

Abstract
Human subtelomeres are polymorphic patchworks of inter-chromosomal segmental duplications at the ends of chromosomes. We provide evidence here that these patchworks arose recently through repeated translocations between chromosome ends. We assess the relative contribution of the major modes of ectopic DNA repair to the formation of subtelomeric duplications and find that non-homologous end-joining predominates. Once subtelomeric duplications arise, they are prone to homology-based sequence transfers as evidenced by incongruent phylogenetic relationships of neighboring sections. Inter-chromosomal recombination of subtelomeres is a potent force for recent change. Cytogenetic and sequence analyses reveal that pieces of the subtelomeric patchwork changed location and copy number during primate evolution with unprecedented frequency. Half of known subtelomeric sequence formed recently through human-specific sequence transfers and duplications. Subtelomeric dynamics result in a gene-duplication rate significantly higher than the genome average and could have both advantageous and pathological consequences in human biology. More generally, our analyses suggest an evolutionary cycle between segmental polymorphisms and genome rearrangements.
The human genome contains an abundance of large DNA segments that duplicated during the last 40 million years1,2. These segmental duplications (SDs) represent ≥5% of the genome2 and are found frequently near centromeres and telomeres3. SDs are emerging as significant factors in chromosomal rearrangements leading to disease4 and rapid gene innovation2, but the mechanisms by which they form are not well understood. Here, we focus on the unusually dense concentrations of inter-chromosomal SDs comprising human subtelomeres, which form the transition zones between chromosome-specific sequence and the arrays of telomeric repeats capping each chromosomal end. Previous cytogenetic studies showed that human subtelomeres are strikingly polymorphic in content – large segments can be present in or absent from normal alleles5 – and that copy number of subtelomeric segments can vary among higher primates6–9. This natural plasticity combined with documented expression of several human subtelomeric genes10,11 suggests that the evolutionary dynamics of subtelomeric regions could contribute to normal phenotypic variation within and between primate species, as is observed in other organisms (reviewed in 5). However, subtle rearrangements of DNA near the ends of chromosomes are observed in association with human disorders, including mental retardation12. Although full sequence coverage is not yet achieved for all chromosome ends, let alone for multiple alleles of each end, much can be learned from available sequence about subtelomere organization, evolution, variation, and function, as well as more generally about the origin and consequences of segmental duplications.
Complex inter-related structures
Our “paralogy map” of subtelomeric SDs (Fig. 1, Table S1) uses all finished sequences of genomic clones submitted to GenBank before April 2003. The map comprises ~2.6 Mbp of sequence present in two or more of 33 human subtelomeres (including three allelic pairs). The seven completely sequenced subtelomeres in the set are bounded distally by 0.5–2.4 kbp of various tandemly repeated units13 called telomere-associated repeats (TAR1) and a short sample of the native telomeric arrays14. Numerous degenerate telomere-like repeats and TAR1 elements are also situated at varying distances from telomeres15,16 (Fig. 1). Notably, these repeats are almost always oriented 5’-3’ towards the telomere.
Figure 1.

Subtelomeric paralogy map. Subtelomeric contigs (Table S1 gives constituent accessions and localization methods) are aligned at telomeres or to maximize alignments of paralogous blocks. Copies of a given block have the same color, width, and number. Only blocks 15 and 40 on 4q, 22 on 3q, 34–37 on 1p, and 38 on 6q are in inverted orientation relative to other corresponding block copies. 2qFS_I and _II represent ancestral telomeres fused head-to-head at 2q13–14; other interstitial paralogies are not displayed or analyzed here. A and B indicate allelic variants. Yq/Xq pseudoautosomal homology extends distal of dotted line.
The paralogy map reveals the complex patchwork of sequence blocks shared by human subtelomeres. Different subtelomeres can show >100 kbp continuous similarity, but a segment shared by a given chromosome set extends only 13 kbp on average before being displaced on at least one subtelomere by a segment with a different chromosomal distribution. In the 33 subtelomeric contigs analyzed, we identify 41 homology blocks larger than 3 kbp (Fig. 1). These blocks occur in 2 to 18 (average, 5) copies, with 88%–99.9% identity (Table S2). Almost all instances of these blocks are in the same orientation and relative order (Fig. 1). PCR analyses of monochromosomal hybrid cell lines confirm block boundaries defined by sequence alignments and identify at least one additional chromosomal copy for 17 of 29 blocks evaluated (Fig. S1, Table S3).
Subtelomeres contain members of 25 small gene families (Fig. 1), with one gene per 30 kbp on average. Eighteen families contain at least one subtelomeric member that encodes a potentially functional protein (Table S4). Thus, gain, loss, or alteration of subtelomeric genes has potential phenotypic effect. Subtelomeric genes have highly varied functions and include odorant and cytokine receptors, tubulins, transcription factors, and genes of unknown function.
Sequence in the paralogy map and duplicates thereof detected in later assemblies and/or by PCR (a total of 0.97 Mbp) account for ≥83% of the estimated subtelomeric terrain in a typical genome16. Approximately 90% of the 490 kbp of finished sequence added to nine ends in the latest genome assembly (Build 35) is >90% identical to sequence already represented in our dataset; only 26 kbp is novel. Thus, our dataset represents a reasonably comprehensive sample from which mechanistic information can be derived.
Mechanisms of sequence transfer
To investigate the mechanisms producing subtelomeric SDs, we considered their evolutionary history as consisting of two phases. The first is duplication to a new chromosome creating a novel structural boundary, and the second involves interactions between existing duplicates. We analyzed the patterns and breakpoints of homology in sequenced subtelomeres to infer the mechanism of inter-chromosomal sequence transfer resulting in the first step. Two primary models were considered that might give rise to subtelomeric SDs: chromosome translocations and DNA transposition.
Several observations argue for the translocation model (Fig. 2) and against transposition. First, subtelomeric blocks do not have characteristic features associated with known transposons or their insertion sites17. Proposed targets for insertions of SDs by a more general transpositional model18 are also not found at subtelomeric homology breakpoints. Second, the preserved centromere-telomere orientation and order of most duplicated blocks and degenerate telomeric repeats and the embedded patterns of shared blocks (Fig. 1) argue against a transpositional model.
Figure 2.
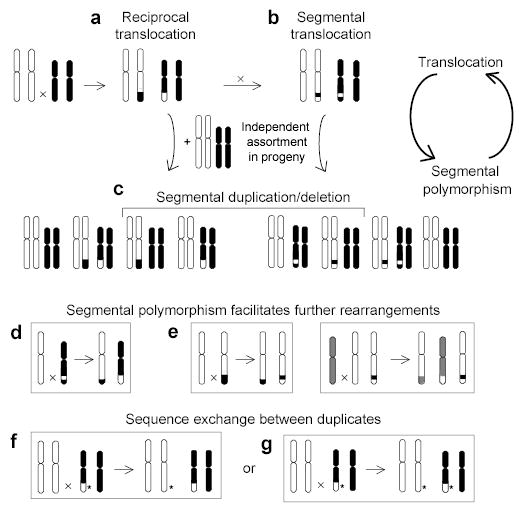
A translocation-based model of segmental duplication and polymorphism. (a) A terminal duplication/deletion can arise if a translocation product and an intact homolog are passed from parent to offspring, creating a segmental polymorphism in (c). (b) A segmental duplication/deletion can arise if a second inter-chromosomal exchange occurs between the translocated chromosomes. Segmental polymorphism can facilitate further rearrangements by (d) promoting translocations through inter-chromosomal homologies, or (e) causing translocation or other rearrangement due to the absence of homology. Both reciprocal and non-reciprocal homology-based sequence transfers (f and g) are possible between duplicates generated by any of the above steps. *, sequence variant.
Instead, the block patterns are consistent with patchwork formation by numerous translocations involving the tips of chromosomes and subsequent transmission of unbalanced chromosomal complements to offspring (Fig. 2). In this model, each translocation event has the potential to create a novel homology boundary and define a new block. Fig. 3 illustrates how two translocations led to the duplication of a subtelomeric segment (block 4 plus 5) and its juxtaposition between different neighbors on chromosomes 15q and 8p. The sequence of events can be inferred from the state of interspersed repeat elements at homology breakpoints. Chromosomes 15q and 16q represent ancestral states, and the intermediate state of 6p reveals temporal separation of the two translocations leading to the block configuration on 8p.
Figure 3.
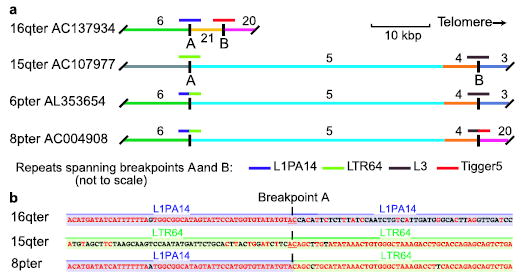
Layers of inter-chromosomal translocations form subtelomeric blocks. (a) Paralogous blocks have shared color and number; short colored lines above indicate different repetitive elements at homology breakpoints A and B, which define two translocations. An intact copy of each repeat is preserved in 16q and 15q sequences spanning the homology breakpoints with 6p and 8p, which contain truncated repeats fused by NHEJ. (b) Only two identical nucleotides (underlined) are found at the point where the original two sequences were joined at breakpoint A to form a hybrid. Aligned matching bases are red.
Translocations can result from aberrant repair by either non-homologous end-joining (NHEJ) or homologous recombination; both are major mechanisms of double-strand break (DSB) repair in mammalian cells19,20. To deduce the relative contribution of NHEJ and non-allelic homologous recombination (NAHR) to subtelomeric block juxtapositions, we examined all homology breakpoints at single-nucleotide resolution. The presence of repetitive elements of the same class (or paralogous genes) at the homology boundary in both aligned junction sequences, often with a transition from high to lower sequence identity within the repeat, is strongly suggestive of homology-based repair (e.g., Fig. S2, where the original state can be recognized by characteristic direct repeats flanking the Alu element). In contrast, the absence of aligned repeats or the presence of a truncated repetitive element (or gene) at the homology boundary in one sequence is indicative of NHEJ (as at breakpoints A and B, Fig. 3).
We identified a complete non-redundant set of 56 junction-sequence alignments, each representing a unique translocation event, in the sequenced subtelomeres. We deduced the repair mechanism in 53 of these cases (Figs. 4, S3; Table S5). The vast majority appears to result from NHEJ (49/53, 92%) (Fig. 4b). We infer repeat-mediated NAHR for only 4 (8%) of the events (Fig. 4a); three involved Alu repeats. In the 15 cases of NHEJ where structures representing both original partners and one translocation derivative are available, we found ≤5 bp of homology between the original sequences at the junction site (e.g., Fig. 3b). Small insertions found at eight junction sites are consistent with NHEJ-mediated translocations; eight cases of apparent large deletions could have formed either by translocation or intra-chromosomal deletion (Table S5, Fig. S4)
Figure 4.

Most subtelomeric homology breakpoints are consistent with NHEJ. For each mechanistic scenario, we diagram both original and derived forms, assuming reciprocal exchange. One derived form would be lacking in non-reciprocal cases. The third column gives a schematic example of each scenario identified in pairwise alignments of subtelomeric homology blocks. Fifty-three of the complete, non-redundant set of 56 homology breakpoints were assigned a mechanistic scenario (details in Table S5, Fig. S3). In some cases, two originals and one hybrid were available for comparison (e.g., NHEJ group 1). Other predicted states were not among surviving, sequenced alleles.
Although duplication borders of SDs were found in genome-wide analyses to be enriched in recently active Alu repeats21,22, interspersed repeats are not enriched at the DSBs leading to subtelomeric SDs. Of a total of 102 independent DSBs (Fig. 4), 45% occurred within a repetitive element (10.8% in Alu elements), close to the frequency expected from subtelomeric repeat content (Table S6). We do, however, find degenerate telomeric repeats at 4% of these DSBs, whereas they occupy 0.5% of subtelomeric sequence (Tables S5,S6). Subtelomeres are notably enriched in degenerate telomeric repeats relative to adjacent single-copy sequence or other genomic regions (~10- and ~100-fold, respectively) (Table S6). These repeats could have been appended during DSB repair, the postulated genesis of other interstitial telomere-like repeats23. Breakpoint 22 is a clear example of such a process. While we cannot rule out a functional role for these repeats, they are likely scars of many past DSB repairs.
Generation of diverse structures by a multiplicity of translocations between chromosome ends (Fig. 2a–c) is just one aspect of subtelomeric dynamics. Once duplicates exist on different chromosomes, they are subject to homology-based reciprocal or non-reciprocal sequence transfers (Fig. 2f,2g). These events do not generate novel block boundaries, but can supplant mutations accrued on one chromosome with those from another copy and spread structures formed by NHEJ to new locations (Fig. 2d). We reasoned that if duplication and subsequent homology-based sequence transfer are separated by sufficient time, the latter could be observed as a significant shift in sequence identity within regions of similarity.
To assay for such events, we evaluated fluctuations in sequence identity along a 60-kbp region, parts of which are shared by seven subtelomeres with 88%–99.5% identity (Fig. 5a). Four computational approaches indicate that homology-based sequence transfers occurred many times between these paralogs. (1) The best-matching pairs, i.e., partners in the most recent transfer events, change ≥5 times along the sequences (Figs. 5b, S5). (2) The phylogenetic relationships of neighboring sections are strikingly incongruent (Fig. 5c). (3) The percent identity between any two subtelomeres shifts significantly multiple times across their alignment (Figs. 5d–e). High similarity is unlikely to result from local selective pressure, because the most similar portions of different sequence pairs do not coincide. (4) Strong statistical support for multiple sequence transfers, ranging from several hundred to several thousand basepairs, is obtained using GeneConv24 (Fig. 5f, Table S7). Thus, subtelomeric blocks on different chromosomes do not evolve independently, but continued inter-chromosomal interactions obfuscate their duplication history. Transfers are also likely to be prevalent among the many subtelomeric blocks that are >98% identical, but only more subtle haplotype analyses might detect these events25.
Figure 5.
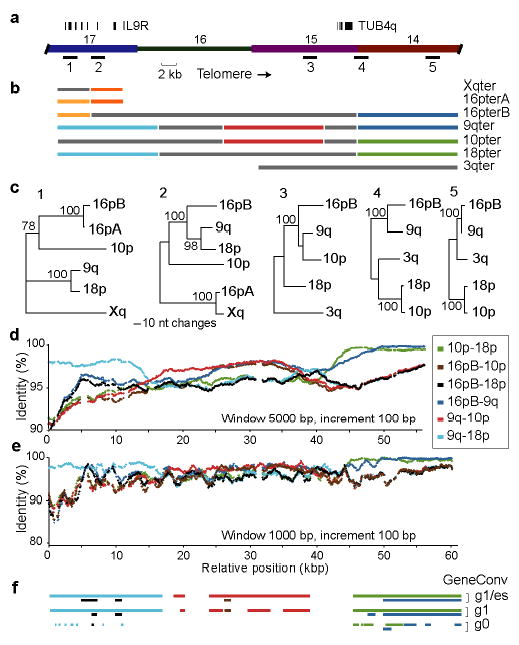
Homology-based sequence transfers between subtelomeres. (a) The region analyzed encompasses four numbered blocks, two multi-exon genes, and five sequences sampled for phylogenetic analyses. (b) Diagram of multiple sequence alignment with colors (excluding gray) indicating the best matching pairs with ≥98% identity in non-overlapping 5-kbp windows. (c) Neighbor-joining trees with bootstrap values (over 1000 replicates) constructed from 2-kbp samples of the alignments. (d) and (e) Plot of percent identity between four subtelomeres in 5-kbp and 1-kbp windows, respectively. Colors indicate alignments of different pairs. (f) The same colors indicate transferred segments found statistically significant by GeneConv with different stringency parameters.
Dynamics of primate subtelomeres
Recent changes in subtelomeric composition can be detected using fluorescence in situ hybridization (FISH) to determine the copy number and location of sequences in chromosomes of different primate species. Previous descriptions of subtelomeric dynamics using this approach were confounded by use of probes encompassing several blocks, each with different chromosomal distribution and evolutionary history5,26. To refine the analysis of structural changes in subtelomeres, we used four small FISH probes that each encompasses a single homology block. This approach reveals an unanticipated degree of recent genomic rearrangement in subtelomeres. Each block varies in copy number and chromosomal location between human individuals (Fig. 6), and FISH detects more chromosomal sites than are evident in the genome assembly or hybrid panel (Table S8). We detect content variation at 14 chromosomal ends using just four blocks on three individuals. Further analyses would undoubtedly uncover more variation.
Figure 6.
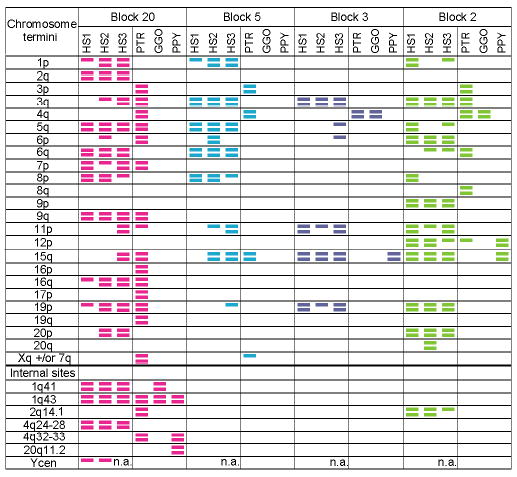
Chromosomal distribution of four subtelomeric blocks. FISH was conducted on three unrelated humans (HS1-3), chimpanzee (PTR), gorilla (GGO), and orangutan (PPY) (see Supplementary Methods). Colored bars indicate sites at which FISH signals were consistently observed on both homologs (two bars) or only one homolog (one bar). Colors correspond to Fig. 1. Chromosome locations are given according to the human karyotype. No signal was observed for block 5 in gorilla and orangutan; its presence was also not detected by PCR (Table S3).
Gross structural polymorphism of human subtelomeres is also evident in finished sequence of allelic pairs (Fig. 1). The two sequenced alleles of 16p are 99.8% identical in chromosome-specific DNA sequence, transition to much lower identity (~93%) within the adjoining block 17, and have no detectable homology in distal sequence (Fig. S6). The 19p alleles also differ grossly in subtelomeric content (Fig. 1). One of the structurally variant 4q alleles (4qA) is found in association with facioscapulohumeral dystrophy27. Other cases of gross allelic variation are revealed by PCR analyses of the hybrid panel (Fig. S1).
Subtelomeric dynamics are not confined to the human lineage. Blocks moved, and copies were lost and gained during primate evolution (Fig. 6). For example, block 20 is present at ≥9 subtelomeric locations in chimpanzee and human, whereas it occurs at only a few interstitial sites in gorilla and orangutan. The odorant receptor gene-containing block 5 was completely lost from the orangutan and gorilla genomes, yet is duplicated in chimpanzee and humans to four or more sites. The high similarity of sequenced human copies of these blocks (Fig.1, Table S2) and the fact that humans have more copies of three of the four blocks than other primates argue that the diversity of these particular block distributions arose primarily by recent duplications, rather than by loss of different subsets of ancestral copies. We estimate that 25 independent events, involving relocation or copy-number change of a total of ~1.2 Mbp, occurred on average to explain the observed differences between chimpanzee and human in the subtelomeric distribution of these blocks. The FISH analyses also suggest that chimpanzees, like humans, exhibit gross variation in subtelomeric content. Future, less anthropocentric analyses will likely reveal subtelomeric blocks that humans have lost, but that were retained and perhaps duplicated in other primates.
Timing and rates of subtelomeric transfers
The very recent nature of the inter-chromosomal events shaping subtelomeres is apparent from the high similarity of paralogous blocks on different chromosomes and from our cytogenetic analyses. For 28 of the 41 blocks, even the most dissimilar copies exceed 97% identity (Fig. 1, Table S2). Assuming a mutation rate of 10−3 substitutions per site per My28, all but the original copy of these 28 blocks must have formed by duplication or been impacted by homology-based sequence transfer in the last 15 My, i.e., during the divergence of humans and great apes.
When all pairs of human subtelomeric blocks are compared, the vast majority has 99%–99.9% identity (Fig. S7). Pairwise comparisons of all inter-chromosomal SDs in the genome peak at ~98%1, indicating that subtelomeric SDs result from more recent events than other SDs. Indeed, we find, after correcting for redundancy, that subtelomeres account for 40% of all duplications in the latest genome assembly3 with a match on another chromosome of ≥98.7% identity. Remarkably, ~1 Mbp (40%) of known subtelomeric terrain has a paralogous match of ≥99.5% identity, often rivaling the similarity of allelic copies.
We conservatively estimate that 49% (1.13 Mbp) of known subtelomeric sequence was generated after humans and chimpanzee diverged (Fig. S8). This amount equates to an observed rate of subtelomeric inter-chromosomal sequence duplication and/or transfer during the last 6.5 My of ~0.075 bases per site per My (Table S9) .We estimate from our cytogenetic analyses of the four subtelomeric blocks that each nucleotide had 0.09 chance per My of being relocated or changed in copy number during that time. The sequence- and cytogenetic-based estimation methods capture slightly different aspects of subtelomeric dynamics and underestimate the true rates of inter-chromosomal sequence transfer. Nevertheless, both estimates yield rates >60-fold that of point mutation28 or bases added by retrotransposon insertion29 in the same evolutionary period.
Given the amount of new subtelomeric sequence apparently created during the last 5 My (1.0 Mbp), we estimate that ~7 gene duplicates arose in human subtelomeres per My in recent times (Table S9). Even if half of these genes are deceptively young due to sequence transfers between pre-existing copies, the rate of gene duplication in subtelomeres (0.04 duplicates per gene per My) is 4-fold higher than the genome-wide average30. The rate of gene creation in subtelomeres is only matched by that in pericentromeric regions, which, like subtelomeres, are hotbeds of segmental duplications31.
Discussion
We demonstrate here that a multitude of mainly NHEJ-mediated translocations led to a complex patchwork of segmental duplications in human subtelomeres that exchange sequence at a remarkably high rate. The extraordinary recent dynamics of subtelomeres complicate the description of the human genomic landscape and its variation. Perhaps no chromosome-specific marker or block organization exists within subtelomeres, as they appear to evolve as a pool of variant allelic and paralogous structures. Moreover, inter-allelic subtelomeric recombination rates may be impossible to quantify due to the high frequency of inter-chromosomal transfers.
Why are subtelomeres so plastic? Copy-number deviations of subtelomeric DNA might be better tolerated than segmental aneuploidy of other genomic regions. Additionally, subtelomeres might be more susceptible to DSBs and/or more readily repaired through inter-chromosomal interactions than other regions32. Telomere clustering in meiotic cells33 might favor exchange of chromosome ends during DSB healing. Gross allelic differences likely make some subtelomeres prone to mispairing at meiosis, catalyzing further change.
Subtelomeric rearrangements might not be restricted to the germline, but could also arise in somatic cells during repair of DSBs or eroded telomeres. The resulting genotypic heterogeneity might affect fitness at the cellular and/or individual levels. Indeed, subtelomeres coalesce with telomeres in DNA-repair foci in naturally senescent cells34,35 and cells with artificially induced telomere dysfunction36. Furthermore, the high level of apparent sister-chromatid exchanges observed at chromosome ends (10−2/Mbp/generation)37 signals a high DSB rate and could subsume inter-chromosomal subtelomeric exchanges.
The results of ectopic repair of subtelomeres could be advantageous beyond healing of a damaged end. With their propensity to duplicate and exchange, subtelomeres could serve as a nursery for new genes and a place where haplotypes can diversify faster than in single-copy genomic regions. Sequence transfer between paralogous genes (as in Fig. 5) has the potential to create advantageous new combinations of sequence variants, aiding adaptive peak shifts38. Indeed, subtelomeric genes are associated with adaptive processes in other organisms39–41 (and citations in 5). Subtelomeric dynamics are a two-edged sword, however. Some DSB-repair events could result in loss or gain of dosage-sensitive genes in the most distal single-copy DNA or in contextual changes with adverse effects on gene regulation. The sequence analyses presented here contributes to a developing framework within which the role of subtelomeric dynamics in normal variation, adaptive change, and clinically manifest disorders can be explored.
The translocation-based model developed here to explain subtelomeric SDs could be broadly applicable to other inter-chromosomal SDs (Fig. 2). The first step in this model is a reciprocal translocation (Fig. 2a), which arises de novo in ~1/2000 concepti42. One in 500 healthy individuals carries a cytogenetically visible, balanced translocation43. A second inter-chromosomal exchange between the translocation derivatives (Fig. 2b) is likely to be selectively favored if it reduces the risk of passing a grossly imbalanced chromosomal complement to gametes. Duplicated segments, particularly when present on just one allele, can in turn promote translocations through NAHR (Fig. 2d). Furthermore, a DSB occurring in a hemizygous region stemming from an unbalanced translocation has increased probability of causing another translocation, inversion or intrachromosomal deletion, due to the absence of homologous template for its repair (Fig. 2e). Thus, segmental polymorphisms predispose to further rearrangements, which in turn lead to new segmentally polymorphic structures. This cycle of segmental polymorphism and gross genomic rearrangement is particularly obvious in subtelomeres and could underlie the structural variation44–46 and genomic disorders4 arising at many other locations in the human genome.
Methods
Additional results and methodological details, including the basis for all rate calculations, are provided as Supplementary Methods.
Sequence collation and analysis
Details of the iterative search for finished subtelomeric sequences are provided in Supplementary Methods. Sequences with continuous overlap of >99.8% nucleotide identity were merged into contigs (Table S1) and assumed to represent the same genomic region or an allelic variant. We used a combination of approaches, including PCR of a monochromosomal hybrid panel (Table S3), FISH (Table S10), and matches to half-YAC vector-insert junction sequences10 (Table S11), to establish or verify the chromosome location of contigs (Table S1). Regions of similarity were identified from pairwise sequence alignments made by BLAST247, without masking repeats. Blocks of paralogy were delineated when one or more contigs showed a break in homology except where paralogy adjoined a gap in available sequence. Block color/number are changed in Fig. 1 if similarity is lost on one or more subtelomeres, except when loss of homology occurs within 3 kbp of another breakpoint. However, all breakpoints were evaluated for mechanistic signatures (see below). Blocks from different chromosomal contigs were aligned using cross_match (http://www.phrap.org/) and MAVID48. Percent identities of block copies were calculated without insertions or deletions and with Jukes-Cantor correction for multiple substitutions. From 1438 alignments (26.8 Mbp total aligned sequence), a best matching partner was identified for each block in each chromosomal contig (Fig. S7). To remove redundancy, only one of the two alignments in cases of reciprocal best matches was included in estimation of the amount of recently generated sequence (see Supplementary Methods, Table S9). We also calculated the sum of non-overlapping inter-chromosomally duplicated bases with paralogous match ≥98.7% in subtelomeres or elsewhere in latest genome assembly (Build 35) as outlined in Supplementary Methods.
Subtelomeric block analysis by PCR and FISH
The subtelomeric content of 24 individual human chromosomes isolated in a hybrid panel was analyzed by PCR using 160 primer pairs (Table S3; Fig. S1). FISH was performed as detailed in Supplementary Methods using block-specific probes generated by long-range PCR (blocks 20, 5, and 2) or cosmid f75016 (block 3) on primary cultures of three unrelated Caucasians (2 males, one female) and cell lines of male chimpanzee, orangutan, or gorilla. The assumptions employed to conservatively estimate the rate with which these blocks changed copy-number or location since human and chimpanzee diverged are given in Supplementary Methods. Note that this rate excludes homology-based sequence transfers among pre-existing copies, whereas the sequence-based estimate includes duplications and homology-based sequence transfers, but not changes in segment location.
Breakpoint analyses
We identified homology breakpoints from all pairwise subtelomeric sequence alignments and evaluated a nonredundant set for mechanistic signatures as described in Supplementary Methods (Table S5). All remaining block junctions in Fig. 1 are nearly identical replicas of members of this junction set due to their duplication within larger segments. The number of independent DSBs was counted as two for each deduced NHEJ event, one for each NAHR event in the non-redundant set. We queried the human genome by BLAT with the 200 bp each NHEJ breakpoint lacking a gene or known repeat and found no novel repeats.
Detection of homology-based transfer
Changes in percent identity along pairwise sequence alignments were determined using the percentIDplot program (E.W. and E.L, unpublished). The best-matching pair in each 5-kbp and 2-kbp window in each sequence was identified from a multiple sequence alignment generated using MAVID48 (Fig. S5). Phylogenetic trees were constructed using PAUP49.
Supplementary Material
Acknowledgments
We are grateful to the many contributors to the Human Genome Project who generated the sequences that made this study possible and the Eichler and Haussler groups for making data on segmental duplications readily accessible. Our work was supported by NIH grant GM57070. We thank Evan Eichler, Harmit Malik, Dan Gottschling, Jennifer Gogarten, Katie Rudd, and Mike Schlador for comments on the manuscript and Joe Felsenstein for advice on phylogenetic analyses.
Footnotes
Supplementary Information accompanies the paper on Nature’s website on www.nature.com/nature.
Competing interest statement. The authors declare that they have no competing financial interests.
References
- 1.Samonte RV, Eichler EE. Segmental duplications and the evolution of the primate genome. Nat Rev Genet. 2002;3:65–72. doi: 10.1038/nrg705. [DOI] [PubMed] [Google Scholar]
- 2.Bailey JA, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–7. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 3.Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–17. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw CJ, Lupski JR. Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet. 2004;13(Spec No 1):R57–64. doi: 10.1093/hmg/ddh073. [DOI] [PubMed] [Google Scholar]
- 5.Mefford H, Trask BJ. The complex structure and dynamic evolution of human subtelomeres. Nature Reviews Genetics. 2002;3:91–102. doi: 10.1038/nrg727. [DOI] [PubMed] [Google Scholar]
- 6.Trask BJ, et al. Members of the olfactory receptor gene family are contained in large blocks of DNA duplicated polymorphically near the ends of human chromosomes. Hum Mol Genet. 1998;7:13–26. doi: 10.1093/hmg/7.1.13. [DOI] [PubMed] [Google Scholar]
- 7.Monfouilloux S, et al. Recent human-specific spreading of a subtelomeric domain. Genomics. 1998;51:165–176. doi: 10.1006/geno.1998.5358. [DOI] [PubMed] [Google Scholar]
- 8.Martin CL, et al. The evolutionary origin of human subtelomeric homologies---or where the ends begin. Am J Hum Genet. 2002;70:972–84. doi: 10.1086/339768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan Y, Linardopoulou E, Friedman C, Williams EM, Trask BJ. Genomic structure and evolution of the ancestral chromosome fusion site in 2q13–2q14.1. Genome Res. 2002;12:1651–1662. doi: 10.1101/gr.337602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riethman HC, et al. Integration of telomere sequences with the draft human genome sequence. Nature. 2001;409:948–51. doi: 10.1038/35057180. [DOI] [PubMed] [Google Scholar]
- 11.Linardopoulou E, et al. Transcriptional activity of multiple copies of a subtelomerically located olfactory receptor gene that is polymorphic in number and location. Hum Mol Genet. 2001;10:2373–83. doi: 10.1093/hmg/10.21.2373. [DOI] [PubMed] [Google Scholar]
- 12.Knight SJ, Flint J. The use of subtelomeric probes to study mental retardation. Methods Cell Biol. 2004;75:799–831. doi: 10.1016/s0091-679x(04)75035-9. [DOI] [PubMed] [Google Scholar]
- 13.Brown WR, et al. Structure and polymorphism of human telomere-associated DNA. Cell. 1990;63:119–132. doi: 10.1016/0092-8674(90)90293-n. [DOI] [PubMed] [Google Scholar]
- 14.de Lange T, et al. Structure and variability of human chromosome ends. Mol Cell Biol. 1990;10:518–527. doi: 10.1128/mcb.10.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flint J, et al. Sequence comparison of human and yeast telomeres identifies structurally distinct subtelomeric domains. Hum Mol Genet. 1997;6:1305–1313. doi: 10.1093/hmg/6.8.1305. [DOI] [PubMed] [Google Scholar]
- 16.Riethman H, et al. Mapping and initial analysis of human subtelomeric sequence assemblies. Genome Res. 2004;14:18–28. doi: 10.1101/gr.1245004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smit AF, Riggs AD. Tiggers and DNA transposon fossils in the human genome. Proc Natl Acad Sci U S A. 1996;93:1443–1448. doi: 10.1073/pnas.93.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eichler EE, Archidiacono N, Rocchi M. CAGGG repeats and the pericentromeric duplication of the hominoid genome. Genome Res. 1999;9:1048–58. doi: 10.1101/gr.9.11.1048. [DOI] [PubMed] [Google Scholar]
- 19.Pfeiffer P, Goedecke W, Obe G. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis. 2000;15:289–302. doi: 10.1093/mutage/15.4.289. [DOI] [PubMed] [Google Scholar]
- 20.Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23:5706–15. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bailey JA, Liu G, Eichler EE. An Alu transposition model for the origin and expansion of human segmental duplications. Am J Hum Genet. 2003;73:823–34. doi: 10.1086/378594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Y, Mishra B. Quantifying the mechanisms for segmental duplications in mammalian genomes by statistical analysis and modeling. Proc Natl Acad Sci U S A. 2005;102:4151–6. doi: 10.1073/pnas.0407957102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nergadze SG, Rocchi M, Azzalin CM, Mondello C, Giulotto E. Insertion of telomeric repeats at intrachromosomal break sites during primate evolution. Genome Res. 2004;14:1704–10. doi: 10.1101/gr.2778904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawyer S. Statistical tests for detecting gene conversion. Mol Biol Evol. 1989;6:526–38. doi: 10.1093/oxfordjournals.molbev.a040567. [DOI] [PubMed] [Google Scholar]
- 25.Mefford HC, Linardopoulou E, Coil D, van den Engh G, Trask BJ. Comparative sequencing of a multicopy subtelomeric region containing olfactory receptor genes reveals multiple interactions between non-homologous chromosomes. Hum Mol Genet. 2001;10:2363–2372. doi: 10.1093/hmg/10.21.2363. [DOI] [PubMed] [Google Scholar]
- 26.Der-Sarkissian H, Vergnaud G, Borde YM, Thomas G, Londono-Vallejo JA. Segmental polymorphisms in the proterminal regions of a subset of human chromosomes. Genome Res. 2002;12:1673–8. doi: 10.1101/gr.322802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemmers RJ, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet. 2002;32:235–6. doi: 10.1038/ng999. [DOI] [PubMed] [Google Scholar]
- 28.Chen FC, Li WH. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet. 2001;68:444–56. doi: 10.1086/318206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu G, et al. Analysis of primate genomic variation reveals a repeat-driven expansion of the human genome. Genome Res. 2003;13:358–68. doi: 10.1101/gr.923303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–5. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 31.She X, et al. The structure and evolution of centromeric transition regions within the human genome. Nature. 2004;430:857–64. doi: 10.1038/nature02806. [DOI] [PubMed] [Google Scholar]
- 32.Ricchetti M, Dujon B, Fairhead C. Distance from the chromosome end determines the efficiency of double strand break repair in subtelomeres of haploid yeast. J Mol Biol. 2003;328:847–62. doi: 10.1016/s0022-2836(03)00315-2. [DOI] [PubMed] [Google Scholar]
- 33.Bass HW. Telomere dynamics unique to meiotic prophase: formation and significance of the bouquet. Cell Mol Life Sci. 2003;60:2319–24. doi: 10.1007/s00018-003-3312-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.d’Adda di Fagagna F, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 35.Zou Y, Sfeir A, Gryaznov SM, Shay JW, Wright WE. Does a sentinel or a subset of short telomeres determine replicative senescence? Mol Biol Cell. 2004;15:3709–18. doi: 10.1091/mbc.E04-03-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–56. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 37.Cornforth MN, Eberle RL. Termini of human chromosomes display elevated rates of mitotic recombination. Mutagenesis. 2001;16:85–9. doi: 10.1093/mutage/16.1.85. [DOI] [PubMed] [Google Scholar]
- 38.Hansen TF, Carter AJ, Chiu CH. Gene conversion may aid adaptive peak shifts. J Theor Biol. 2000;207:495–511. doi: 10.1006/jtbi.2000.2189. [DOI] [PubMed] [Google Scholar]
- 39.Halme A, Bumgarner S, Styles C, Fink GR. Genetic and epigenetic regulation of the FLO gene family generates cell-surface variation in yeast. Cell. 2004;116:405–15. doi: 10.1016/s0092-8674(04)00118-7. [DOI] [PubMed] [Google Scholar]
- 40.Fabre E, et al. Comparative genomics in hemiascomycete yeasts: evolution of sex, silencing, and subtelomeres. Mol Biol Evol. 2005;22:856–73. doi: 10.1093/molbev/msi070. [DOI] [PubMed] [Google Scholar]
- 41.De Las Penas A, et al. Virulence-related surface glycoproteins in the yeast pathogen Candida glabrata are encoded in subtelomeric clusters and subject to RAP1- and SIR-dependent transcriptional silencing. Genes Dev. 2003;17:2245–58. doi: 10.1101/gad.1121003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet. 1991;49:995–1013. [PMC free article] [PubMed] [Google Scholar]
- 43.Genetics & Public Policy Center. Genetics Information: Translocations. http://www.dnapolicy.org/genetics/translocations.jhtml (2004).
- 44.Wong Z, Royle NJ, Jeffreys AJ. A novel human DNA polymorphism resulting from transfer of DNA from chromosome 6 to chromosome 16. Genomics. 1990;7:222–34. doi: 10.1016/0888-7543(90)90544-5. [DOI] [PubMed] [Google Scholar]
- 45.Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–8. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 46.Iafrate, A. J. et al. Detection of large-scale variation in the human genome. Nat Genet (2004). [DOI] [PubMed]
- 47.Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999;174:247–50. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- 48.Bray N, Pachter L. MAVID multiple alignment server. Nucleic Acids Res. 2003;31:3525–6. doi: 10.1093/nar/gkg623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swofford, D. L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). (Sinauer Associates, Sunderland, MA, 2000).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.