

Abstract
The structure of the lipid-enveloped Sindbis virus has been determined by fitting atomic resolution crystallographic structures of component proteins into an 11-Å resolution cryoelectron microscopy map. The virus has T=4 quasisymmetry elements that are accurately maintained between the external glycoproteins, the transmembrane helical region, and the internal nucleocapsid core. The crystal structure of the E1 glycoprotein was fitted into the cryoelectron microscopy density, in part by using the known carbohydrate positions as restraints. A difference map showed that the E2 glycoprotein was shaped similarly to E1, suggesting a possible common evolutionary origin for these two glycoproteins. The structure shows that the E2 glycoprotein would have to move away from the center of the trimeric spike in order to expose enough viral membrane surface to permit fusion with the cellular membrane during the initial stages of host infection. The well-resolved E1-E2 transmembrane regions form α-helical coiled coils that were consistent with T=4 symmetry. The known structure of the capsid protein was fitted into the density corresponding to the nucleocapsid, revising the structure published earlier.
Three-dimensional, near-atomic resolution studies of viruses have been possible for a wide variety of nonenveloped icosahedral viruses because of their ability to form well-diffracting crystals. Apart from the remarkable exception of the bacteriophage PRD1 (D. I. Stuart and D. H. Bamford, personal communication), it has not been possible to produce useful crystals of enveloped viruses. However, with advances in cryoelectron microscopy (cryoEM), it is now feasible to determine the structures of noncrystallizable viruses to 11-Å resolution or better. These maps can sometimes be interpreted to the rough equivalent of atomic resolution when higher-resolution structures of their components are available and when specific markers, such as glycosylation sites, have been mapped by cryoEM (41, 45, 58, 63). Here, we use such a combination of X-ray crystallography and cryoEM to present a more detailed view and interpretation of an enveloped virus than has previously been possible.
Alphaviruses are among the simplest of enveloped viruses on account of both their relatively small genome and their icosahedral symmetry. They have a number of structural and functional similarities to flaviviruses, suggesting a possible common primordial origin of some of their components (24, 27, 37, 52), although their genomes differ significantly. Alphaviruses have been favored for structural studies because of the higher yields compared with other enveloped RNA virus propagations. Many alphaviruses have the further advantage for laboratory studies that they mostly produce inapparent or mild symptoms in human hosts, compared with the frequently severe effects of flavivirus infections.
Alphaviruses have a positive-strand RNA genome with about 11,700 bases enclosed in a protein core consisting of 240 identical subunits. A subgenomic mRNA encodes a polyprotein that is posttranslationally cleaved into five individual proteins: capsid, E3, E2, 6K, and E1 (6, 17, 40, 49). Glycoproteins E1 and E2 assemble into heterodimers that are glycosylated in the Golgi and are transported to the plasma membrane. During membrane trafficking, the E1-E2 heterodimers organize themselves into 80 (E1-E2)3 trimeric spikes to form an icosahedral scaffold on the surface of the virus (Fig. 1) (14, 27, 37). The nucleocapsid cores are assembled in the cytoplasm of infected cells and are then transported to the plasma membrane, where they interact with the cytoplasmic domains of the glycoprotein spikes. During the budding process of the virion from the cell, the glycoproteins and a part of the plasma membrane are used to form the viral envelope to produce virions in which both the internal core and external glycoproteins have matching T=4 icosahedral quasisymmetry (7, 8, 15, 33, 57). The mature Sindbis virus is composed only of the E1, E2, and capsid proteins.
FIG. 1.

CryoEM density distribution for Sindbis virus. Top left, surface of the virus at 20-Å resolution. The red triangle marks the boundary of an icosahedral asymmetric unit. The numbers show the positions of icosahedral two-, three-, and fivefold axes limiting the asymmetric unit. Note that there is one trimeric spike associated with an icosahedral threefold axis plus a second trimeric spike in a general position in the icosahedral asymmetric unit. Top right, cross section through the 11-Å map along the black line shown in the top left and bottom right panels. Note the clearly defined lipid bilayer and the transmembrane domains crossing the membrane. The nucleocapsid (NCP) is seen inside the membrane. Bottom left, a central cross-section through the 11-Å resolution map, showing the glycoproteins (blue), the lipid bilayer (green), the nucleocapsid (red), the mixed RNA-protein region (orange), and the internal RNA (magenta). The orientation of the icosahedral (two-, three-, and fivefold) as well as quasi-threefold (q3) axes is shown in yellow. Below the cross section is shown the radially averaged mean density (blue) and the root mean square deviation of the density from the mean (magenta). Right bottom are radial sections, for the 11-Å resolution map, at r1 = 199 Å (nucleocapsid core), r2 = 243 Å (transmembrane), r3 = 261 Å (base of the glycoproteins), r4 = 298 Å (includes the base of the spike), and r5 = 324 Å (leaf-like structure of E2) with grey scale used to denote density levels, black being the highest density. Note the T=4 organization of the transmembrane region in r2 and r3.
Structural studies of alphaviruses have consisted primarily of cryoEM analyses of the whole virus and X-ray crystallographic examination of the component structural proteins. The structure of the 151-amino-acid carboxy-terminal domain of the capsid protein (9) verified earlier suggestions (20) that the protein was derived from a chymotrypsin-like proteinase, which had the function of cleaving itself from a polyprotein prior to becoming a capsid subunit. The 25-Å resolution cryoEM density map of Ross River virus (8) was interpreted in terms of the capsid's carboxy-terminal domain structure and showed that the 113 amino-terminal residues were intertwined with the genomic RNA, leaving only the 151 carboxy-terminal residues to perform the function of a capsid protein. This organization of the assembled nucleocapsid viral core was later supported by a study of Semliki Forest virus (30). Other studies suggested that the cytoplasmic region of E2, at residues Tyr400 and Leu402, interacts with a binding site on the surface of the nucleocapsid (26, 50), thereby correlating the icosahedral symmetry of the glycoprotein scaffold with that of the nucleocapsid core (37).
The structure of a large fragment of the ectodomain component of E1 has been determined and was suggested to be homologous to the E glycoprotein found in flaviviruses (27), although there is no remaining trace of any common heritage in the amino acid sequence. The overall structure of the flavivirus tick-borne encephalitis virus E protein has three domains (39) (Fig. 2A). The second domain (DII) can be described as a six-stranded β-barrel with one of its end loops being a peptide required for fusion with the host cell (23, 28). The third domain (DIII) has an immunoglobulin-like β-barrel fold and may be important for binding to cellular receptors.
FIG. 2.

Comparison of the backbone structures of Semliki Forest virus (SFV) E1 with tick-borne encephalitis virus (TBEV) E. (A) Ribbon diagrams, with domain I in red, domain II in yellow, fusion peptides in green, and domain III in blue. (B) Stereodiagram showing superposition of Semliki Forest virus E1 (blue) on tick-borne encephalitis virus E (red). (C) Alignment of Sindbis virus (SINV) and Semliki Forest virus E1 with tick-borne encephalitis virus E, based on the structural superposition of Semliki Forest virus E1 on tick-borne encephalitis virus E. Residues of tick-borne encephalitis virus E that do not match the structure of Semliki Forest virus E1 have been omitted. Residues in domains I, II, and III are colored red, yellow, and blue, respectively, consistent with the coloring scheme used above and throughout. Shown is the complete amino acid sequence of Sindbis virus and just those residues of tick-borne encephalitis virus that could be structurally aligned with Semliki Forest virus E1. Sequences underlined with a solid bar are β-strands, and those underlined with dashed bars are α-helices. Secondary structural nomenclature is taken from Rey et al. (39).
The positions of glycosylation sites on E1 and E2 in alphaviruses have been determined by using cryoEM difference maps between wild-type and deglycosylated mutant viruses (37). These sites showed that E1 was positioned roughly tangentially to the surface of the virus, similar to the structure of the E glycoprotein in flaviviruses (39). The carbohydrate positions also demonstrated that E2 was positioned more or less radially, forming the external broad ends of the spikes associated with the cellular attachment site (51). These orientations of E1 and E2 were consistent with the cryoEM density maps of Semliki Forest virus (27) and of a deglycosylated mutant of Sindbis virus (37).
Here, we report a systematic analysis of an 11-Å resolution cryoEM map of a deglycosylated mutant of Sindbis virus. The program EMfit (8, 42, 45) was used to orient and position the known structures of Semliki Forest virus E1 (27), of the Sindbis virus capsid protein (25), and of the GCN4 dimeric coiled coil (12) into the cryoEM map. A difference map showed that the molecular envelope of the E2 monomer is remarkably similar to that of the E1 monomer, suggesting a possible similarity in fold. This difference map also demonstrated the close association of E1 and E2 in the formation of a heterodimer and organization of the trimeric spike. The virus map showed clearly the dimensions of the lipid bilayer and the coiled-coil structure of E1 and E2 in the transmembrane region. When put together, these observations give a fairly complete look at an RNA-enveloped virus structure in quasiatomic detail, allowing some insight into the assembly and fusion processes. The virus structure is one of the first visualizations of a lipid bilayer in situ and also shows that the external icosahedral symmetry of the glycoproteins is in register with the internal symmetry of the nucleocapsid.
MATERIALS AND METHODS
Virus preparation.
The structure of Sindbis virus described here is that of a partially deglycosylated form of the virus produced by a mutation at residue 318 of E2 from Asn to Gln (37). The mutational strategy and procedure for virus production were those described elsewhere (37).
Electron microscopy.
Small (2.8-μl) aliquots of purified N318Q mutant Sindbis virus sample at a concentration of approximately 6 mg/ml equilibrated in TNE buffer (50 mM Tris-HCl [pH 7.5], 200 mM NaCl, and 1 mM EDTA) were adhered to holey carbon-coated grids and frozen in liquid ethane (3). Images were recorded on Kodak SO-163 films in a Philips CM200 field emission gun transmission electron microscope (Philips, Eindhoven, The Netherlands) under low-dose conditions (≈18 e−/Å2) at a nominal magnification of ×38,000. Micrographs were digitized on a Zeiss SCAI scanner with 7-μm intervals, and sets of four pixels were averaged to give effective sampling steps of 3.68 Å at the specimen.
A total of 10,868 particles were selected from 27 micrographs whose defocus levels ranged from 1.10 to 2.58 μm underfocus. The self-common-lines method (16) was used to determine an initial model of the virus. The orientation and origin of each particle image were determined through use of the model-based, polar-Fourier transform method (2). Orientations were further refined with a modified cross-common-lines procedure (16), which compares phase differences between the model projections and viral images. The microscope contrast transfer function (CTF) was determined for each viral image (see reference 3 for the definition of CTF). Corrections to compensate in part for the effects of the CTF on each image were applied in Fourier space by modifying structure factors (obs) computed from the image according to the following formula:
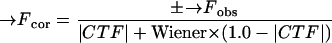 |
where F→cor is the corrected structure factor, F→obs is the original structure factor calculated from a viral image, ± signifies that phases are reversed at spatial frequencies where the sign of the CTF is positive, and the Wiener factor was assumed to be 0.2. The CTF values were then used to weight the corresponding linear equations required to establish the three-dimensional image reconstruction. A heuristic factor, WCTF
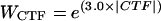 |
which depends on the value of the CTF for each F→cor, is used to weight the terms on both sides of the linear equation
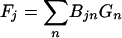 |
(see reference 10 for details) in the reconstruction program.
The resolution of the final reconstruction density map was estimated by comparing the structure factors computed from two reconstructions, each based on independent halves of the whole image data set. All densities in the two reconstruction maps were used for comparison. The Fourier shell correlation coefficient dropped from 0.6 to 0.29 between 11.4- and 11.1-Å resolution. The phase residues at these resolutions were ≈40 and 58°, respectively. The final map was calculated from 4,931 viral images to a Fourier limit of 10.0 Å with a parallelized version (C. Xiao, unpublished data) of the EM3DR reconstruction program, which uses the original Fourier-Bessel procedure of Crowther (10). The density map was sharpened through the application of an inverse temperature factor of 1,000 Å2 (21). Some features of the map had negative densities as a result of the modifications and omission of the zero-order Fourier term.
Interpretation of the 11-Å map.
All fitting of X-ray crystallographic structures of components was performed with the EMfit program (8, 42, 45). The final atomic coordinates were deposited in the PDB under accession number 1LD4.
RESULTS AND DISCUSSION
E1 glycoprotein.
The density outside the easily recognizable lipid bilayer (Fig. 1) has large positive and negative excursions between 261-Å and 365-Å radii, representing the glycoproteins E1 and E2. Pletnev et al. (37) determined the positions of the carbohydrate sites in Sindbis virus with respect to the viral icosahedral axes within an experimental error of about 2 Å. These sites have now been used to restrain the fitting of the Sindbis virus E1 structure into the 11-Å resolution Sindbis virus map. The Sindbis virus E1 structure was modeled from the homologous Semliki Forest virus E1 crystal structure, with which it has 52% amino acid identity.
The fitting process (Table 1) depended upon finding the orientation and position of the Sindbis virus E1 model that simultaneously maximized the fit of the atoms into all four quasiequivalent cryoEM densities and minimized the distance between the center of the glycosylation sites in the map and the corresponding glycosylated Asn residues in the atomic model (45). Both in positioning E1 and also in positioning the nucleocapsid protein (see below), only the Cα atoms were used initially, but all atoms were used for the final fitting (Table 1). The positions of the Sindbis virus E1 glycosylation sites for residues 139 and 245 were used as constraints by minimizing their distance from the respective Cα atoms when merely fitting the Cα backbone or with respect to the Asn Nδ atoms when fitting all atoms of the Sindbis virus E1 model. The known positions of the glycosylation site for residue 141 in Ross River virus and Semliki Forest virus were also used as a restraint for fitting the Sindbis virus E1 model (37). In this case, the restraint was made with respect to the Cα atoms both for fitting the Cα backbone and for fitting the complete Sindbis virus E1 model.
TABLE 1.
Results obtained by fitting the crystallographic structure components into the cryoEM map
| Protein | Modela | Sumf (%) | Clash (%) | Density (%) | Avg distance (Å) | Orientation
|
Center
|
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| θ1 | θ2 | θ3 | x | y | z | ||||||
| E1 | SFV E1 (Cα) | 43.4 | 0.7 | 0.1 | 17.2 | 189.8 | 85.5 | 171.0 | 28.4 | 77.3 | 283.5 |
| SINV E1 (Cα) | 41.6 | 1.2 | 1.0 | 16.2 | 189.0 | 85.8 | 170.8 | 27.9 | 77.3 | 283.5 | |
| TBEV E (Cα) | 29.3 | 0.0 | 14.1 | 18.7 | 153.5 | 89.2 | 49.5 | 27.4 | 69.8 | 283.5 | |
| SINV E1 (all) | 37.4 | 0.5 | 4.4 | 14.5 | 187.0 | 85.2 | 170.8 | 28.4 | 78.8 | 283.5 | |
| E2 | SFV E1 (Cα) | 30.3 | 1.4 | 8.1 | 21.3 | 22.5 | 55.5 | 46.1 | 12.0 | 92.8 | 289.8 |
| TM | GCN4 (Cα, trunc) | 31.9 | 0.0 | 1.8 | 16.3 | 132.2 | 78.0 | 106.2 | 4.5 | 48.8 | 238.0 |
| GCN4 (Cα, ext) | 26.1 | 0.0 | 13.6 | — | 132.2 | 78.0 | 106.2 | 3.0 | 54.3 | 253.2 | |
| CP | CP (Cα) | 45.1 | 1.2 | 0.3 | — | 99.0 | 106.5 | 140.5 | 8.7 | 44.0 | 192.0 |
| CP + pep (Cα) | 45.0 | 0.6 | 0.3 | 9.5 | 99.5 | 107.2 | 140.0 | 8.7 | 44.0 | 192.5 | |
| CP (all) | 40.1 | 0.1 | 3.6 | — | 97.5 | 108.0 | 139.5 | 8.7 | 44.5 | 191.5 | |
| CP + pep (all) | 40.2 | 0.1 | 3.6 | 9.9 | 98.5 | 108.0 | 138.5 | 8.2 | 44.0 | 192.0 | |
| CP (old fit, all) | 35.1 | 5.8 | 8.0 | — | — | 8.2 | 41.5 | 192.0 | |||
Cα indicates that only Cα atoms were used; all indicates that main chain and side chain atoms were used; trunc indicates that only residues 250 to 277 were used for fitting; ext indicates that the entire GCN4 structure was used; + pep shows that a peptide (residues 108 to 110) was added to the capsid protein (CP) structure in the binding pocket. The previously determined orientation from Ross River virus (8) was used without change for the CP (old fit, all). However, the positions were refined to account for a slightly different estimated pixel size. SFV, Semliki Forest virus; SINV, Sindbis virus; TBEV, tick-borne encephalitis virus. Results were obtained by using the EMfit program (45).
Other criteria were also used to obtain the best fit of the Sindbis virus E1 model to the map. These included minimizing the number of steric clashes between neighboring subunits, minimizing the number of atoms in negative density, and avoiding clashes between fitted E1 atoms and glycosylation sites belonging to E2 (45). The quasisymmetry elements were defined in terms of radial quasi-twofold axes between the icosahedral threefold and radial quasi-threefold axes (Fig. 3). This definition emphasized the threefold nature of the trimeric spikes, as opposed to the pentameric and hexameric associations of subunits seen in the nucleocapsid core. The latter had been used for fitting the known Sindbis virus capsid protein structure into an earlier cryoEM Ross River virus reconstruction (8). The quasisymmetry axial positions were refined by searching for orientations that optimized the quality of fit (Table 2).
FIG. 3.
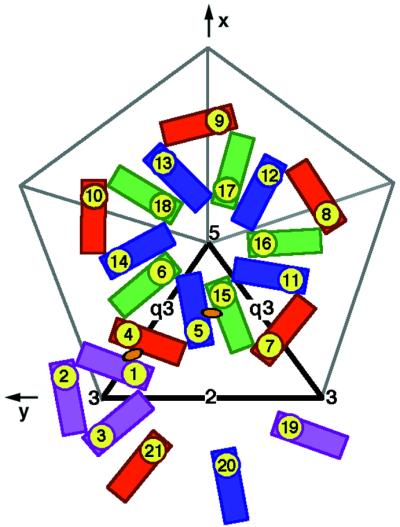
T=4 quasi-symmetry generation depicted diagrammatically in terms of the E1 glycoprotein. E1 molecules related by icosahedral symmetry are all of the same color (purple, red, blue, or green). Quasi-symmetry operators relate molecules of different color. The initial molecule is placed in position 1. The adjacent icosahedral threefold axis (marked by a 3) then generates molecules 2 and 3, and the adjacent quasi-twofold axis (marked by an orange oval) generates molecule 4. A quasi-threefold axis (marked by q3) generates molecules 5 and 6 from molecule 4. Subsequently, the icosahedral fivefold axis (marked by a 5) generates molecules 7 through 18 from molecules 4, 5, and 6, and the icosahedral twofold axis (marked by a 2) generates molecules 19, 20, and 21 from molecules 1, 5, and 7, respectively. Note that molecules 1, 5, 7, 19, 20, and 21 form a hexamer and molecules 6, 15, 16, 17, and 18 form a pentamer, as in the nucleocapsid. The thick black outlined triangle marks one of the 60 icosahedral asymmetric units.
TABLE 2.
Polar angles defining quasisymmetry axesa
| Symmetry axis | Psi | Phi | Kappa |
|---|---|---|---|
| Quasi-threefold between molecules 4, 5, and 6 | 79.6 | −72.6 | 120 |
| Quasi-twofold between molecules 1 and 4 | 73.9 | −81.2 | 180 |
The fitted E1 molecule puts domain II and the fusion peptide (residues 79 to 96) furthest away from the viral membrane. Domains I and II of the E1 molecule have a slant of roughly 35° relative to the viral surface, consistent with the observations of Lescar et al. (27). Three E1 molecules are organized as a trimer around a central patch of membrane shielded by the overhanging density belonging to E2 (Fig. 4), although the E1 molecules are not in contact with each other. Domain III of E1 ends near the outer leaflet of the membrane (see below).
FIG. 4.


Stereodiagrams showing the glycoproteins E1 and E2. (A) Surface-shaded representation of the E1-E2 glycoprotein spike at the quasi-threefold axis. Note the pore at the center of the spike, consistent with the results of Parades et al. (34). (B) Fit of E1 monomer Cα backbone into density (top view) around the quasi-threefold axis, viewed as in A. The molecules, represented by their Cα backbone, are colored as in Fig. 3. The cryoEM density is grey. (C) E1 i3 and q3 trimers (top view), color coded and oriented as in Fig. 3. Part of the triangular icosahedral asymmetric unit is outlined. The E1 carbohydrate difference densities (139 grey, 245 brown) are also shown, as well as their corresponding Cα atoms (black circle for 139 and black dot for 245). (D) Heterodimer showing the E1 structure and the E2 difference density (side view). The density corresponding to the lipid bilayer is shown in green. (E) The E2 difference density for molecule 5 (purple) fitted with the E1 Cα backbone (black). Note the central hole in the E2 density visible in the mauve orientation, probably as a result of an α-helix. (F) E2 difference density (side view) around a q3 axis colored blue (molecule 4), mauve (molecule 5), and brown (molecule 6). The E2 carbohydrate difference densities (196 mauve, 318 red) are also shown. The lipid bilayer density is shown in green. The fitted E1 molecules are shown as their Cα backbones, colored as in Fig. 3.
E1 makes two types of contacts with other E1 molecules. The first, involving residues in domain II, are few in number and are between neighboring trimeric spikes related by quasi-twofold symmetry. There are more of the second type of contacts and they occur between neighboring trimeric spikes around the quasi-sixfold axes (coincident with the icosahedral twofold axes) and around the icosahedral fivefold axes, involving mostly domain III. E1 also makes extensive contacts with E2.
The results of the restrained fitting procedure were essentially the same whether only the Cα atoms or all atoms were used (Table 1). There was a remarkably good agreement between the height of the cryoEM density at quasiequivalent positions (Table 3), showing that the properties of the T=4 symmetry operators had been well optimized. The correlation of densities at Cα atoms between pairs of quasiequivalent molecules varied from 0.84 to 0.92. The quality of fit, measured in terms of the average density of the fitted atoms (Sumf), was roughly the same for each of the three E1 domains in each of the four quasiequivalent positions (Table 3), demonstrating that the crystallographically determined Semliki Forest virus structure could be fitted to the cryoEM density without resorting to adjustment of interdomain hinge angles. Independent verification of these results was obtained by using the SITUS program (64).
TABLE 3.
Sumf values of the separate domains when fitting T=4-equivalent molecules into the cryoEM mapa
| Protein | Model | Molecule 1
|
Molecule 4
|
Molecule 5
|
Molecule 6
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D1 | D2 | D3 | D1 | D2 | D3 | D1 | D2 | D3 | D1 | D2 | D3 | ||
| E1 | SFV E1 (Cα) | 41.3 | 44.4 | 42.3 | 42.2 | 43.6 | 43.6 | 42.7 | 44.8 | 42.1 | 41.9 | 44.3 | 41.4 |
| SINV E1 (Cα) | 39.2 | 42.1 | 42.5 | 40.0 | 41.9 | 43.4 | 39.6 | 43.1 | 41.4 | 39.0 | 42.2 | 40.6 | |
| TBEV E (Cα) | 23.7 | 31.5 | 33.8 | 21.2 | 30.3 | 37.2 | 26.7 | 31.9 | 30.6 | 23.5 | 30.5 | 29.0 | |
| SINV E1 (all) | 33.9 | 38.1 | 39.6 | 33.9 | 37.4 | 40.5 | 35.5 | 39.1 | 39.2 | 33.4 | 38.0 | 37.2 | |
| TM | GCN4 (Cα, trunc) | 30.6 | 32.5 | 31.7 | 33.5 | 34.8 | 24.3 | 36.9 | 31.3 | ||||
| GCN4 (Cα, ext) | 26.6 | 31.6 | 17.4 | 25.1 | 28.3 | 23.8 | 29.8 | 26.4 | |||||
| CP | CP (Cα) | 47.2 | 44.7 | 43.9 | 44.6 | ||||||||
| CP + pep (Cα) | 46.8 | 44.5 | 43.9 | 44.8 | |||||||||
| CP (all) | 40.8 | 39.7 | 40.7 | 39.1 | |||||||||
| CP + pep (all) | 41.7 | 40.4 | 29.8 | 38.8 | |||||||||
| CP (old fit, all) | 36.0 | 35.4 | 34.9 | 34.0 | |||||||||
Each glycosylation site (corresponding to residues E1 139, E1 245, E2 196, and E2 318 in Sindbis virus and E1 141, E2 200, and E2 262 in Semliki Forest virus and Ross River virus) has four quasiequivalent positions. The positions of the glycosylation sites permitted the accurate determination of the positions and orientations of the quasisymmetry axes. The center of mass of quasi-threefold-related glycosylation sites provided a trace of the corresponding quasi-threefold axis. Similarly, the center of mass between the glycosylation sites related by a quasi-twofold axis provided a trace of the corresponding quasi-twofold axis. These vectors were extended by including the centers of the transmembrane regions (see below).
Plots (Fig. 5) confirmed the exact orientation of the quasi-symmetry axes and showed that the transmembrane regions obeyed, within experimental error, the same quasisymmetry as the E1 and E2 surface glycoproteins. These plots also showed that the quasi-threefold axes and quasi-twofold axes were almost radial. The quasisymmetry axes used for the fitting operations (Table 1) were assumed to be radial in order to minimize the number of parameters. Although this assumption is not completely accurate, the very small deviation shown by the plots (Fig. 5) can have only a very small impact on the fitting procedure.
FIG. 5.

Plots of x and y versus z, representing the trace of the quasi-threefold and quasi-twofold axes provided by the positions of the carbohydrate and transmembrane sites. Coordinates are for the mass center of sets of three (tracing the quasi-threefold axis) and two (tracing the quasi-twofold axis) carbohydrate sites and the transmembrane region. Equations for the least-squares fitted lines are shown. The appropriate Sindbis virus carbohydrate sites (318, 245, 139, and 196) and transmembrane site are shown at the top.
The E1 structure was fitted into all four quasiequivalent positions (molecules 1, 4, 5, and 6 in Fig. 3) in the cryoEM map by using the T=4 symmetry operators (PDB accession no. 1LD4). The average distance between each glycosylation site and its corresponding Cα or Nδ atom was 15.9 Å and 13.7 Å, respectively. These distances are reasonable to account for about two well-ordered sugar moieties at each glycosylation site.
E2 glycoprotein.
The cryoEM density associated with E2 was calculated by setting the density to zero at every grid point within a 4.5 Å radius of every atomic position, as determined by the restrained fitting of the E1 structure to the 11-Å resolution Sindbis virus map. Thus, the remaining density outside the lipid bilayer should belong to the E2 glycoprotein. It was immediately obvious that E2 was a long, thin molecule, somewhat similar in shape to and twined around E1 (Fig. 4), but the boomerang-shaped E2 molecule is far more radial in its disposition than is E1. E2 is known to contain the receptor attachment site near residue 218, situated in the large, protruding, external leaf-like surface (51). The external surface also contains the alphavirus glycosylation site 196 (in Sindbis virus) and 200 (in Semliki Forest virus and Ross River virus). The glycosylation site at residue 262 (in Semliki Forest virus and Ross River virus) is at the beginning of the stem leading from the external leaf-like surface. The Sindbis virus glycosylation site at residue 318, nearer the carboxy terminus of E2, is close to the membrane surface (37). Thus, all the E2 amino-terminal residues to at least residue 218 probably form the external large, leaf-like domain. From about residue 218 to the transmembrane region, the E2 polypeptide forms a long, narrow stem running roughly parallel to E1 and entering the viral membrane on the inside of the E1 trimer. The highly exposed leaf-like surface of E2 is followed by the narrower stem, which twists around the more tangentially disposed E1 molecule.
E1 and E2 are in contact with each other at both ends of the heterodimer, leaving a small gap between the two contact regions (Fig. 4D). Three E1-E2 heterodimers lean against each other, making a three-start, right-handed helix in the formation of each spike. The distal end of one heterodimer covers the proximal end of the neighboring heterodimer within the helical spike organization. The leaf-like structures of the three distal ends of the E2 molecules cover the fusion peptides of the three E1 molecules within the trimer. The only E2-to-E2 contacts are between the E2 leafs. There is a gap of about 30 Å between the base of neighboring E1-E2 heterodimers (Fig. 4), which would allow E2 to move out of the center of the spike at low pH to permit the exposure of viral membrane in the middle of each spike during fusion, as suggested by Kuhn et al. (24). At the same time, the three E1 molecules within each spike are reorganized into a trimer (19, 59), altering the surface-accessible residues of both E1 and E2. The fit of E1 is consistent with the suggested conformational changes as probed by proteolytic digestions and antibody accessibility (1, 36). The organization of the trimeric structure has some similarities to that of the flavivirus E and membrane (M) proteins in tick-borne encephalitis virus recombinant subviral particles (13).
Although the model of Sindbis virus E1 based on the Semliki Forest virus Cα atoms is unlikely to be accurate, it should be sufficient to determine, at least roughly, which residues of E1 make contact with E2. As described above, a map showing the E2 density was calculated with an atomic radius of 4.5 Å for each of the atoms in the Sindbis virus E1 homology model. In the next step, the average density within a radius of 6.5 Å was calculated for each atom. Those atoms of E1 with large average densities must thus be in contact with E2. The E1-to-E2 contacts at the membrane-proximal ends of the heterodimer involve charged residues in domain III of E1. The E1-to-E2 contacts between heterodimers involve primarily charged residues in domain II. This may account for the conformational changes that occur prior to fusion in an acidic environment (11, 15).
Frequently, a primordial gene is tandemly duplicated, and then each gene evolves separately. A typical example is the three tandem viral proteins VP2, VP3, and VP1 of picornaviruses, which have similar three-dimensional structures but no remaining, detectable sequence similarity (44). Thus, the remarkably similar long, thin shapes of E1 and E2, as well as their tandem organization in alphavirus genomes, might suggest that E1 and E2 have similar folds. Based on this speculation, a three-dimensional orientation search was made with the EMfit program for the best fit of the available Semliki Forest virus E1 backbone structure into the difference density representing E2. The fit to the density was good (Fig. 4E), with a mean Cα atom density of 30.3% of the maximum map density (Table 1). However, the tip of domain II penetrates the outer leaflet of the lipid bilayer. The central portion of the density ascribed to E2 looks like a donut in which the loops surround areas of low density (Fig. 4D, E, and F). The fit of the assumed E2 structure placed an α-helix into one of the loops. The same helix, when fitted to the E1 density, showed a similar but less distinct feature. The model fitted into the E2 density had the opposite direction from E1, with domains I and III forming much of the broad leaf-like external surface of E2 and the thin and long domain II forming the lower end, near the viral membrane. This proposed orientation is significant because domain III in tick-borne encephalitis virus E protein functions as the receptor attachment site. Domain III would form the leaf-like structure of E2 on the outside of the virus, a position known to be associated with alphavirus cell recognition and attachment (51). Furthermore, domain II, with the equivalence of a fusion peptide at the tip of domain II, is now inserted into the outer leaflet of the viral membrane.
At 11-Å resolution, the fit of Semliki Forest virus E1 to the E2 density establishes similarity not of fold, but only of shape. Indeed, as the carboxy-terminal domain III of the E1 structure fitted into the E2 density forms much of the external leaf-like structure, and as it has already been established that the leaf-like structure consists of the amino-terminal section of E2, there must be some differences in fold between E1 and E2. Nevertheless, the glycosylation sites corresponding to E2 residues 196, 200, 262, and 318 align approximately with residues 21, 23, 153, and 225 on the fitted Semliki Forest virus structure, respectively. The correspondence between E2 glycosylation sites and residues on the fitted Semliki Forest virus E1 structure suggests that the sequence of the domains on E2 is DIII-DI-DII, placing at least 110 amino acids of a probable immunoglobulin-like domain at the amino end of the E2 polypeptide, although the spatial arrangement of the domains in E1 and E2 is the same. Similar domain rearrangements are found in other protein structures (5, 47). Verification of the proposed E2 structure will require determination of the atomic resolution structure of E2 by X-ray crystallography.
Structural similarity between Semliki Forest virus E1 and tick-borne encephalitis virus E.
It has long been established that the rate of change of amino acids is usually much faster than that of the three-dimensional fold in the evolution of proteins (48). Therefore, it is not surprising that there is no obvious sequence similarity between Semliki Forest virus E1 and tick-borne encephalitis virus E, although the folds are clearly rather similar (27). A rough scale of similarity can be established by measuring the number and percentage of Cα atoms that can be sequentially aligned in three dimensions (22, 31, 56).
Structural alignment based on the Cα backbone (38, 43) showed that 63% of the residues in domain I and 61% of the residues in domain II of the Semliki Forest virus E1 structure could be equivalenced to residues in the tick-borne encephalitis virus E structure, accounting for the superposition of nearly all the secondary structural elements in domains I and II (Fig. 2). However, only 14% of the residues in domain III could be structurally aligned. Separate alignments of the domains produced essentially the same alignment for domains I and II, demonstrating that these form a single rigid body. Separate alignment of domain III made it possible to increase the number of equivalenced residues to 23%, a level of similarity still well below a significant structural relationship (31). In contrast, in a comparison of the viral proteins VP1, VP2, and VP3 of picornaviruses, the number of residues that can be aligned structurally is between 50 and 70%, although there is no easily recognizable amino acid sequence similarity (29).
The absence of a significant similarity between Semliki Forest virus E1 and tick-borne encephalitis virus E in domain III suggests that it has had a different history or is evolving faster than the other two domains. Thus, these domains may have been derived from different sources for these two families of viruses. This would also be consistent with domain III being fused to the amino instead of the carboxy end of E2 (see above).
The fitting procedure was also used to position tick-borne encephalitis virus E (instead of Semliki Forest virus E1) into the 11-Å cryoEM map of Sindbis virus with only the Cα atoms. Restraints were established by equivalencing the alphavirus E1 glycosylation sites with the tick-borne encephalitis virus flavivirus E amino acid sequence, based on the structural alignment of tick-borne encephalitis virus with Semliki Forest virus E1 (Fig. 2). This showed that the tick-borne encephalitis virus E structure resulted in an average density (Sumf) for Cα atoms of only 29%, as opposed to 43% for the Semliki Forest virus E1 structure (Table 1), demonstrating how well the Sindbis virus map is able to differentiate between the distantly similar tick-borne encephalitis virus E structure and the closely related Semliki Forest virus E1 structure.
Lipid membrane bilayer.
The most striking feature in the 11-Å resolution cryoEM map is the characteristic appearance of the lipid bilayer between 217-Å and 261-Å radii (Fig. 1). The pair of high, roughly spherical, uniform densities centered at 222 Å and 256 Å are generated by the phosphate head groups and are separated by much lower density, centered around a radius of 239 Å, produced by the aliphatic region associated with the phosphate layers. The mean density of the phosphate head groups is about 75% of the average density in the glycoproteins, a ratio closely similar to that observed in a 24-Å resolution cryoEM map of dengue virus (24).
The membrane is penetrated by a number of higher-density regions (Fig. 1 and 6), whose location is consistent with the T= 4 symmetry of the virus. The three transmembrane densities related by a quasi-threefold axis (Fig. 3 and 6) can be superimposed on themselves after a ≈120° rotation to within a root mean square deviation of 1.9 Å. These, in turn, can be superimposed onto the three icosahedrally related transmembrane densities with a root mean square deviation of 1.6 Å. These densities locate the positions of the quasi-twofold and quasi-threefold axes, which are in excellent agreement with the quasi-threefold and quasi-twofold symmetry axes defined by the site of the previously determined carbohydrate positions (Fig. 5) (37).
FIG. 6.

(A) Side view of one of the four T=4 related transmembrane regions fitted with a 28-residue coiled-coil segment derived from the GCN4 structure (Cα, trunc, in Table 1). (B) Sequence of the E1 and E2 glycoproteins fitted to the A and B chains of GCN4, respectively. Ectodomain residues are in blue, residues in phospholipid leaflets are in green, residues in the aliphatic center of the membrane are in yellow, cytoplasmic residues are in red, and residues that bind to capsid protein (CP) are in black.
Each of the four T=4 related transmembrane densities is connected by rods of higher density to the carboxy termini of the ectodomains of E1 and E2 (Fig. 6). On the cytoplasmic side of the membrane, one strand of the transmembrane densities is connected to the surface of the nucleocapsid core subunit. The transmembrane regions themselves show resolved helices, as has also been suggested by Mancini et al. (30), twisting around each other in a left-handed manner, appropriate for a dimeric coiled coil (Fig. 6).
The heterodimeric coiled-coil component of GCN4 (PDB accession number 1YSA) was used as a model to fit into the four quasiequivalent transmembrane densities (Table 1) with the program EMfit. The fitting process was constrained by the established T=4 symmetry (Table 2). The fitting was also restrained by minimizing the distances between the amino terminus of the truncated GCN4 A chain and the carboxy-terminal residue 380 of E1. The fit shows a good agreement between the coiled-coil component of GCN4 and the Sindbis virus transmembrane densities (Fig. 6). The amino ends of the A and B chains of the GCN4 structure are well separated from each other but, nevertheless, are in or near α-helix-like density. The amino end of helix B is close to the density representing the E2 glycoprotein. Thus, the GCN4 structure is a mimic of E1 and E2 in the vicinity of the viral membrane.
There are 28 amino acids in the membrane section in both the A and B chains, with 17 residues of each chain associated with the central aliphatic region of the membrane (Fig. 6). Building of the E1 transmembrane amino acid sequence into the A chain of GCN4 shows that this region extends roughly from residues Thr409 to Ser435, leaving four residues on the cytoplasmic side, in contrast to previous suggestions (4). The extension of E1 and especially E2 towards the nucleocapsid is clearly visible in the cryoEM map. Furthermore, the B chain has nine residues in the cytoplasmic region of E2, emerging from the viral membrane at residue Cys390 and finishing close by the amino terminus of the peptide (residues 400-Tyr-Ala-Leu; see below) associated with the capsid protein binding site (26), consistent with expectations. Thus, chain A of GCN4 runs from near the carboxy terminus of the fitted E1 fragment (probably residue Thr390; see below) to the E1 carboxy terminus at Arg438 on the cytoplasmic side of the viral membrane, whereas chain B runs from the vicinity of the E2 ectodomain to the E2 binding site on the nucleocapsid.
There are 28 amino acids between the carboxy terminus of the fitted E1 fragment at residue 380 and the entrance of the E1 polypeptide into the membrane at residue 409. However, the amino end of the fitted GCN4 A chain is only about 10 Å away from the final E1 ordered carboxy-terminal residue Cys380. There are 18 residues in the GCN4 A chain before it enters the transmembrane region. Thus, there are about 10 amino acids between the end of E1 and the beginning of the GCN4 A helix, for which there is no density and little available space.
The Sindbis virus E1 model was based on the crystallographic determination of the homologous Semliki Forest virus E1 structure. The carboxy terminus of the crystallized Semliki Forest virus E1 had been determined to be at Tyr390 (62), although the final 10 residues (381 to 390) were found to be disordered. In view of the difficulty encountered in assigning the amino acid sequence to the X-ray electron density (27) in domain III, it is possible that the final short, disconnected amino acid sequence might have been incorrectly assigned and that the residues leading to Semliki Forest virus E1 Cys380 really should have been residues leading to Tyr390.
Nucleocapsid.
The density on the inside of the lipid bilayer, between 220-Å and 180-Å radii, also has large positive and negative density values, resulting in the largest root mean square deviations in the map, corresponding to the nucleocapsid core region (Fig. 1). The average density of the core is almost equal to the average density in the glycoprotein annulus, demonstrating good structural order in both. In comparison, the core region in dengue virus is only half the height of the glycoprotein (24), demonstrating a very different structural character of the nucleocapsid in these two families of enveloped RNA viruses.
The X-ray crystallographic structure of Sindbis virus capsid protein (9) was fitted into the 11-Å resolution cryoEM map of Sindbis virus (Table 1) with the EMfit program. The procedure was the same as had been used to interpret the capsid structure of Ross River virus at 24-Å resolution (8) except for the resolution of the maps and the parameters used to define the quasisymmetry (see above). The orientation of the capsid protein was found to be completely different from that reported previously (Table 1). Whereas the new orientation has a quality of fit similar to that of placing the E1 glycoprotein into the 11-Å map, the old orientation gives a significantly worse fit (Table 1). The new orientation fills more of the cryoEM density, including the intersubunit region, has few clashes between neighboring subunits, and is consistent with the higher-resolution details of the 11-Å map. Furthermore, in the new orientation, the surface binding pocket (26) is close to the carboxy end of the GCN4 B chain. Thus, a final fit was established by adding a peptide (corresponding to E2 residues 400-Tyr-Ala-Leu) to the binding pocket of the search model and by minimizing the distance between the amino end of the bound peptide and the carboxy end of the fitted GCN4 B chain (Table 1 and Fig. 7).
FIG. 7.

Fit of the capsid protein (CP) crystal structure into the cryoEM map. (A) Top view, showing the hexamer and pentamer around an icosahedral twofold axis and a fivefold axis, respectively. Molecules are colored as in Fig. 3. The electron density is colored grey. (B) Side view of one capsid protein crystal structure (black) fitted into the cryoEM density. The membrane density is colored green. Tyr180 in the peptide binding pocket is shown in blue. (C) CryoEM difference density after subtracting the density due to the capsid protein. Note that the amino-terminal residues 106 to 114 are out of density but that there is an obvious density region where these residues are positioned in the virus as opposed to the crystal structure. This density leads from the mixed protein-RNA region into the capsid protein shell.
The revised orientation of the capsid protein places the amino end (residue 114) of the chymotrypsin-like C-terminal domain on the inner surface of the capsid, where it might be expected to join with the disordered N-terminal domain associated with the genomic RNA. Indeed, there is additional, uninterpreted density, equivalent to an extended polypeptide of about a dozen residues, leading from the RNA region towards residue 114, running close and parallel to residues 106 to 114 (Fig. 7). The polypeptide chain between residues 106 to 114 is associated with neighboring molecules in the crystal lattice of various capsid protein crystal structures (26). Hence, it is not surprising that in the virion these residues are more closely associated with the C-terminal domain than they are in crystal structures.
The charge distribution on the pentameric and hexameric rings of capsid protein subunits is largely positive on the external side facing the negatively charged membrane surface, confirming that the present orientation of the capsid protein is chemically reasonable. The internal face of these oligomeric rings, facing the RNA and the positively charged amino-terminal capsid domain, is more neutral.
The revised capsid structure places the NG atom of Lys250 on the inside of the capsid hexamers and pentamers, which would allow adjacent subunits within the same ring to be cross-linked by a 12-Å-long dimethyl suberimidate cross-linker. It has been shown that Sindbis virus cores can be self-assembled in vitro in the presence of suitable RNA or DNA oligomers and isolated capsid protein (53). Furthermore, assembly can occur from dimers of cross-linked capsid dimers (54, 55). This made sense in terms of the previous interpretation of the capsid protein orientation, which placed Lys250 on the outside of hexamers and pentamers separated by a distance suitable for cross-linking between hexamers and pentamers. The capsid orientation that now fits the density better would make it impossible to assemble pentamers from sets of dimers. Presumably, therefore, the in vitro-assembled cores, with pairwise cross-linked protein subunits, would be imperfect, missing one of five subunits in each of the 12 pentamers. However, a deficiency of 12 monomers out of a total of 240 monomers per particle would probably have gone undetected in the electron microscopy used to assay the assembly reaction. It is noteworthy that only about 20% of dimers could be isolated when in vitro-assembled cores or cores isolated from cytoplasm were cross-linked (54).
Previous experiments that introduced site-specific mutations in the residues thought to create the interface between subunits did not eliminate virus particle formation (K. E. Owen and R. J. Kuhn, unpublished data). Thus, our finding an alternative solution to the structure of the nucleocapsid was consistent with the previous negative results. The new structure has a remarkably small subunit-to-subunit interface, suggesting that formation of the interface is not the major driving force that creates the observed pentamers and hexamers. Instead, it is probably the association of RNA with the amino-terminal domain (residues 1 to 113), as well as the formation of a coiled-coil association between the amino-terminal domain of the capsid proteins (35), that drives the assembly. Nevertheless, the presently proposed structure does place the guanidinium group of Arg174 within 2.3 Å of the carboxy group of Glu259 in the neighboring subunit. Arg174 is a conserved basic residue, and Glu259 is completely conserved in all alphaviruses sequenced, suggesting that this interaction might be important for assembly.
RNA structure.
Within the core, between a radius of 150 and 180 Å, is a region whose average density is only half the height of the glycoprotein or nucleocapsid densities and whose character is more uniform, with a smaller root mean square deviation (Fig. 1). As mentioned above, the difference density produced by subtracting the fitted capsid protein clearly shows the path of the capsid protein residues from about 100 to 114 leading from this layer of partially disordered structure to the beginning of the well-ordered capsid structure (Fig. 7).
It has been shown that residues 76 to 113 interact specifically with RNA (32, 61), whereas residues 1 to 75 in the N-terminal domain of the capsid protein interact nonspecifically with the predominantly negatively charged RNA (18, 60). This is consistent with the assumption that the lower, more uniform density inside the ordered capsid structure is a mixed protein-RNA region. The ratio of the volume of the capsid protein density compared to the volume of the mixed protein-RNA region is 2.0. However, the ratio of the number of amino acids in the corresponding regions is 1.50 (assuming that all the 100 amino acids of the capsid's amino-terminal domain are in the mixed region). Thus, the mixed protein-RNA region has space for an ample amount of RNA as well as all the protein in the amino-terminal region of the capsid.
The lack of extensive capsid-capsid contacts suggests that the RNA-capsid association has to play a critical role in assembly, as was also recognized by the requirement of nucleic acid for the in vitro self-assembly of cores (53). The density distribution of the mixed protein-RNA region features higher densities running towards the quasi- and icosahedral-threefold axes (Fig. 7C). Thus, each quasi-threefold axial position associates one capsid protein pentamer with two hexamers, and each icosahedral threefold position associates three capsid hexamers. This implies that the association of pentamers and hexamers is determined in the same way by their relationships with the quasi- and icosahedral spikes as they are with their association with each other in forming a complex with the genomic RNA.
Finally, inside the 150-Å radius is a region of moderately uniform density, corresponding to the remainder of the genomic RNA. The features in the RNA region are long and narrow, similar to the RNA region in dengue virus (24).
Acknowledgments
The first two authors contributed equally to this work.
We thank Robert Ashmore for his RobEM graphic user interface program, Chuan Xiao for writing several programs that helped to speed up the calculations, and Sharon Wilder as well as Cheryl Towell for help in the preparation of the manuscript. We are grateful to Paul Chipman, who participated in some of the cryoEM data collection.
The work was supported by an NIH Program Project Grant (AI45976) to T.S.B., R.J.K., and M.G.R. as well as NIH grants to T.S.B. (GM33050) and to R.J.K. (GM5627).
REFERENCES
- 1.Ahn, A., M. R. Klimjack, P. K. Chatterjee, and M. Kielian. 1999. An epitope of the Semliki Forest virus fusion protein exposed during virus-membrane fusion. J. Virol. 73:10029-10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker, T. S., and R. H. Cheng. 1996. A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J. Struct. Biol. 116:120-130. [DOI] [PubMed] [Google Scholar]
- 3.Baker, T. S., N. H. Olson, and S. D. Fuller. 1999. Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol. Mol. Biol. Rev. 63:862-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barth, B. U., M. Suomalainen, P. Liljeström, and H. Garoff. 1992. Alphavirus assembly and entry: role of the cytoplasmic tail of the E1 spike subunit. J. Virol. 66:7560-7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bashton, M., and C. Chothia. 2002. The geometry of domain combination in proteins. J. Mol. Biol. 315:927-939. [DOI] [PubMed] [Google Scholar]
- 6.Cancedda, R., L. Villa-Komaroff, H. F. Lodish, and M. Schlesinger. 1975. Initiation sites for translation of Sindbis virus 42S and 26S messenger RNAs. Cell 6:215-222. [DOI] [PubMed] [Google Scholar]
- 7.Caspar, D. L. D., and A. Klug. 1962. Physical principles in the construction of regular viruses. Cold Spring Harbor Symp. Quant. Biol. 27:1-24. [DOI] [PubMed] [Google Scholar]
- 8.Cheng, R. H., R. J. Kuhn, N. H. Olson, M. G. Rossmann, H. K. Choi, T. J. Smith, and T. S. Baker. 1995. Nucleocapsid and glycoprotein organization in an enveloped virus. Cell 80:621-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi, H. K., L. Tong, W. Minor, P. Dumas, U. Boege, M. G. Rossmann, and G. Wengler. 1991. Structure of Sindbis virus core protein reveals a chymotrypsin-like serine proteinase and the organization of the virion. Nature (London) 354:37-43. [DOI] [PubMed] [Google Scholar]
- 10.Crowther, R. A., D. J. DeRosier, and A. Klug. 1970. The reconstruction of a three-dimensional structure from projections and its application to electron microscopy. Proc. R. Soc. London A317:319-340. [DOI] [PubMed]
- 11.Edwards, J., E. Mann, and D. T. Brown. 1983. Conformational changes in Sindbis virus envelope proteins accompanying exposure to low pH. J. Virol. 45:1090-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellenberger, T. E., C. J. Brandl, K. Struhl, and S. C. Harrison. 1992. The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted α helices: crystal structure of the protein-DNA complex. Cell 71:1223-1237. [DOI] [PubMed] [Google Scholar]
- 13.Ferlenghi, I., M. Clarke, T. Ruttan, S. L. Allison, J. Schalich, F. X. Heinz, S. C. Harrison, F. A. Rey, and S. D. Fuller. 2001. Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol. Cell 7:593-602. [DOI] [PubMed] [Google Scholar]
- 14.Forsell, K., L. Xing, T. Kozlovska, R. H. Cheng, and H. Garoff. 2000. Membrane proteins organize a symmetrical virus. EMBO J. 19:5081-5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuller, S. D., J. A. Berriman, S. J. Butcher, and B. E. Gowen. 1995. Low pH induces swiveling of the glycoprotein heterodimers in the Semliki Forest virus spike complex. Cell 81:715-725. [DOI] [PubMed] [Google Scholar]
- 16.Fuller, S. D., S. J. Butcher, R. H. Cheng, and T. S. Baker. 1996. Three-dimensional reconstruction of icosahedral particles — the uncommon line. J. Struct. Biol. 116:48-55. [DOI] [PubMed] [Google Scholar]
- 17.Garoff, H., A. M. Frischauf, K. Simons, H. Lehrach, and H. Delius. 1980. Nucleotide sequence of cDNA coding for Semliki Forest virus membrane glycoproteins. Nature (London) 288:236-241. [DOI] [PubMed] [Google Scholar]
- 18.Geigenmüller-Gnirke, U., H. Nitschko, and S. Schlesinger. 1993. Deletion analysis of the capsid protein of Sindbis virus: identification of the RNA binding region. J. Virol. 67:1620-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibbons, D. L., A. Ahn, P. K. Chatterjee, and M. Kielian. 2000. Formation and characterization of the trimeric form of the fusion protein of Semliki Forest virus. J. Virol. 74:7772-7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hahn, C. S., and J. H. Strauss. 1990. Site-directed mutagenesis of the proposed catalytic amino acids of the Sindbis virus capsid protein autoprotease. J. Virol. 64:3069-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Havelka, W. A., R. Henderson, and D. Oesterhelt. 1995. Three-dimensional structure of halorhodopsin at 7 Å resolution. J. Mol. Biol. 247:726-738. [DOI] [PubMed] [Google Scholar]
- 22.Holm, L., and C. Sander. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233:123-138. [DOI] [PubMed] [Google Scholar]
- 23.Kielian, M., M. R. Klimjack, S. Ghosh, and W. A. Duffus. 1996. Mechanisms of mutations inhibiting fusion and infection by Semliki Forest virus. J. Cell Biol. 134:863-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhn, R. J., W. Zhang, M. G. Rossmann, S. V. Pletnev, J. Corver, E. Lenches, C. T. Jones, S. Mukhopadhyay, P. R. Chipman, E. G. Strauss, T. S. Baker, and J. H. Strauss. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee, S., R. J. Kuhn, and M. G. Rossmann. 1998. Probing the potential glycoprotein binding site of Sindbis virus capsid protein with dioxane and model building. Proteins 33:311-317. [PubMed]
- 26.Lee, S., K. E. Owen, H. K. Choi, H. Lee, G. Lu, G. Wengler, D. T. Brown, M. G. Rossmann, and R. J. Kuhn. 1996. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure 4:531-541. [DOI] [PubMed] [Google Scholar]
- 27.Lescar, J., A. Roussel, M. W. Wein, J. Navaza, S. D. Fuller, G. Wengler, G. Wengler, and F. A. Rey. 2001. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105:137-148. [DOI] [PubMed] [Google Scholar]
- 28.Levy-Mintz, P., and M. Kielian. 1991. Mutagenesis of the putative fusion domain of the Semliki Forest virus spike protein. J. Virol. 65:4292-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo, M., G. Vriend, G. Kamer, I. Minor, E. Arnold, M. G. Rossmann, U. Boege, D. G. Scraba, G. M. Duke, and A. C. Palmenberg. 1987. The atomic structure of Mengo virus at 3.0 Å resolution. Science 235:182-191. [DOI] [PubMed] [Google Scholar]
- 30.Mancini, E. J., M. Clarke, B. Gowen, T. Rutten, and S. D. Fuller. 2000. Cryo-electron microscopy reveals the functional anatomy of an enveloped virus, Semliki Forest virus. Mol. Cell 5:255-266. [DOI] [PubMed] [Google Scholar]
- 31.Matthews, B. W., and M. G. Rossmann. 1985. Comparison of protein structures. Methods Enzymol. 115:397-420. [DOI] [PubMed] [Google Scholar]
- 32.Owen, K. E., and R. J. Kuhn. 1996. Identification of a region in the Sindbis virus nucleocapsid protein that is involved in specificity of RNA encapsidation. J. Virol. 70:2757-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paredes, A. M., D. T. Brown, R. Rothnagel, W. Chiu, R. J. Schoepp, R. E. Johnston, and B. V. V. Prasad. 1993. Three-dimensional structure of a membrane-containing virus. Proc. Natl. Acad. Sci. USA 90:9095-9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paredes, A. M., H. Heidner, P. Thuman-Commike, B. V. V. Prasad, R. E. Johnston, and W. Chiu. 1998. Structural localization of the E3 glycoprotein in attenuated Sindbis virus mutants. J. Virol. 72:1534-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perera, R., K. E. Owen, T. L. Tellinghuisen, A. E. Gorbalenya, and R. J. Kuhn. 2001. Alphavirus nucleocapsid protein contains a putative coiled coil α-helix important for core assembly. J. Virol. 75:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phinney, B. S., K. Blackburn, and D. T. Brown. 2000. The surface conformation of Sindbis virus glycoproteins E1 and E2 at neutral and low pH, as determined by mass spectrometry-based mapping. J. Virol. 74:5667-5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pletnev, S. V., W. Zhang, S. Mukhopadhyay, B. R. Fisher, R. Hernandez, D. T. Brown, T. S. Baker, M. G. Rossmann, and R. J. Kuhn. 2001. Locations of carbohydrate sites on Sindbis virus glycoproteins show that E1 forms an icosahedral scaffold. Cell 105:127-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rao, S. T., and M. G. Rossmann. 1973. Comparison of super-secondary structures in proteins. J. Mol. Biol. 76:241-256. [DOI] [PubMed] [Google Scholar]
- 39.Rey, F. A., F. X. Heinz, C. Mandl, C. Kunz, and S. C. Harrison. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature (London) 375:291-298. [DOI] [PubMed] [Google Scholar]
- 40.Rice, C. M., and J. H. Strauss. 1981. Synthesis, cleavage and sequence analysis of DNA complementary to the 26 S messenger RNA of Sindbis virus. J. Mol. Biol. 150:315-340. [DOI] [PubMed] [Google Scholar]
- 41.Roseman, A. M. 2000. Docking structures of domains into maps from cryo-electron microscopy with local correlation. Acta Crystallogr. Sect. D 56:1332-1340. [DOI] [PubMed] [Google Scholar]
- 42.Rossmann, M. G. 2000. Fitting atomic models into electron microscopy maps. Acta Crystallogr. Sect. D 56:1341-1349. [DOI] [PubMed] [Google Scholar]
- 43.Rossmann, M. G., and P. Argos. 1975. A comparison of the heme binding pocket in globins and cytochrome b5. J. Biol. Chem. 250:7525-7532. [PubMed] [Google Scholar]
- 44.Rossmann, M. G., E. Arnold, J. W. Erickson, E. A. Frankenberger, J. P. Griffith, H. J. Hecht, J. E. Johnson, G. Kamer, M. Luo, A. G. Mosser, R. R. Rueckert, B. Sherry, and G. Vriend. 1985. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature (London) 317:145-153. [DOI] [PubMed] [Google Scholar]
- 45.Rossmann, M. G., R. Bernal, and S. V. Pletnev. 2002. Combining electron microscopic with X-ray crystallographic structures. J. Struct. Biol. 136:190-200. [DOI] [PubMed]
- 46.Rossmann, M. G., and D. M. Blow. 1962. The detection of subunits within the crystallographic asymmetric unit. Acta Crystallogr. 15:24-31. [Google Scholar]
- 47.Rossmann, M. G., A. Liljas, C. I. Brändén, and L. J. Banaszak. 1975. Evolutionary and structural relationships among dehydrogenases, p. 61-102. In P. D. Boyer (ed.), The enzymes, 3rd ed., vol. XI. Academic Press, New York, N.Y.
- 48.Rossmann, M. G., D. Moras, and K. W. Olsen. 1974. Chemical and biological evolution of a nucleotide-binding protein. Nature (London) 250:194-199. [DOI] [PubMed] [Google Scholar]
- 49.Schlesinger, M. J., and S. Schlesinger. 1986. Formation and assembly of alphavirus glycoproteins, p. 121-148. In S. Schlesinger and M. J. Schlesinger (ed.), The togaviridae and flaviviridae. Plenum Publishing Corp., New York, N.Y.
- 50.Skoging, U., M. Vihinen, L. Nilsson, and P. Liljeström. 1996. Aromatic interactions define the binding of the alphavirus spike to its nucleocapsid. Structure 4:519-529. [DOI] [PubMed] [Google Scholar]
- 51.Smith, T. J., R. H. Cheng, N. H. Olson, P. Peterson, E. Chase, R. J. Kuhn, and T. S. Baker. 1995. Putative receptor binding sites on alphaviruses as visualized by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 92:10648-10652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strauss, J. H., and E. G. Strauss. 2001. Virus evolution: how does an enveloped virus make a regular structure? Cell 105:5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tellinghuisen, T. L., A. E. Hamburger, B. R. Fisher, R. Ostendorp, and R. J. Kuhn. 1999. In vitro assembly of alphavirus cores by using nucleocapsid protein expressed in Escherichia coli. J. Virol. 73:5309-5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tellinghuisen, T. L., and R. J. Kuhn. 2000. Nucleic acid-dependent cross-linking of the nucleocapsid protein of Sindbis virus. J. Virol. 74:4302-4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tellinghuisen, T. L., R. Perera, and R. J. Kuhn. 2001. In vitro assembly of Sindbis virus core-like particles from cross-linked dimers of truncated and mutant capsid proteins. J. Virol. 75:2810-2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Todd, A. E., C. A. Orengo, and J. M. Thornton. 2001. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 307:1113-1143. [DOI] [PubMed] [Google Scholar]
- 57.Vénien-Bryan, C., and S. D. Fuller. 1994. The organization of the spike complex of Semliki Forest virus. J. Mol. Biol. 236:572-583. [DOI] [PubMed] [Google Scholar]
- 58.Volkmann, N., K. J. Amann, S. Stoilova-McPhie, C. Egile, D. C. Winter, L. Hazelwood, J. E. Heuser, R. Li, T. D. Pollard, and D. Hanein. 2001. Structure of Arp2/3 complex in its activated state and in actin filament branch junctions. Science 293:2456-2459. [DOI] [PubMed] [Google Scholar]
- 59.Wahlberg, J. M., R. Bron, J. Wilschut, and H. Garoff. 1992. Membrane fusion of Semliki Forest virus involves homotrimers of the fusion protein. J. Virol. 66:7309-7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weiss, B., U. Geigenmüller-Gnirke, and S. Schlesinger. 1994. Interactions between Sindbis virus RNAs and a 68 amino acid derivative of the viral capsid protein further defines the capsid binding site. Nucleic Acids Res. 22:780-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weiss, B., H. Nitschko, I. Ghattas, R. Wright, and S. Schlesinger. 1989. Evidence for specificity in the encapsidation of Sindbis virus RNAs. J. Virol. 63:5310-5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wengler, G., G. Wengler, and F. A. Rey. 1999. The isolation of the ectodomain of the alphavirus E1 protein as a soluble hemagglutinin and its crystallization. Virology 257:472-482. [DOI] [PubMed] [Google Scholar]
- 63.Wriggers, W., and P. Chacón. 2001. Modeling tricks and fitting techniques for multiresolution structures. Structure 9:779-788. [DOI] [PubMed] [Google Scholar]
- 64.Wriggers, W., R. A. Milligan, and J. A. McCammon. 1999. Situs: a package for docking crystal structures into low-resolution maps from electron microscopy. J. Struct. Biol. 125:185-189. [DOI] [PubMed] [Google Scholar]