon interaction with ATP, the βγ-subunit of heterotrimeric G-proteins, phosphatidylinositol 4,5-bisphosphate, or protons, or due to a change in the transmembrane electric potential difference (4–6). The molecular architecture of the ion-conduction pathway of KcsA provides a wealth of information on how K+ ion selection and conductance may occur (1,7,8). The gating of the KcsA K+ channel, however, is less well understood. Electrophysiological data indicate an increased open probability of KcsA at acidic pH (9–11). The gating transitions occurring upon KcsA opening have been studied by equilibrium spectroscopic techniques (12,13), leading to the proposal of a rotation and tilt mechanism of both transmembrane helices. At present, the question remains whether this rotation and tilt mechanism or the outwardly directed hinge-bending mechanism deduced from the open pore conformation of the Ca2+-gated MthK K+ channel (14,15) correctly describes the gating transitions of KcsA.
In this study, we characterize the full-length architecture of the KcsA K+ channel by neutron and x-ray solution scattering. We show that a standard shape-reconstruction analysis can be applied to this detergent-protein two-component system. The low-resolution structure of full-length KcsA is in good agreement with the proposed model of full-length KcsA deduced from EPR spectroscopy (3). In addition, our solution scattering data can also be used to characterize the KcsA channel at acidic pH, a condition that is known to induce conformational rearrangements leading to an increased open probability of the channel (13,10). We show that full-length KcsA forms a stable dimer at pH 5.0 and that dimerization only occurs in the presence of the C-terminal intracellular domain.
MATERIALS AND METHODS
Protein purification
The C-terminally truncated version of KcsA was prepared as described (1). The full-length version of KcsA was expressed as a hexa-Histidine tagged protein. The His-tag was removed after metal affinity chromatography by thrombin digest. A thrombin cleavage site was engineered between the C-terminus of the KcsA sequence and the His-tag and 1 mg full-length KcsA were digested with 0.5 units of thrombin for 3 h at room temperature (RT). The final purification step was gel filtration on a Pharmacia S200 analytical gel filtration column. Both KcsA species were purified in decyl-β-D-maltopyranoside as detergent (chemical formula, C22H42O11; critical micellar concentration, 1.8 mM in 150 mM NaCl (Anatrace, Maumee, OH)).
Small-angle neutron scattering
The final concentrated protein samples were dialyzed against detergent-containing buffer for a period of 5–10 days before analysis. Full-length KcsA was at 7 mg/ml and truncated KcsA was at 8 mg/ml concentration. Samples analyzed in D2O containing buffer were equilibrated for four days before analysis. The buffer composition for all experiments was 50 mM TrisHCl/DCl, pH 7.5, 150 mM KCl, 3 mM dithiothreitol, and 5 mM decyl-β-D-maltopyranoside (DM). Low-pH buffer contained 50 mM potassium acetate buffer, pH 5.0, instead of TrisHCl. Samples analyzed at low pH were purified in TrisHCl, pH 7.5, buffer and dialyzed against 100 times the sample volume of low-pH buffer for 2 h at RT before analysis. The scattering data were collected at beamline D22 (Institut Laue-Langevin, Grenoble, France). Samples analyzed at pH 7.5 were investigated at a camera length of 3.5 m with a collimation at 5.5 m. Low-pH data were collected at 5 and 2 m camera length with collimation at 5.6 and 8.2 m. The maximum scattering vector measured at pH 7.5 was 0.28 Å−1 and 0.47 Å−1 at pH 5.0. Samples were measured in a 1 mm quartz cuvette at 6 Å wavelength. Experiments were performed at 21°C. Scattering patterns were radially averaged and corrected for buffer scattering and detector response using GRASANSP by C. Dewhurst (Institut Laue-Langevin, Grenoble, France). The scattering from a 1-mm-path length H2O sample was used to place the data on an absolute scale. The total exposure time for each sample was between 30 and 120 min. Zero-angle scattering intensities were determined from Guinier analysis using PRIMUS (17).
Solution x-ray scattering
Small-angle x-ray scattering (SAXS) experiments were performed with protein concentrations of 7.2 and 5.6 mg/ml for full-length and truncated KcsA. Both protein species were also measured at half the concentration to exclude the possibility of aggregation effects. Data were collected at beamline 2.1 at the U.K. Synchrotron Radiation Source (SRS) at Daresbury. High and low-angle scattering data were collected in mica cells at camera lengths of 1 and 3.25 m and an x-ray wavelength of 1.54 Å with beam currents between 120 and 200 mA. Each sample was exposed for 25 min in 30 s frames. Frames at the beginning and the end of each data collection were compared to exclude the possibility of protein aggregation. Scattering data were collected to a maximum scattering vector, q, of 0.735 Å−1. The data reduction involved radial integration, normalization of the one-dimensional data to the intensity of the transmitted beam, correction for detector artifacts, and subtraction of buffer scattering (OTOKO, SRS, Daresbury). The q-range was calibrated with an oriented specimen of wet rat tail collagen (diffraction spacing 670 Å) and silver behenate (diffraction spacing 58.38 Å).
Low-pH SAXS data were recorded after both protein samples were dialyzed against 100 times the sample volumes of 50 mM potassium acetate buffer, pH 5.0, 150 mM KCl, 3 mM dithiothreitol, and 5 mM DM for 2 h at RT.
Data analysis
The initial scattering-intensity data were normalized with respect to the protein concentration. Transformation of the intensity data into a distance distribution was done using GNOM (17). If not stated otherwise, D(r) was assumed to be zero at rmin. The maximum dimension was systematically adjusted until D(rmax) reached zero. The distance distributions were normalized by dividing D(r) by 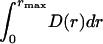 . Guinier analysis was performed according to
. Guinier analysis was performed according to
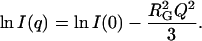 |
Alternatively, radii of gyration were calculated from the distance distribution using GNOM. Data are represented using the Origin software (OriginLab, Northampton, MA). Shape reconstruction of full-length KcsA was performed using the program DAMMIN (19). DAMMIN was preferred to other reconstruction software as it allows the application of molecular symmetry and does not require the number of amino acids of the scattering particle. Furthermore, it has been used successfully in applications using both SAXS and small-angle neutron scattering (SANS). Due to the known fourfold symmetry of the KcsA membrane-spanning region, shape models at pH 7.5 were calculated by predefining a P4 point symmetry. A P42 point symmetry was defined for shape reconstruction at pH 5.0. The disconnectivity penalty weight was increased twofold to select for densely packed models. The shape reconstructions were performed with a dummy atom radius of 3.4 Å. Dummy atom models are represented using Aesop (M. E. Noble, University of Oxford). DAMMIN-derived shape models and x-ray structure alignments were performed in supcomb13 (21). Theoretical and experimental scattering data were compared using CRYSON25 and CRYSOL25, (22). A complete full-length KcsA model including all side chains was generated from the KcsA model derived from EPR spectroscopy (3) using MaxSprout (www.ebi.ac.uk/maxsprout) without energy minimization.
RESULTS
Small-angle neutron scattering analysis
Neutron scattering experiments were performed on the detergent-solubilized KcsA channel at different D2O solvent concentrations. Detergent solubilization and purification of integral membrane proteins yields a three-component system, namely the protein with its surrounding detergent micelle and the solvent. The variation of the solvent in neutron scattering, due to variation of the D2O/H2O mole fraction, allows us to specifically contrast-match one component of a multicomponent system; thereby making it invisible. Hence, contrast variation can be used to separate the detergent micelle from the solubilized proteinaceous species in a neutron scattering experiment.
Solution SANS experiments were designed to compare structural parameters of full-length KcsA and its C-terminally truncated version. SANS data were collected and analyzed at 0%, 22%, and 100% D2O solvent concentration. The radii of gyration, RG, and maximum dimensions, rmax, of both KcsA species were calculated and compared for each D2O solvent concentration. On the basis of published detergent scattering match points, we estimated the detergent scattering match point for decyl-β-D-maltopyranoside (C22H42O11) at ∼22% D2O solvent concentration (24), thereby minimizing the contribution of the solubilizing detergent micelle to the observed neutron scattering. However, the fluctuation of scattering length of the DM detergent molecule must be stressed. This is due to the individual scattering match points of the polar maltoside headgroup of ∼46% vs. ∼3% D2O solvent concentration for the hydrophobic decyl side chain. The overall match point at 22% D2O solvent concentration, minimizing the scattering contribution of both parts of the molecule, is but an average of the individual contrast match points. Fig. 1 illustrates the different components of the KcsA-micelle system and portrays the effect of contrast variation for neutron scattering experiments.
FIGURE 1.

Schematic with length scales of the KcsA-micelle system and its components along with their neutron scattering characteristics. KcsA (Protein Data Bank (pdb) 1BL8) is shown together with decyl-β-D-maltopyranoside molecules indicating the region presumably covered by detergent molecules. Regions with similar neutron scattering length are shaded accordingly. The protein as well as the detergent maltoside headgroup D2O match point is at ∼41–43% D2O solvent concentration. The hydrophobic decyl detergent side chain is contrast-matched at ∼3% D2O solvent concentration; therefore, the match point for the entire DM detergent micelle is at ∼22% D2O solvent concentration. It was assumed that a DM molecule consists of an ∼10 Å long decyl side chain and a maltoside headgroup of similar length. Indicated approximate dimensions: radius of the KcsA channel within the periplasmic side of the lipid bilayer, 24 Å; radius of the KcsA channel plus the decyl detergent side chain, 34 Å; radius of the KcsA-micelle complex, 44 Å; and thickness of the micelle covered region of KcsA, 20 Å. (Lower panels) Cartoon illustration of the relative scattering contribution of the KcsA-micelle complex at the D2O solvent concentration used in this study.
SANS data analysis
SANS intensity data were collected in a q-range from 0.0024 to 0.28 Å−1, with q = 4π λ−1 sinθ and a scattering angle of 2θ, for both KcsA channel species. The SANS intensity data shown in Fig. 2, A and B, were radially averaged and reduced as described in Materials and Methods.
FIGURE 2.

SANS data of full-length and truncated KcsA at pH 7.5. SANS data were collected over a q-range from 0.0024 to 0.28 Å−1. (A and B) Intensity data of full-length and truncated KcsA at 0%, 22%, and 100% D2O solvent concentration. (C) Guinier analysis of full-length and truncated KcsA at 0% (RG, 36.1 ± 0.4/29.7 ± 0.3 Å), 22% (RG, 38.8 ± 0.3/32.6 ± 0.9 Å) and 100% (RG, 35.7 ± 0.3/28.2 ± 0.3 Å) D2O solvent concentration. (D) Square root of the normalized zero-angle scattering intensity of full-length and truncated KcsA at 0%, 22%, 41%, and 100% D2O solvent concentration. This analysis yields the match point for the protein-detergent complex. (E) Stuhrmann analysis of both KcsA species at pH 7.5. The reciprocal of the contrast is plotted against the square of the radius of gyration (RG) with α = 4.9 and 2.1 × 10−12 and β for full-length KcsA is 1.3 × 10−26. Data derived at 41% D2O solvent concentration were collected as part of the low-pH analysis of full-length KcsA described hereafter (Table 2) and included for statistical reasons.
The scattering-intensity data were transformed to obtain distance distributions, D(r), for both KcsA species at the respective D2O solvent concentrations (Fig. 3). The differences in the maximum dimensions, rmax, obtained for full-length and truncated KcsA are between 37 and 55 Å, depending on the D2O solvent concentration used. At 0% D2O concentration, the full-length KcsA channel is characterized by an rmax of 125 Å vs. 82 Å for truncated KcsA. These data compare to 125 vs. 88 Å at 22% and 135 vs. 80 Å at 100% D2O concentration. The distance distribution function describes the summation over all distances between point scatterers in the molecule. The most common distances observed are 37.5, 48.8, and 28.7 Å for full-length KcsA at 0%, 22%, and 100% D2O solvent concentration (Fig. 3 A). These maxima compare to 36.1, 47.5, and 27.2 Å, respectively, for the truncated version of KcsA (Fig. 3 B), peaks M1–M3. The KcsA K+ channel has an outer diameter of ∼48 Å within the periplasmic leaflet of the lipid bilayer. A DM detergent molecule is ∼20 Å in length, consisting of a 9.0 Å decyl side chain and a maltoside headgroup ∼11 Å in length (Fig. 1). The detergent side chain is almost contrast-matched at 0% D2O solvent concentration, giving rise to a detectable KcsA-micelle structure consisting of the protein itself and maltoside detergent headgroups surrounding an “empty” torus of ∼9 Å diameter. The decyl detergent side chain, on the other hand, is at maximum contrast when 100% D2O solvent concentration is used. In this situation, the detergent side chains contribute most to the observed particle scattering. Therefore, the difference of 9 Å between the M1 and M2 peaks in the distance distributions at 0% and 100% D2O solvent concentration of full-length and truncated KcsA (Fig. 3, A and B) can be explained by the difference in scattering-length density of the detergent headgroups and side chains. At 22% D2O solvent concentration the detergent-protein complex can be described as consisting of an inner core of ∼19 Å radius with positive scattering density (protein) surrounded by a shell of negative scattering density (decyl side chains) extending to a diameter of ∼68 Å and an overall maximum correlation length of 88 Å (owing to the maltoside headgroups on the outside (Figs. 1 and 3 B)). Depending on the solvent scattering length, the possibility of negative contrast can give negative regions in the distance distribution D(r) (17). As a result of similar contributions from positive and negative contrast regions at 22% D2O, D(r) becomes negative for truncated KcsA (Fig. 3 B). This scattering behavior is analogous to the one seen for, e.g., SDS micelles (25). Two peaks (M3 at ∼48 Å and M4 at ∼8 Å (Fig. 3)) arise from interference of pairs of volume elements with both volume elements situated in the same scattering contrast (M3, shell of decyl side chains, and M4, protein core). Due to the neutron scattering density fluctuation within the DM detergent molecule, we were unable to completely contrast-match the DM detergent micelle (26).
FIGURE 3.

Distance distribution of full-length and truncated KcsA at 0%, 22%, and 100% D2O solvent concentration and pH 7.5. (A) Full-length KcsA. (B) Truncated KcsA. M1–M4 highlight maxima corresponding to the most abundant separation of point scatterers within the channel species (full-length KcsA: 28.7, 37.5, 48.8, and 8.8 Å; truncated KcsA: 27.2, 36.1, 47.5, and 7.9 Å, respectively).
The Guinier analysis of the scattering data was confined to the q-range of 0.017–0.08 Å−1 and shows the strict linearity within this region (Fig. 2 C). Consistent with a difference in rmax, the radii of gyration for both KcsA channel species as determined from the distance distribution suggest significant length differences between the two species. These differences in RG are 8.2 (± 0.2) Å at 0% D2O and 5.8 (± 1.1) and 10.8 (± 0.1) Å at 22% and 100% D2O solvent concentration. All structural parameters derived from the distance distribution analysis of both KcsA species are summarized in Table 1.
TABLE 1.
Structural parameters of KcsA derived from SANS data at pH 7.5
| D2O | Full-length | Truncated | rmax (Å) | RG (Å) |
|---|---|---|---|---|
| 0% | x | 125 | 36.9 ± 0.4 | |
| 0% | x | 82 | 28.7 ± 0.3 | |
| 22% | x | 125 | 40.8 ± 1.0 | |
| 22% | x | 88 | 35.0 ± 0.5 | |
| 100% | x | 135 | 36.5 ± 0.3 | |
| 100% | x | 80 | 25.7 ± 0.2 |
The radii of gyration, RG, were calculated from the distance distribution using GNOM. rmax, maximum dimension of the macromolecule. The error in the determination of rmax with GNOM is generally of the order of 5%.
The contrast variation neutron scattering analysis further provides the possibility to assess the shape of the protein-micelle complex. Determining the contrast match point for the protein-detergent micelle complex provides useful information on the number of detergent molecules associated with the protein. Fig. 2 D shows the square root of the zero-angle scattering intensity, I(0), of full-length and truncated KcsA normalized by the transmission, total concentration, and sample thickness relative to the H2O/D2O solvent concentration. Both KcsA species (solubilized in a DM detergent micelle) are contrast matched at 28% D2O solvent concentration. Recall that the decyl-maltoside detergent has an inhomogeneous scattering-length density. The maltoside detergent headgroup is contrast matched at ∼46%, whereas the aliphatic decyl side chain is contrast matched at ∼3% D2O solvent concentration (24). Similarly, the protein-only scattering match point can be estimated at a concentration of 41% D2O (24). Therefore, the experimental neutron scattering contrast match point must be interpreted as the H2O/D2O ratio that yields the lowest protein-micelle scattering possible. This experimental scattering match point allows us to calculate the number of detergent molecules associated with the KcsA channel according to the Timmins et al. (24) formula
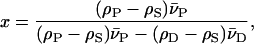 |
where x represents the mass fraction of detergent bound to the protein, ρ the scattering-length density, and 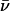 the partial specific volume of protein (P: 0.72 cm3/g) and detergent (D: 0.8 cm3/g) (27). The scattering-length density of the protein and detergent can be calculated from its chemical composition as a function of the H2O/D2O mole fraction. It was assumed that 80% of the total exchangeable protein protons are replaced by deuterium and that all six exchangeable protons of the maltoside detergent headgroups are replaced (26). This calculation yields an approximate number of 180 detergent molecules associated with the KcsA channel at pH 7.5, 21°C, and a detergent molecular weight of 482.6 g/mole.
the partial specific volume of protein (P: 0.72 cm3/g) and detergent (D: 0.8 cm3/g) (27). The scattering-length density of the protein and detergent can be calculated from its chemical composition as a function of the H2O/D2O mole fraction. It was assumed that 80% of the total exchangeable protein protons are replaced by deuterium and that all six exchangeable protons of the maltoside detergent headgroups are replaced (26). This calculation yields an approximate number of 180 detergent molecules associated with the KcsA channel at pH 7.5, 21°C, and a detergent molecular weight of 482.6 g/mole.
The determination of radii of gyration at different contrasts provides useful information on the shape and distribution of the different components of an inhomogeneous particle. Stuhrmann analysis utilizes the relationship between the reciprocal of the contrast Δρ versus the square of the radius of gyration (28,29):
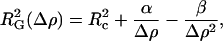 |
where Rc represents the RG at infinite contrast, α is positive if the higher scattering density within an inhomogeneous particle is found on the outside of the particle, and β corresponds to the displacement of the scattering centers within the particle as the contrast is varied. β is zero if the scattering centers of the particle components superimpose (26). Fig. 2 E shows the Stuhrmann analysis for the truncated and full-length KcsA channel. α is positive for full-length and truncated KcsA (4.9 ± 1.4 and 2.1 ± 0.2 × 10−12), and β is 1.3 ± 0.7 × 10−26 for the full-length channel, causing the nonlinearity of the fit (Fig. 2 E). Rc is 35.6 (± 2.3) Å and 25.6 (± 0.2) Å for full-length and truncated KcsA. This suggests only a slight displacement of the scattering centers with varying contrast within the full-length KcsA-micelle particle. Thus, one may assume that the detergent micelle is surrounding the transmembrane spanning region of KcsA and that the intracellular C-terminal domain protrudes from this globular headgroup. However, the positive slope of the Stuhrmann plot is unexpected. The theoretical neutron contrast match point calculated for the full-length KcsA channel according to its chemical composition and 80% maximum proton exchange corresponds to 39.5% D2O solvent concentration. The scattering match point for the maltoside detergent headgroup corresponds to 48% D2O. A torus of detergent molecules surrounding the transmembrane spanning region of KcsA would generate a detergent belt with higher scattering density on the solvent-facing periphery compared to the interior of the ring (Fig. 1). On this basis we assume that the difference in scattering-length density between the maltoside headgroup and the KcsA protein is sufficient to give rise to the observed positive slope of the Stuhrmann plot (Fig. 2 E).
Small-angle x-ray scattering analysis
Complementary to SANS, a SAXS analysis of full-length and truncated KcsA was performed. X-rays are generally of higher intensity than neutron beams and allow the collection of scattering data to higher resolutions. Additionally, x-ray scattering allows one to estimate the hydration shell of the macromolecule under investigation.
The SAXS scattering data were collected in a q-range from 0.0025 to 0.79 Å−1 at 21°C. Fig. 4 A compares the recorded scattering intensities for both KcsA species. The inset shows the Guinier analysis of both datasets in a q-range from 0.002 to 0.093 Å−1 with RG values of 43.8 (± 0.4) Å and 36.6 (± 1.7) Å for full-length and truncated KcsA.
FIGURE 4.

SAXS data of full-length and truncated KcsA at pH 7.5. (A) Scattering intensity data and Guinier analysis (inset) performed over a q-range from 0.002 to 0.093 Å−1 with radii of gyration of 43.8 ± 0.4 and 36.6 ± 1.7 Å. (B) Distance distribution of full-length and truncated KcsA. The full-length KcsA channel extends its truncated version by 40 Å. M1, M2, and M3 are 13, 41, and 46.2 Å, respectively. (C) Comparsion of SAXS and 0% and 100% SANS scattering-intensity data. The arrows point to the onset of the first shoulder observable in each profile, which is shifted toward larger scattering-length vectors from SAXS to 0% SANS and 100% SANS data. Error bars are omitted for clarity.
Similar to the neutron data analysis, the SAXS intensity data were transformed to yield a distance distribution, D(r) (Fig. 4 B). D(r) distributions for both full-length and truncated KcsA show a prominent shoulder at 13 Å (Fig. 4 B, M1), a feature that might represent the high α-helical contents of the K+ channel (30). In agreement with the SANS data, the full-length KcsA channel extends the C-terminally truncated version in its maximum dimension by ∼40 Å. In addition to the M1 peaks observed in the distance distributions, secondary peaks at 46 and 41 Å (Fig. 4 B, M2 and M3) are observed for full-length and truncated KcsA. These distances relate to the ∼48 Å diameter of the protein part of the channel, whereas the shoulder at higher distances originates from the detergent micelle extending the separation of scattering centers to ∼68 Å. The radii of gyration and maximum dimensions of full-length and truncated KcsA are summarized in Table 2. Due to a similar solvent-solute contrast, the neutron scattering data recorded at 0% D2O solvent concentration can be compared to the SAXS scattering data. In general, the structural parameters derived from SANS data analysis are smaller than the equivalent SAXS data (Tables 1 and 2). The most common separation of point scatterers for full-length KcsA according to the SANS data is 37.5 Å vs. 42 Å determined by SAXS. In addition, the maximum dimensions determined for full-length KcsA are 125 Å and 145 Å as derived from SANS and SAXS data analysis, respectively. These discrepancies can be explained by considering the hydration status of the macromolecule. In general, the observed scattering intensity is proportional to the particle contrast, Δρ, given by the difference in solvent and solute scattering lengths (26). In an x-ray scattering experiment, the difference between solvent and solute (protein) scattering-length density is small; therefore, a hydration layer surrounding the protein surface would contribute significantly to the observed x-ray scattering (22). In a neutron scattering experiment, however, the scattering length of H2O is slightly negative and the neutron scattering length of D2O is much higher than that of a protein, so that the protein is measured at negative contrast at 100% and positive contrast at 0% D2O solvent concentration. Therefore, a hydration shell around the protein with higher density than the bulk solvent would in both cases reduce the apparent size of the particle. A comparison of the SAXS and SANS data derived at 0% and 100% D2O solvent concentration illustrates this hydration effect (Fig. 4 C). The onset of the first shoulder observed in the scattering intensity profiles is shifted toward higher scattering-length vectors following the order SAXS, 0% SANS, and 100% SANS. These experimental observations can be interpreted as a shrinking of the measured particle due to the reduced detection of the hydration shell in the scattering-intensity data.
TABLE 2.
Comparison of structural parameters of full-length KcsA at pH 7.5 and 5.0
| SAXS: full-length KcsA
|
SANS: full-length KcsA
|
|||||
|---|---|---|---|---|---|---|
| pH | RG (Å) | rmax (Å) | D2O | pH | RG (Å) | rmax (Å) |
| 7.5 | 41.9 ± 0.4 | 145.0 | 0% | 7.5 | 36.9 ± 0.4 | 125.0 |
| 5.0
|
68.1 ± 0.7
|
225.0
|
0% | 5.0 | 63.9 ± 0.6 | 200.0 |
| SAXS: truncated KcsA
|
22% | 7.5 | 40.8 ± 1.0 | 125.0 | ||
| pH | RG (Å) | rmax (Å) | 22% | 5.0 | 64.3 ± 1.2 | 200.0 |
| 7.5 | 34.5 ± 0.2 | 105.0 | 41% | 7.5 | 20.5 ± 0.1 | 68.0 |
| 5.0 | 34.6 ± 0.3 | 105.0 | 41% | 5.0 | 68.9 ± 0.9 | 209.0 |
| 100% | 7.5 | 36.5 ± 0.3 | 135.0 | |||
| 100% | 5.0 | 67.9 ± 0.7 | 205.0 | |||
All data were derived from distance distribution analysis using GNOM.
It is likely that the packing of the maltoside headgroups on the surface of the detergent micelle belt surrounding the KcsA channel allows the intercalation of water molecules. This water-rich layer may hence explain the prominent hydration effect observed in the SAXS data.
KcsA shape reconstruction
The analysis of the neutron and x-ray scattering data suggests that the full-length version of KcsA extends its C-terminally truncated counterpart by ∼40–50 Å. The DAMMIN program utilizes a simulated annealing procedure to generate low-resolution models from small-angle scattering data (19). Bearing in mind that such models consist of single density spheres (so-called dummy atoms), assessment of shape reconstructions belonging to different scattering contrasts nevertheless allows pinpointing structural commonalities. Therefore, dummy-atom full-length KcsA models were calculated from the 0% and 100% D2O SANS and SAXS scattering data. Between 5 and 10 individual models were calculated for each dataset, with almost no variation. Data collected at 22% D2O solvent concentration did not generate reproducible results, presumably due to the low signal/noise ratio at this low-contrast condition. The dummy-atom model obtained from the 0% D2O scattering data has an oval shape (Fig. 5 a). The structure is 122 × 53 Å in dimension and forms a constriction of 27 Å at one pole. The full-length KcsA model derived from 100% D2O scattering data is 116 Å in length and can be divided in two domains, a compact headgroup and an hourglass-shaped extension (Fig. 5 b). The headgroup is 45 Å in length and 30 Å wide; the hourglass-shaped extension is ∼70 Å long. A prominent constriction occurs midway between the end of the head domain and the tip of the hourglass-shaped extension.
FIGURE 5.

Dummy atom model of full-length KcsA derived from SANS and SAXS intensity data at pH 7.5. Shape models derived from 0% (a) and 100% (b) D2O SANS data are compared the model derived from x-ray scattering (c). (d) A surface representation of the C-terminally truncated KcsA channel (red surface, pdb code 1BL8) aligned with the head domain of the full-length KcsA model derived from the 100% D2O SANS data. The dummy atom radius was reduced to 1.5 Å for clarity. (e) Shape reconstruction from 100% D2O SANS of truncated KcsA at pH 7.5.
In comparison, the shape reconstruction obtained for the C-terminally truncated KcsA version at 100% D2O solvent concentration lacks the hourglass-shaped extension and is only 68 Å in its longest dimension (Fig. 5 e).
In accordance with the SANS data analysis, a dummy-atom shape model was also calculated from the full-length KcsA SAXS intensity data. The model consists of a 48 × 50 Å globular head domain extended by an hourglass-shaped structure 54 Å in length (Fig. 5 c). The extension is divided in a conical structure directly after the head domain and a loosely packed domain proximal to it. The head domain encloses a central cavity of 40 × 25 Å. Fig. 5 d compares the membrane-spanning region of KcsA derived from x-ray crystallography (1,2) with the shape models derived from solution scattering.
The x-ray scattering of DM-detergent micelles lacking protein gives rise to a prominent minimum in I(q) at q = 0.1 Å−1 (data not shown). Equally, the SAXS protein-detergent convoluted scattering function exhibits a local minimum near q = 0.1 Å−1 (Fig. 4), suggesting a considerable convolution of protein- and detergent-derived intensity data. This effect was not observed with the SANS intensity data and may substantiate the shape differences of the SANS and SAXS models within the detergent-surrounded KcsA head domain.
pH-induced conformational transition of KcsA
The gating of ion channels proceeds via conformational changes that enable or block the passage of ions along their conduction pathways. KcsA has been reported to increase its open probability at acidic pH (10,11). We investigated the possibility of detecting changes in the conformation of KcsA upon acidification by means of solution neutron and x-ray scattering.
SAXS analysis at pH 5.0
The buffer conditions for full-length and truncated KcsA were adjusted to pH 5.0, a pH that is well known to increase the open probability of the KcsA channel (10,11). Significant pH-induced changes in the SAXS scattering profile of full-length KcsA are detectable over a q-range from 0.0025 to 0.35 Å−1 (Fig. 6 A). The radius of gyration of full-length KcsA as determined from the distance distribution increases from 41.9 (± 0.4) to 68.1 (± 0.7) Å. The corresponding maximum dimension also expands from 145 to 225 Å (Fig. 6 B). The distance distribution calculated for full-length KcsA at pH 5.0 shows significant differences compared to the distribution obtained at pH 7.5 (Fig. 6 B). The M1 shoulder at 13 Å is still present, most likely reflecting the mainly α-helical fold of the KcsA channel. A second and third peak exists at 46 Å (M2) and 67 Å (M3). A similar M2 peak was already observed in the distance distribution calculated at pH 7.5 (Figs. 4 B and 6 B); at low pH, however, this peak separates into two maxima and a broad shoulder at ∼110 Å (M4). The appearance of the M4 shoulder indicates that the newly formed complex consists of two separated domains that are joined rather inflexibly. Interestingly, a molecular dimension of 110 Å is consistent with the full-length KcsA model as derived from EPR spectroscopy (3). On the basis of these results, it is tempting to propose a pH-induced dimerization of full-length KcsA. This idea is supported by the ∼2-fold increase in the zero-angle scattering intensity of full-length KcsA at pH 5.0 (Fig. 6 A).
FIGURE 6.

X-ray scattering analysis of full-length KcsA at pH 7.5 and 5.0. (A) The zero-angle scattering intensity of full-length KcsA increases ∼2-fold upon acidification. The intensity profiles at pH 7.5 and 5.0 do not show any detectable difference past a scattering vector of 0.4 Å−1 (inset). (B) The distance distribution of full-length KcsA at pH 7.5 and 5.0 reveals an increase in rmax of 80 Å upon acidification. (C) Superposition of high- and low-pH scattering data of truncated KcsA.
These observations are directly correlated with the presence of the C-terminal domain of KcsA, as the scattering profile of the C-terminally truncated KcsA species fails to exhibit any changes upon acidification (Fig. 6 C).
SANS analysis at pH 5.0
Accordingly, the zero-angle neutron scattering intensity at the detergent scattering match point (22% D2O solvent concentration) increases ∼2-fold upon acidification of full-length KcsA. A similar increase is also observed at the estimated protein scattering match point at 41% D2O solvent concentration (Fig. 7 A) (26,31). The doubling of zero-angle scattering intensity with decreasing pH suggests the dimerization of the tetrameric full-length KcsA channel. Strikingly, all low-pH SANS intensity data show a prominent shoulder at q = 0.04 Å−1 (Fig. 7 A), indicating a dumbbell shape for the protein complex formed (32,33). No differences between the scattering profiles at pH 7.5 and 5.0 are detectable for scattering vectors >0.1 Å−1.
FIGURE 7.

Comparison of full-length KcsA SANS data at pH 7.5 and 5.0. (A) Increase in zero-angle scattering intensity of full-length KcsA at pH 5.0. (B and C) Guinier analysis and distance distribution at 0%, 22%, 41%, and 100% D2O solvent concentration. D(rmin) = D(rmax) = 0 was applied. (D) Protein-detergent match point at pH 5.0 (28% D2O). (E) Stuhrmann plot for full-length KcsA at pH 5.0. (F) Gel filtration analysis of full-length KcsA. Full-length KcsA was gel-filtered on a Shodex analytical gel filtration column at pH 7.5 and 5.0, respectively. Dashed line, pH 7.5; dotted line, pH 5.0 and buffer exchange on the gel filtration column; solid line, pH 5.0 and sample equilibration at pH 5.0 for 90 min before gel filtration. The void volume of this column is at 5.5 ml.
At pH 5.0 the radius of gyration as well as the maximum dimension of full-length KcsA increases significantly at all D2O solvent concentrations measured (Fig. 7 and Table 2).
At 0% D2O solvent concentration, the most common distance of 38 Å observed in the distance distribution at pH 7.5 (Fig. 3), splits into a broad peak with two maxima at 50 and 107 Å at pH 5.0 (Fig. 7 C). Equally, at 100% D2O solvent concentration and pH 5.0, two peaks are observed in the full-length KcsA distance distribution at 34 and 100 Å, respectively. This distribution of distances between scattering centers in the newly formed particle suggests two domains with a maximum radius of ∼30–50 Å separated by ∼200 Å (Fig. 7 C, M1 and M2). At 41% D2O solvent concentration, the distance distribution is split in a distribution around 18 Å with a prominent shoulder at 46 Å and a second broad distribution with peaks at 83, 105, 125, and 144 Å. Imagining a minimal protein scattering contribution at this condition and a dimerization of full-length KcsA via its C-terminal intracellular domain would place two detergent micelle tori ∼80–100 Å apart from each other (assuming an intracellular domain of KcsA ∼40–50 Å in length). The distance distribution would then reflect the properties of one torus with a thickness of ∼40 Å (determined by the detergent dimensions) as well as the dimeric arrangement of two micelle tori with a total dimension of ∼210 Å, as estimated from the maximum dimension measured at 41% D2O solvent concentration and pH 5.0 (Fig. 7 C). At 22% D2O solvent concentration, the detergent scattering match point, the distance distribution peak at 48 Å observed at pH 7.5 (Fig. 3) splits into two main peaks at 50 and 68 Å with a shoulder at 106 Å and a sharp peak at 6 Å (Fig. 7 C). This distribution further supports the interpretation of two domains inflexibly connected to each other.
The protein-micelle match point for full-length KcsA at pH 5.0 was determined at 28% D2O solvent concentration, consistent with the corresponding match point at pH 7.5 (Figs. 7 D and 2 D). Hence, the protein/detergent ratio has not changed at low pH. All structural parameters of full-length KcsA determined at pH 7.5 and 5.0 are compared in Table 2.
Stuhrmann analysis of the full-length KcsA channel at acidic pH reveals a negative slope (α, −1.644 ± 0.25 × 10−12) and an Rc of 67.33 (± 0.3) Å (Fig. 7 E). A negative value of α is usually indicative of a macromolecular arrangement with higher scattering density in its core relative to its periphery (26). A dimeric KcsA channel joined together via its C-terminal intracellular domain can be envisioned as such. However, this is in contrast to the slightly positive slope observed for full-length KcsA at pH 7.5 (Fig. 2 E) and remains unclear.
Gel filtration analysis of full-length KcsA at pH 7.5 and 5.0 allows one to estimate the extent of channel oligomerization at pH 5.0 (Fig. 7 F). Full-length KcsA was subjected to size exclusion chromatography on a Shodex KW-803 gel filtration column at pH 7.5 and 5.0. The pH 7.5 elution volume of 10.5 ml shifted to ∼9.3 ml at pH 5.0. Oligomerization of KcsA at low pH is very fast. The sample at pH 7.5 loaded onto the column equilibrated at pH 5.0 almost exclusively eluted at a higher molecular weight elution volume, suggesting that channel dimerization occurred during the chromatography run (Fig. 7 F).
Low-pH conformation of full-length KcsA
Dummy-atom shape models were calculated for full-length KcsA at pH 5.0 from the 0% and 100% D2O SANS and the SAXS data. The dummy-atom molecular shape model derived from the 0% D2O SANS data is ∼188 × 50 Å in dimension (Fig. 8 a). The fourfold symmetry axis runs along the longest dimension. The shape model is of 48 Å thickness along the twofold symmetry axis, which compares to a 27 Å constriction observed in the 0% D2O SANS data model calculated at pH 7.5 (Fig. 5 a). Its two poles are rotated relative to each other along the fourfold axis by ∼35 degrees. Similarly, the dummy atom shape model based on the 100% D2O SANS data is 172 × 45 Å in dimension, with the fourfold symmetry axis running along its maximum dimension (Fig. 8 b). The model is 43 Å wide along the twofold symmetry axis. This thickness compares to a 10-Å constriction observed in the corresponding DAMMIN model at pH 7.5 (Fig. 5 b). Both poles of the elongated molecular shape are rotated relative to each other by 35°. The shape reconstruction of the full-length KcsA complex based on SAXS data leads to a more open structure. The model consists of two poles of 80 × 68 Å connected by four pillars 67 Å long and 10 Å wide (Fig. 8 c). The structure encloses a central cavity of ∼80 Å diameter. The two poles are rotated relative to each other by ∼45 degrees and each pole encloses a cavity 29 × 36 Å. This splayed open structure of the full-length KcsA complex at pH 5.0 is in contrast to its close packing at pH 7.5 (Fig. 5 c).
FIGURE 8.

Low-pH shape model of full-length KcsA. Dummy atom models were calculated with P42 point symmetry for 0% (a) and 100% (b) D2O solvent concentration. (c) The KcsA model derived from SAXS data. Structural domains related by a twofold symmetry operation are shown in light and dark gray in a side and top view.
DISCUSSION
The primary goal of this study was the characterization of an integral membrane protein by x-ray and neutron small-angle scattering. Due to their fragile nature and the generally limited amounts of sample available, structural characterization of membrane proteins is notoriously difficult. Techniques such as x-ray and neutron scattering circumvent the need for well ordered three-dimensional crystals, providing powerful tools complementing structural methods in membrane protein molecular biology.
The C-terminally truncated bacterial K+ channel KcsA from S. lividans has been characterized by x-ray crystallography (1,2). Comparison of the structural parameters obtained from small-angle scattering of full-length KcsA and its C-terminally truncated version used for crystallization identifies a significant length difference between the two species (Table 1). Neutron scattering experiments performed at 0%, 22%, and 100% D2O solvent concentration reveal variations in the radii of gyration, RG, between full-length and truncated KcsA from 8.1 to 10.8 Å. The corresponding differences in the maximum dimension, rmax, between the macromolecules are 43, 37, and 55 Å. Both KcsA species reveal a difference in RG of 7.4 Å and of 40 Å in rmax in the equivalent x-ray scattering experiment. These results suggest a stably folded intracellular domain of KcsA extending the membrane-spanning region by ∼40–50 Å.
Even though the scattering investigation of a detergent-solubilized membrane protein does not represent a single phase system, the consistency of shapes for the full-length KcsA channel reconstructed with the dummy-atom annealing procedure (19) from neutron and x-ray scattering data points to the reliability of the determined overall conformation. The models are between 108 and 122 Å in length, consisting of a prominent globular domain and an elongated extension (Fig. 5). The globular domain is assigned to the membrane-spanning region of KcsA surrounded by a belt-shaped detergent micelle (34). Strikingly, the extending domains are of an hourglass shape, with a central constriction. This suggests a two-domain organization of the intracellular domain of KcsA. This organization of full-length KcsA is supported by EPR spectroscopy data proposing a mainly α-helical intracellular domain with closest proximity of residue Thr-141 for each of the four subunits (3).
This alternative model of full-length KcsA (hereafter referred to as the “EPR model”) suggests an intracellular C-terminal extension of the full-length channel of ∼50 Å (3). Fig. 9 compares the experimentally determined full-length KcsA scattering data used for shape reconstruction with calculated scattering intensities derived from this EPR model including all amino acid side chains. Theoretical scattering intensities were calculated and evaluated using CRYSON25 for SANS and CRYSOL25 for SAXS data (22). The EPR model fits the SANS data reasonably well, yielding the best fit at the highest neutron contrast (100% D2O solvent concentration). The x-ray derived scattering data, however, show the highest discrepancy between experimental and theoretical data. Variations between theoretical and experimental data are ascribed first to the use of only single density spheres to describe a two-component system (protein and detergent micelle), and second to the absence of the detergent micelle in the template used for scattering-profile simulations. The protein-micelle particle is at highest solute-solvent contrast at 100% D2O solvent concentration (24). Unexpectedly, the best fit obtained between the experimental and theoretical scattering intensity profiles is at this particular solvent composition when using a protein-only template structure. Although it is difficult to substantiate this phenomenon in detail, one explanation might be heterogeneity with respect to size and shape of the detergent micelle surrounding the KcsA membrane spanning region together with a continuous exchange of detergent molecules between neighboring micelles. This heterogeneity may lead to a reduced scattering contribution of the detergent micelle. In contrast, the x-ray scattering data deviate more strongly from the theoretically determined scattering profile. A likely reason for this lies in the higher sensitivity of x-rays for ordered solvent structure, e.g., around the exterior of the detergent micelle interacting with the DM maltoside headgroups, which will significantly contribute to the hydration of the protein-detergent complex. A more comprehensive analysis of this effect ought to be assessed by modeling KcsA-detergent complexes and comparing those to experimental scattering data.
FIGURE 9.

Comparison of experimental and theoretical scattering data at pH 7.5. The theoretical scattering intensities of the full-length KcsA EPR model (solid line) are compared to the experimental intensities at 0% (A) and 100% (B) D2O solvent concentration and to the x-ray derived scattering data (C). Single density spheres were used to model the experimental curves.(D) Side and top views of the KcsA EPR model (pdb code 1F6G), with N- and C-terminal domains indicated.
In a next step, neutron and x-ray small-angle scattering was applied to characterize conformational changes of full-length KcsA upon acidification. Neutron scattering performed at 22% and 41% D2O solvent concentration and pH 5.0 reveals a twofold increase in zero-angle scattering intensity, I(0), of full-length KcsA (Fig. 7 A). The 22% D2O scattering data are only minimally affected by the decyl-β-D-maltopyranoside detergent micelle scattering, whereas the 41% D2O scattering data mainly report on the detergent component (24,26,31). A 1:2 ratio of zero-angle scattering intensity suggests dimerization of the full-length KcsA channel upon pH decrease. The radii of gyration and maximum dimensions of full-length KcsA increase accordingly upon acidification. Similar changes are detected for the SAXS-derived structural parameters of full-length KcsA at pH 7.5 and 5.0. These observations are directly linked to the presence of the 35 C-terminal amino acids of KcsA as the C-terminally truncated KcsA species does not exhibit any observable structural changes upon acidification (Fig. 6 C). Gel filtration analysis at low pH confirms the dimerization of the full-length KcsA channel. Consistently, shape reconstruction of the pseudo-octameric full-length KcsA complex generates elongated structures with a fourfold symmetry axis running along the maximum dimension and a twofold axis perpendicular to it. The 27 Å central constriction observed in the KcsA intracellular domain in the 0% D2O SANS shape model increases to ∼48 Å in the corresponding low-pH model (Fig. 8). This compares to a transition from 10 to 43 Å identified in the low-pH 100% D2O SANS model. The full-length KcsA model at pH 5.0 derived from SAXS data is splayed open with two densely packed poles connected by four pillars. The space between the two poles is ∼80 Å in diameter. Although we cannot make any certain conclusions on the conformation of the membrane spanning helices of KcsA at acidic pH and the exact gating mechanism (hinge-bending versus rotation and tilt), our results indicate a widening of the diameter of the intracellular domain by ∼20–30 Å. These intracellular rearrangements most likely form the basis for the observed channel dimerization at low pH.
It is interesting to note that the truncated KcsA channel does not appear to respond to the low-pH environment by a global structural change since the x-ray scattering data for pH 7.5 and pH 5.0 measured to a nominal Bragg resolution of up to 8 Å superimpose neatly (Fig. 6 C). It is conceivable that the intracellular C-terminal domain of KcsA stabilizes the closed conformation of the channel at neutral pH. Removal of this domain might enable more conformational flexibility within the transmembrane region of KcsA, leading to an equilibrium distribution of open and closed channels at pH 7.5 and 5.0 in detergent. This interpretation is further supported by rubidium uptake assays performed with reconstituted full-length and truncated KcsA in lipid vesicles (11). The intrinsic flexibility of the C-terminally truncated KcsA channel is also evident from cocrystallization experiments of KcsA and tetrabutyl-ammonium ions (35). These experiments require the incorporation of bulky molecular structures in the ion-conduction pathway from the intracellular side of the channel in the absence of any driving force, such as a transmembrane potential. The fact that tetrabutyl-ammonium ions can diffuse into the ion-conduction pathway of the truncated KcsA channel under equilibrium conditions and neutral pH suggests that the hydrophobic ion-conduction pathway (located within the inner bilayer leaflet) must undergo conformational rearrangements allowing these molecules to pass. An equilibrium distribution of truncated KcsA channels in different conformational states at pH 7.5 and 5.0 might explain the failure to detect any differences between the low- and high-pH states of truncated KcsA by solution scattering.
The activation of the KcsA K+ channel at acidic pH indicates the contribution of amino acid side chains with corresponding pKa values, such as Glu and Asp residues (36). With respect to the results obtained, it is tempting to speculate that acidification of the detergent-solubilized KcsA channel leads to a destabilization of packing interactions within its intracellular domain. This destabilization could enable an increased thermal motion of the KcsA transmembrane helices leading to an open structure. Consequently, the C-terminal intracellular domain of KcsA could rearrange and move outward, forming a cylinder-shaped intracellular extension.
We point out that the observed dimerization of the detergent-solubilized KcsA K+ channel at low pH most certainly is due to an in vitro effect. However, the observed dimerization is very likely stabilizing an open conformation of the KcsA channel which in vivo is achieved by other interaction partners. Since the biological function and regulation of the KcsA channel is poorly understood, it cannot be excluded that KcsA requires additional intracellular factors to open. These may stabilize a single KcsA channel in a conformation similar to the low-pH structure observed in vitro.
Acknowledgments
We are very grateful to Peter Timmins for helpful discussion and support at the data collection at Institut Laue-Langevin, Grenoble, France.
This work was funded by a Wellcome Trust Research Career Development Fellowship awarded to D.A.D. J.Z. was supported by the Boehringer Ingelheim Fonds.
Jochen Zimmer's present address is Dept. of Cell Biology, Harvard Medical School, 240 Longwood Ave., Boston, MA 02115.
Declan A. Doyle's present address is Structural Genomics Consortium, University of Oxford, Botnar Research Centre, Oxford OX3 7LD, UK.
References
- 1.Doyle, D. A., J. Morais Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 2.Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]
- 3.Cortes, D. M., L. G. Cuello, and E. Perozo. 2001. Molecular architecture of full-length KcsA: role of cytoplasmic domains in ion permeation and activation gating. J. Gen. Physiol. 117:165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reimann, F., and F. M. Ashcroft. 1999. Inwardly rectifying potassium channels. Curr. Opin. Cell Biol. 11:503–508. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong, C. M., and F. Bezanilla. 1974. Charge movement associated with the opening and closing of the activation gates of the Na channels. J. Gen. Physiol. 63:533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sigworth, F. J. 1994. Voltage gating of ion channels. Q. Rev. Biophys. 27:1–40. [DOI] [PubMed] [Google Scholar]
- 7.Morais-Cabral, J. H., Y. Zhou, and R. MacKinnon. 2001. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 414:37–42. [DOI] [PubMed] [Google Scholar]
- 8.Roux, B., and R. MacKinnon. 1999. The cavity and pore helices in the KcsA K+ channel: electrostatic stabilization of monovalent cations. Science. 285:100–102. [DOI] [PubMed] [Google Scholar]
- 9.Heginbotham, L., M. LeMasurier, L. Kolmakova-Partensky, and C. Miller. 1999. Single Streptomyces lividans K(+) channels: functional asymmetries and sidedness of proton activation. J. Gen. Physiol. 114:551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LeMasurier, M., L. Heginbotham, and C. Miller. 2001. KcsA: it's a potassium channel. J. Gen. Physiol. 118:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuello, L. G., J. G. Romero, D. M. Cortes, and E. Perozo. 1998. pH-dependent gating in the Streptomyces lividans K+ channel. Biochemistry. 37:3229–3236. [DOI] [PubMed] [Google Scholar]
- 12.Liu, Y. S., P. Sompornpisut, and E. Perozo. 2001. Structure of the KcsA channel intracellular gate in the open state. Nat. Struct. Biol. 8:883–887. [DOI] [PubMed] [Google Scholar]
- 13.Perozo, E., D. M. Cortes, and L. G. Cuello. 1999. Structural rearrangements underlying K+-channel activation gating. Science. 285:73–78. [DOI] [PubMed] [Google Scholar]
- 14.Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- 15.Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- 16.Reference deleted in proof.
- 17.Svergun, D. I. 1992. Determination of regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25:495–503. [Google Scholar]
- 18.Reference deleted in proof.
- 19.Svergun, D. I. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76:2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reference deleted in proof.
- 21.Kozin, M., and D. I. Svergun. 2001. Automated matching of high- and low resolution structural models. J. Appl. Crystallogr. 34:33–41. [Google Scholar]
- 22.Svergun, D. I., S. Richard, M. H. Koch, Z. Sayers, S. Kuprin, and G. Zaccai. 1998. Protein hydration in solution: experimental observation by x-ray and neutron scattering. Proc. Natl. Acad. Sci. USA. 95:2267–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reference deleted in proof.
- 24.Timmins, P., E. Pebay-Peyroula, and W. Welte. 1994. Detergent organization in solution and in crystals of membrane proteins. Biophys. Chem. 53:27–36. [DOI] [PubMed] [Google Scholar]
- 25.Steenstrup, S., and S. Hansen. 1994. The maximum-entropy method without the positivity constraint: applications to the determination of the distance-distribution function in small-angle scattering. J. Appl. Crystallogr. 27:574–580. [Google Scholar]
- 26.Perkins, S. J. 1988. X-ray and neutron solution scattering. In Modern Physical Methods in Biochemistry, Part B. Elsevier Science Publishers, Amsterdam.
- 27.Friesen, R. H., J. Knol, and B. Poolman. 2000. Quaternary structure of the lactose transport protein of Streptococcus thermophilus in the detergent-solubilized and membrane-reconstituted state. J. Biol. Chem. 275:33527–33535. [DOI] [PubMed] [Google Scholar]
- 28.Ibel, K., and H. Stuhrmann. 1975. Comparison of neutron and X-ray scattering of dilute myoglobin solutions. J. Mol. Biol. 93:255–265. [DOI] [PubMed] [Google Scholar]
- 29.Svergun, D. I., N. Burkhardt, J. S. Pedersen, M. H. Koch, V. V. Volkov, M. B. Kozin, W. Meerwink, H. B. Stuhrmann, G. Diedrich, and K. H. Nierhaus. 1997. Solution scattering structural analysis of the 70 S Escherichia coli ribosome by contrast variation. I. Invariants and validation of electron microscopy models. J. Mol. Biol. 271:588–601. [DOI] [PubMed] [Google Scholar]
- 30.Heller, W. T., N. L. Finley, W. J. Dong, P. Timmins, H. C. Cheung, P. R. Rosevear, and J. Trewhella. 2003. Small-angle neutron scattering with contrast variation reveals spatial relationships between the three subunits in the ternary cardiac troponin complex and the effects of troponin I phosphorylation. Biochemistry. 42:7790–7800. [DOI] [PubMed] [Google Scholar]
- 31.Perkins, S. J. 1986. Protein volumes and hydration effects. The calculations of partial specific volumes, neutron scattering matchpoints and 280-nm absorption coefficients for proteins and glycoproteins from amino acid sequences. Eur. J. Biochem. 157:169–180. [DOI] [PubMed] [Google Scholar]
- 32.Glatter, O., and O. Kratky. 1982. Small Angle X-ray Scattering. Academic Press, London.
- 33.Fujisawa, T., T. Ueki, and Y. Inoko. 1987. X-ray scattering from a troponin C solution and its interpretation with a dumbbell shaped molecule model. J. Appl. Crystallogr. 20:349–355. [Google Scholar]
- 34.Pebay-Peyroula, E., R. M. Garavito, J. P. Rosenbusch, M. Zulauf, and P. A. Timmins. 1995. Detergent structure in tetragonal crystals of OmpF porin. Structure. 3:1051–1059. [DOI] [PubMed] [Google Scholar]
- 35.Zhou, M., J. H. Morais-Cabral, S. Mann, and R. MacKinnon. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]
- 36.Karshikoff, A., V. Spassov, S. W. Cowan, R. Ladenstein, and T. Schirmer. 1994. Electrostatic properties of two porin channels from Escherichia coli. J. Mol. Biol. 240:372–384. [DOI] [PubMed] [Google Scholar]