

Abstract
Mucopolysaccharidosis I (MPS I, α-l-iduronidase deficiency disease) is a heritable lysosomal storage disorder involving multiple organs, including the heart. Malfunction of the heart is also a major manifestation in the mouse model of MPS I, progressing in severity from 6 to 10 months (of a one-year life span). In comparisons of MPS I with wild type mice, the heart was found enlarged, with thickened septal and posterior walls, primarily because of infiltration of the muscle by storage-laden cells. Heart valves were enlarged and misshapen, and contained large numbers of highly vacuolated interstitial cells. The thickened aortic wall contained vacuolated smooth muscle cells and interrupted elastic fibers. Hemodynamic measurements and echocardiography revealed reduced left ventricular function as well as mitral and aortic regurgitation. But despite these abnormalities, free-roaming MPS I mice implanted with radio telemetry devices showed surprisingly normal heart rate and blood pressure, though their electrocardiograms were abnormal. An incidental finding of the telemetry studies was a disturbed circadian rhythm in the MPS I mice. Restoration of enzyme activity in the heart of one mouse, by transplantation of retrovirally modified bone marrow, resulted in normalization of left ventricular function as well as loss of storage vacuoles in myocytes and endothelial cells, though not in valvular interstitial cells. This study demonstrates the usefulness of the mouse model for in-depth studies of the cardiovascular component of MPS I.
Keywords: Mucopolysaccharidosis, Hurler, α-l-iduronidase, lysosomal storage, heart, echocardiography, electrocardiography, ventricular function, circadian rhythm
Introduction
Mucopolysaccharidosis I (MPS I) is an autosomal recessive disorder caused by mutation of the IDUA gene; the resulting deficiency of the lysosomal enzyme α-l-iduronidase causes accumulation of its substrates, dermatan sulfate and heparan sulfate [1]. The disorder is clinically heterogeneous, the clinical spectrum ranging from the very severe Hurler syndrome to the attenuated Scheie syndrome, with a diverse group of intermediate severity known as Hurler/Scheie (OMIM # 67014, 67016 and 67015, respectively). Heart disease, corneal clouding, organomegaly, skeletal malformations and joint stiffness are present in varying degrees in all forms of MPS I, while significant mental retardation is present in the severe form only. Life span is generally limited to childhood in the severe form and early adulthood in the intermediate form, but can be normal in the most attenuated form. Molecular heterogeneity is the probable cause of the clinical variability, with a combination of mutant alleles determining the clinical phenotype. Eighty nine mutations of the IDUA gene are listed in the Human Gene Mutation Data Base [2], only a few of which are common.
Cardiovascular disease is a prominent feature of Hurler syndrome and had been noted in early descriptions of the disease (reviewed in [3]). Myocardial thickening, occlusion of coronary arteries, and valvular disease are common in all forms of MPS I [4–7]. Systemic or pulmonary hypertension occurs in some MPS I patients [4,8–11]. Congestive heart failure is a frequent cause of death in the severe form of MPS I [9], while valve replacement is often necessary in the attenuated forms [1].
Animal models of MPS I are being used to study pathogenesis and develop therapy. These include the naturally occurring MPS I dog [12] and cat [13] models, and two very similar knockout mouse models [14–16]. We have used the mouse model developed in our laboratory [16] to study cardiac malfunction; a preliminary account of this work has been presented in abstract form [17].
Materials and methods
Mice
The mouse model of MPS I was developed by disruption of the Idua gene, followed by repeated backcrossing to place the mutant Idua gene on a C57BL/6 background [16]. The mutant mice live for about one year. Studies of cardiovascular pathology and function were conducted at 6 and 10 months of age, unless stated otherwise, in order to provide intermediate and near-terminal evaluations. All surgical and animal care procedures conformed to USPHS guidelines in protocols approved by the UCLA Office for Protection of Research Subjects.
Biochemical and morphologic studies
Assays for α-l-iduronidase and for soluble glycosaminoglycans were performed as described in [18]. For gene therapy, bone marrow of male mice was transduced with the MND vectors previously described (with inserts of human IDUA cDNA or of eGFP for treated and sham-treated mice respectively) prior to transplantation into female recipient mice as described [18] except for irradiation with 137Cs rather than 60Co. The mice were transplanted at 6–8 weeks of age and studied 8 months later. Tissue was collected for morphologic studies after euthanasia, fixed in 4% formaldehyde for embedding in paraffin, and in 2% glutaraldehyde for embedding in Spurr resin for electron microscopy (30 nm ultrathin sections) or light microscopy (1.5 μm semi-thin sections).
Ultrasound Echocardiography
Mice were sedated with Avertin (2,2,2, tribromoethanol, Sigma-Aldrich Chemical Corp., St. Louis, MO) administered i.p. at 0.25 mg/g,, and imaged ultrasonically with an Acuson Sequoia C256 using 15L8 probe (Siemens Medical Solutions Inc, Mountain View, CA). Long-axis, short-axis, and apical views were used to obtain two-dimensional, M-mode, and spectral Doppler images of the heart and aorta [19]. Measurements of chamber dimensions and function were made using the Siemens and AccessPoint software (FreelandSystems.com). Mouse electrocardiograms (ECG) were simultaneously obtained in the standard lead II arrangement with subcutaneous Pt electrodes (Grass Instruments, Warwick, RI).
Hemodynamic Assessment
Mice were anesthetized with a mixture of Ketamine (115 :g/gm body mass), Xylazine (29 :g/gm) and Buprenorphine (3.5 :g/gm). To obtain left ventricular (LV) hemodynamic data, the right carotid artery was cannulated with a 1.4Fr catheter (Millar Instruments, Houston, TX) advanced into the left ventricle. Hemodynamic data were recorded continuously for at least 30 minutes and analyzed with HEM V3.3 software (Notocord Systems, Croissy sur Seine, France).
Implantation of Radio Telemetry Transducers for Ambulatory Monitoring
The ECG, body temperature, arterial pressure and bio-activity were measured in awake, freely moving mice by implanting radio telemetry devices (TA10ETA-F20 or TA11PA-C20, Data Sciences Intl., St Paul, MN) under halothane (1.5–2.0%) anesthesia (Marro et al, 2000; Mills et al, 2000)[20,21]. Ten seconds of data were collected every 5 minutes for the duration of the study which ranged from 10 to 32 days in the six mice examined. These data were analyzed with the telemetry software (Data Sciences Inc., St. Paul, MN) to determine pressures, temperatures, heart rates, ECG wave heights, and other temporal parameters during both the light (inactive) and dark (active) circadian cycles. The coefficient of variance (CV, %) was determined from over 2000 R-R intervals in both the light and dark cycles as described [22]. Raw ECG waveforms from all mice were individually scanned for arrhythmias by two independent observers.
Statistical Methods
Data are shown as means and standard errors. Statistical significance between groups was determined by using two-tailed t-tests and ANOVA with Instat V3.05 statistical software (GraphPad Inc, San Diego, CA).
Results
Glycosaminoglycan storage
The heart and aorta of the MPS I mice (Idua −/−) were devoid of α-l-iduronidase activity, and as a consequence glycosaminoglycan was stored in those tissues (Fig. 1). The pool of soluble glycosaminoglycan, presumed to be the lysosomal pool, was found to be 3.2 ± 0.2 μg/mg dry weight for heart and 7.1 ± 1.2 μg/mg dry weight for aorta of the MPS I mice, in contrast to barely detectable pools in the corresponding WT tissues.
Fig. 1.
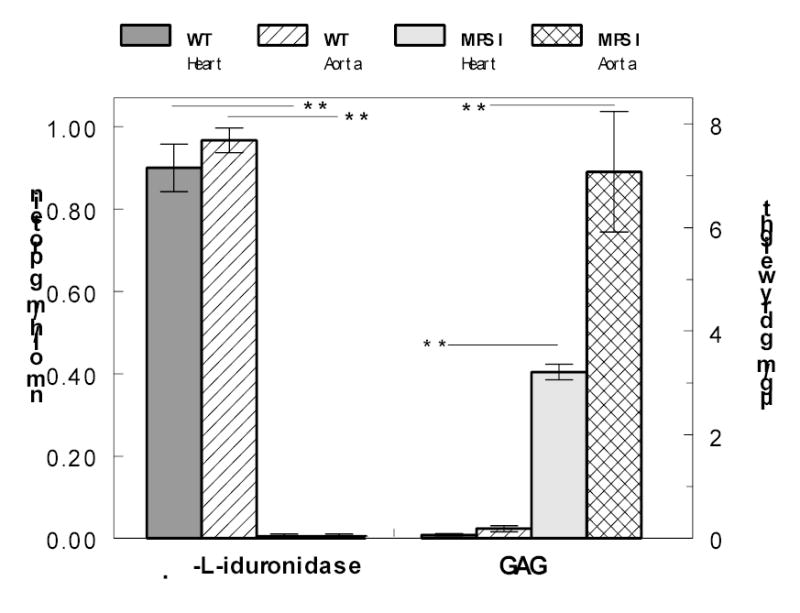
Activity of α-l-iduronidase and accumulation of soluble GAG in heart muscle and aorta of WT and MPS I mice. Bars indicate means ± S.E.M; n = 3 for α-l-iduronidase determination, and 8–10 for determination of GAG. Heparan sulfate was used as standard for GAG determinations.
Morphologic changes
Examination of the heart by light microscopy showed the morphologic consequences of α-l-iduronidase deficiency and GAG storage. Sample sections are presented in Fig. 2, showing the contrast between different structures of the heart of the WT mouse (Fig. 2A, 2C, 2E and 2G) and the corresponding areas of the heart of the MPS I mouse (Fig. 2B, 2D, 2F and 2H). In the myocardium of the MPS I mouse, the interstitial space between myocardial cells was distended by aggregates of vacuolated interstitial cells and deposits of collagen (Fig. 2B). The endocardium was likewise abnormal (Fig. 2D); endothelial cells were swollen, subjacent myocardial fibers were discontinuous because of aggregates of vacuolated cells between them, and perinuclear vacuoles were present in many myocytes. The aortic valve was grossly enlarged and misshapen (Fig. 2F), as were the mitral and tricuspid valves (not shown). The aortic wall contained highly vacuolated smooth muscle cells (Fig. 2F and 2H). The elastic fibers, which are continuous in the aorta of the normal mouse (Fig. 2G) showed splitting and interruptions in the aorta of the MPS I mouse (Fig. 2H).
Fig. 2.

Morphologic comparison of the heart and aorta and Doppler echocardiograms of the MPS I and WT mice. The WT mouse is represented in the left panels and the MPS I mouse in the right. A and B: left ventricular myocardium. Note the collagen deposits (blue) and aggregates of interstitial cells (arrow) in the myocardium of the mutant mouse (Masson Trichrome stain). C and D: left ventricular endocardium. Black arrows point to the myocardium, clear arrows to the endocardium, while the blue arrow in Panel D points to subendocardial vacuolated cells (H&E stain). E and F: aortic valve and aortic wall. Note the markedly thickened valve leaflet in the mutant mouse, as well as the vacuolation and distention of smooth muscle fibers of the medial layer of the aortic wall. The Masson Trichrome stains collagen light blue and elastic fibers dark blue. G and H: higher magnification of a section of the aortic wall shown above. Arrows in panel H show discontinuity in the elastic fibers. I and J. 2D Doppler echo images of the left ventricle (LV) during diastole. The green arrowheads point to the aortic outflow tract and the white arrows to the aortic valve. Note the turbulence, indicating regurgitation, and enlarged left ventricular outflow tract in the MPS I mouse. The red to yellow colors on the calibration bar indicate blood flow toward, and the blue colors away from, the ultrasonic transducer.
Electron microscopy of the myocardium of the MPS I mouse showed numerous empty vacuoles within myocytes (Fig. 3A). The vacuoles varied in size and were lined up along the mitochondria. The structure of sarcomeres appeared normal; Z-disks were well-defined and filaments aligned as in WT mice. Intercalated disks also appeared normal (not shown). In the mitral valve, endothelial cells were vacuolated to a moderate degree (Fig. 3B), whereas interstitial cells were very heavily vacuolated (Fig. 3C).
Fig. 3.
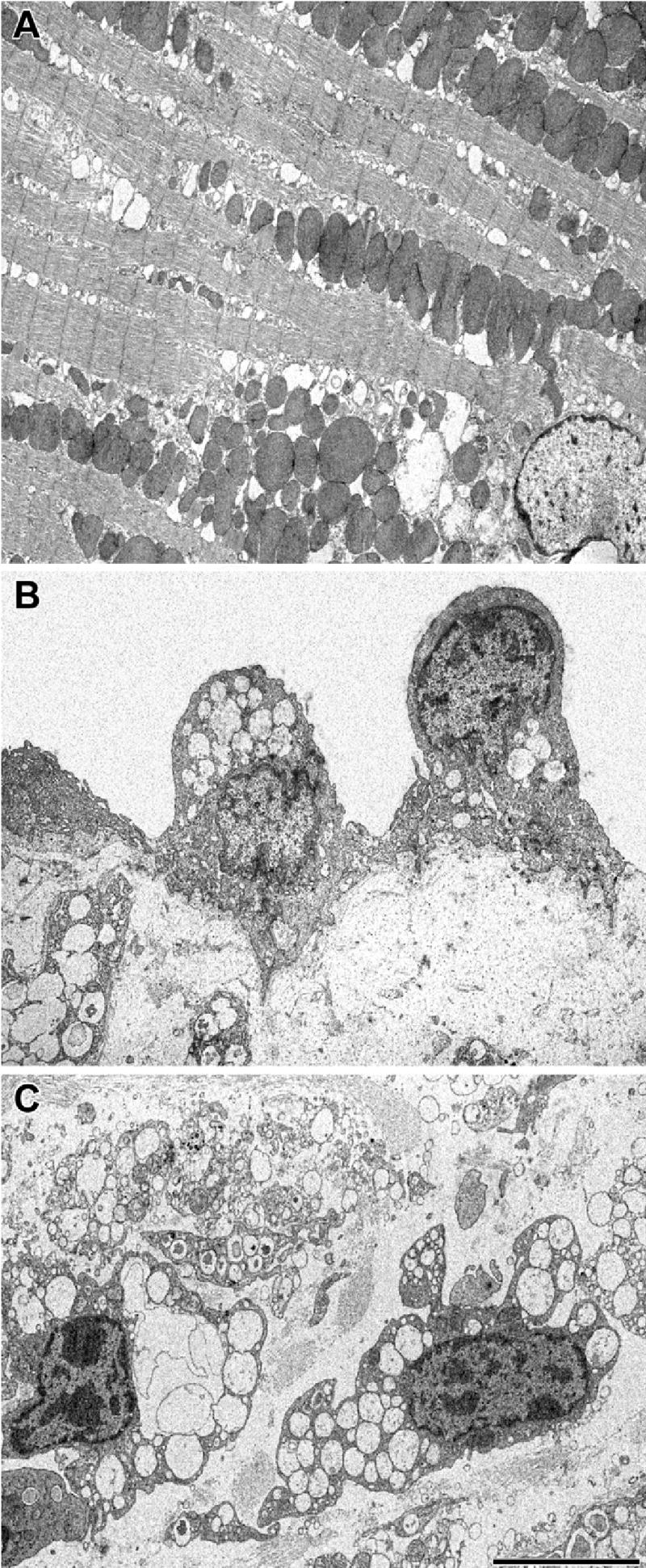
Electron microscopic appearance of cells in the MPS I heart. A: myocyte in the apex of the heart. B: endothelial cell on the surface of the mitral valve. C: interstitial cell in the mitral valve. The scale bar, which applies to all three panels, is 5 μm.
Cardiac enlargement
The MPS I mice had enlarged hearts, as shown post-mortem by morphometry and in vivo by echocardiography of left ventricular chamber dimensions. The heart weight relative to either body weight or to tibia length was significantly larger in the MPS I mice than in the WT controls (Table 1). Only female mice were used in these measurements because of the propensity of normal male C57BL/6 mice to become obese. The enlargement was more marked at 10 months than at 6 months of age. Echocardiography showed that both the septal and posterior walls were significantly thicker in the 10 months-old MPS I mice than in wild type controls, without enlargement of the inner chamber diameters (Table 2). The left ventricular outflow tracts (LVOT) of 10-months-old MPS I mice were significantly enlarged relative to the controls (Table 2).
Table 1.
Enlargement of the heart in MPS I mice
| Genotype | WT | MPS I | ||
|---|---|---|---|---|
| Age |
6 months |
10 months |
6 months |
10 months |
| Number measured | 6 | 6 | 12 | 10 |
| Body Weight (g) | 30.2±1.1 | 29.4±1.8 | 27.4±0.4* | 28.0±0.5 |
| Heart Weight (mg) | 115±3 | 126±6 | 138±9 | 168±11* |
| Tibia Length (mm) | 18.2±0.1 | 17.8±0.3 | 18.0±0.1 | 18.1±0.1 |
| HW/BW (mg/g) | 3.84±0.16 | 4.32±0.17 | 5.03±0.26** | 5.95±0.33** ¶ |
| HW/TL (mg/mm) | 6.35±0.22 | 7.07±0.29 | 7.68±0.41* | 9.27±0.60*¶ |
HW/BW = heart weight to body weight ratio; HW/TL = heart weight to tibia length ratio
= P<0.05 to WT at same age
= P<0.001 to WT at same age
= P<0.05 between age groups
Table 2.
Echocardiographic evaluation of MPS I mice
| Genotype | WT | MPS I | ||
|---|---|---|---|---|
| Age |
6 months |
10 months |
6 months |
10 months |
| Number measured | 10 | 13 | 11 | 12 |
| VST (mm) | 0.68±0.02 | 0.64±0.02 | 0.80±0.05* | 0.83±0.04** |
| PWT (mm) | 0.67±0.01 | 0.66±0.02 | 0.77±0.06 | 0.75±0.03* |
| EDD (mm) | 3.84±0.08 | 4.13±0.11 | 4.06±0.13 | 3.92±0.13 |
| ESD (mm) | 2.73±0.06 | 2.88±0.08 | 2.97±0.11 | 2.97±0.15 |
| LVOT (mm) | - | 1.57±0.02 | - | 2.18±0.06** |
VST = Ventricular Septal Thickness; PWT = Posterior Wall Thickness; EDD = End Diastolic Dimension; ESD = End Systolic Dimension; LVOT = Left Ventricular Outflow Tract diameter;
= P<0.05 to WT at same age;
= P<0.001 to WT at same age
Cardiac dysfunction
The MPS I mice exhibited ventricular dysfunction via echocardiography as early as 6 months of age. By 10 months, both Vcf (an index of contractile function) and ejection fraction were significantly reduced (Fig. 4A). Catheterization of the left ventricle in anesthetized mice showed significant reduction in +dP/dT (an index of contractility) and –dP/dT (an index of relaxation) at both ages studied (Fig. 4B), confirming the dysfunction seen by echocardiography.
Fig. 4.

Evaluation of ventricular and valvular dysfunction in MPS I mice. A. The velocity of circumferential fiber shortening (Vcf) and ejection fraction (EF) from echocardiography are plotted in WT mice (n = 10, 13) and MPS I mice (n = 11, 12) at 6 and 10 months of age. B. Contractility (dP/dT) and relaxation (-dP/dT) from catheterizations are plotted from similar groups of WT (n = 6, 6) and MPS I mice (n = 12, 10). C. Doppler traces across the mitral valve show abnormally long back-flow profiles following valve closure (arrows) indicative of regurgitation in MPS I mice. D. The blood flow velocity (Vmax) and peak pressure gradient (PPG) across the aortic valve are plotted in WT mice (n = 10, 13) and MPS I mice (n = 11, 12) at 6 and 10 months of age. For all plots, * = P < 0.05 and ** = P < 0.001.
Valvular dysfunction is MPS I mice is seen in Doppler echocardiography images as both mitral and aortic regurgitation. Mitral valve regurgitation in MPS I mice is indicated by the large prolonged back-flow of blood (upward direction at arrows) after the valve closes (Fig. 4C). The 2-D color Doppler image shows a large amount of aortic regurgitation during diastole along with a dilated left ventricular outflow tract in the MPS I mice (Fig. 2 J). The compromised valve function in the mutant mice resulted in significantly longer ejection times (65±2ms MPS I; 56±2ms WT; P<0.001), a reduction in maximum blood flow velocity (Vmax) and in peak pressure gradient (PPG) across the aortic valve (Fig. 4D).
Cardiac and circadian parameters in ambulatory mice
Despite the marked decrease in ventricular function and the valvular regurgitation, free-roaming MPS I mice implanted with radio telemetry devices exhibited surprisingly normal heart rates and blood pressures as compared to WT controls at 10 months of age (Table 3). As expected, the systolic and diastolic blood pressures were greater during the active (dark cycle) period in both MPS I and WT mice. But within a light cycle, MPS I mice exhibited a modest, but statistically significant 6–13 mm Hg increase in systolic blood pressure over WT mice. Despite similar heart rates, the heart rate variability (CV %) as determined from the variance in the ECG R-R interval was significantly reduced in the MPS I mice at all times (Table 3).
Table 3.
Telemetric evaluation of MPS I mice
| Genotype | WT | MPS I | ||
|---|---|---|---|---|
|
Light Cycle |
Light |
Dark |
Light |
Dark |
| Systolic Blood Pressure | 113±1 | 122±1¶ | 117±1** | 132±1**¶ |
| Diastolic Blood Pressure | 84±1 | 90±1¶ | 92±1** | 103±1**¶ |
| Heart Rate (bpm) | 652±2 | 676±1 ¶ | 641±2 | 667±3 ¶ |
| CV (%) | 3.23±0.49 | 3.31±0.34 | 1.81±0.19 * | 1.96±0.18 * |
| Cage Activity (arb. units) | 4.91±0.23 | 12.96±0.32 ¶ | 8.07±0.25** | 13.62±0.35 ¶ |
| Body Temperature (ºC) | 36.94±0.02 | 37.76±0.02 ¶ | 35.85±0.03** | 37.12±0.03* ¶ |
| ECG S/R amplitude ratio | 0.53 | 0.53 | 0.35 | 0.34 |
| P wave width (ms) | 9.27±0.05 | 9.37±0.07 | 12.10±.10** | 11.94±0.09** |
| QRS-Tri width (ms) | 21.68±0.35 | 19.76±0.39 ¶ | 30.60±0.25** | 29.84±0.23** |
CV (%) = coefficient of variance of the R-R interval of the ECG. The pressure data is determined from 125 independent measures from one mouse of each genotype. The ECG, temperature and activity data are determined from 25 independent measures from 1 WT and 3 MPS I mice.
= P<0.05 to WT in same light cycle
= P<0.001 to WT in same light cycle
= P<0.05 between light-dark cycles within genotype
Figure 5A plots a 24 hour period of heart rates, body temperatures and cage activity averaged from data taken at the same time of day over 20 days from un-anaesthetized MPS I and WT mice. The circadian (diurnal) cycle is clearly defined in all three parameters of the WT mice with the active (dark) period showing higher activity, body temperature and heart rates (Table 3). Though all these values were within the physiological range, the MPS I mice also had significantly lower body temperatures at all times and higher activity during the light (sleep) cycle than the WT mice (Table 3). Additionally, Figure 5A reveals that the cardiac parameters in MPS I mice fluctuated more and did not maintain as well defined a circadian cycle as in WT mice.
Fig. 5.

Telemetry recordings comparing heart rate, the ECG, body temperature and cage activity from WT and MPS I mice. A. Plots of the heart rate from the ECG (upper traces), body temperature (middle traces) and cage activity (lower traces) from WT and MPS I mice. These data are averaged from raw data taken at the same time of day over a 20 day period and re-plotted over a 24 hour day. The ordinate scales are identical in both plots with the heart rates from 600–720 bpm, temperature from 35.5 to 38ºC and activity from 0–24 arbitrary counts/min. The horizontal lines in the plots represent the dark (active) portion of light cycle for the mice. B. Telemetric ECG recordings of a 0.3 second duration from WT and MPS I mice. The unfiltered waveforms are labeled in the WT panel. The horizontal bar in each panel designates the duration of the QRS-Tri waves. The arrows in the MPS I panel indicate the wide P wave doublet.
MPS I mice had abnormal electrocardiograms (ECG). Fig. 5B shows unfiltered waveforms from WT and MPS I mice, illustrating all the typical waves for mice (P, Q, R, S Tri and negative going T [23]. Though the PR and QT intervals were similar and within the normal range, all three MPS I mice exhibited altered wave amplitudes and widths (duration). The S wave amplitudes were diminished while the Tri wave amplitudes were larger. The S wave amplitude (normalized to the R wave) was diminished by 1/3 in both the light and dark cycles (Table 3). The P and QRS-Tri wave widths were both prolonged and sometimes exhibited multiple peaks (Figure 5B & Table 3). Only 23 arrhythmias (pre-ventricular contractions) were noted from the 4 mice over their 131 days of cumulative recordings; there were no differences between the WT and MPS I mice in their rate of arrhythmias.
Gene therapy by retrovirally modified bone marrow
Previous work [18] had shown that transplantation of 6–8 week-old MPS I mice with bone marrow transduced with a retroviral vector bearing the human IDUA cDNA provided high levels of α-l-iduronidase to many organs, reduced the storage of GAG and normalized cellular morphology. The heart was not studied at that time. In the current study, we repeated the transplantation of genetically modified bone marrow, but of 10 mice so treated, only one showed substantial enzyme activity in the heart (0.65 nmol/h/mg, about 2/3 of the WT mean shown in Fig. 1) when sacrificed 8 months later; five others had a trace of activity in the liver and no activity in the heart, and the remainder had no activity in either tissue. The low success rate in this experiment, compared to the previous series [18], is attributed to loss of vector from the transducing cells, which had been amplified but not reselected. Data are reported for the mouse with substantial activity in the heart as well as for 5–6 sham-treated mice.
The successfully treated mouse showed restoration of left ventricular function to levels similar to that of wild type mice (Fig. 6). The sham-treated mice had significantly reduced function, similar to that of untreated MPS I mice for circumferential fiber shortening and ejection fraction (Fig. 6A). In catheterization measurements, contractility was similar in sham and untreated, but the relaxation in the sham-treated appeared intermediate between MPS I and normal (Fig 6B). The reason for this is not known, but it should be mentioned that the blood vessels of the treated animals, all of which had undergone irradiation at the time of bone marrow transplantation, were stiffer and harder to catheterize than those of untreated MPS I mice
Fig. 6.

Ventricular function recovery after gene therapy. A. The velocity of circumferential fiber shortening (Vcf) and ejection fraction (EF) from echocardiography are plotted in 10 month old WT mice (n = 13), MPS I mice (n = 12), sham treated MPS I mice (n = 5) and a mouse successfully treated with genetically modified bone marrow. B. Contractility (dP/dT) and relaxation (-dP/dT) from catheterizations are plotted from similar groups of WT (n = 6) and MPS I mice (n = 10, 2, 1). For both plots, * = P < 0.05 and ** = P < 0.001.
Electron microscopic examination of myocytes in the successfully treated animal showed that the storage vacuoles had largely disappeared (Fig. 7A). The endothelial cell from the mitral valve of the treated mouse had a normal appearance, with no vacuoles (Fig. 7B). On the other hand, the underlying interstitial cells remained heavily vacuolated (Fig. 7B) and indistinguishable from the interstitial cells of an untreated mouse (Fig. 3). The morphology of all cell types in sham treated mice (not shown) was the same as in the untreated ones (Fig. 3).
Fig. 7.
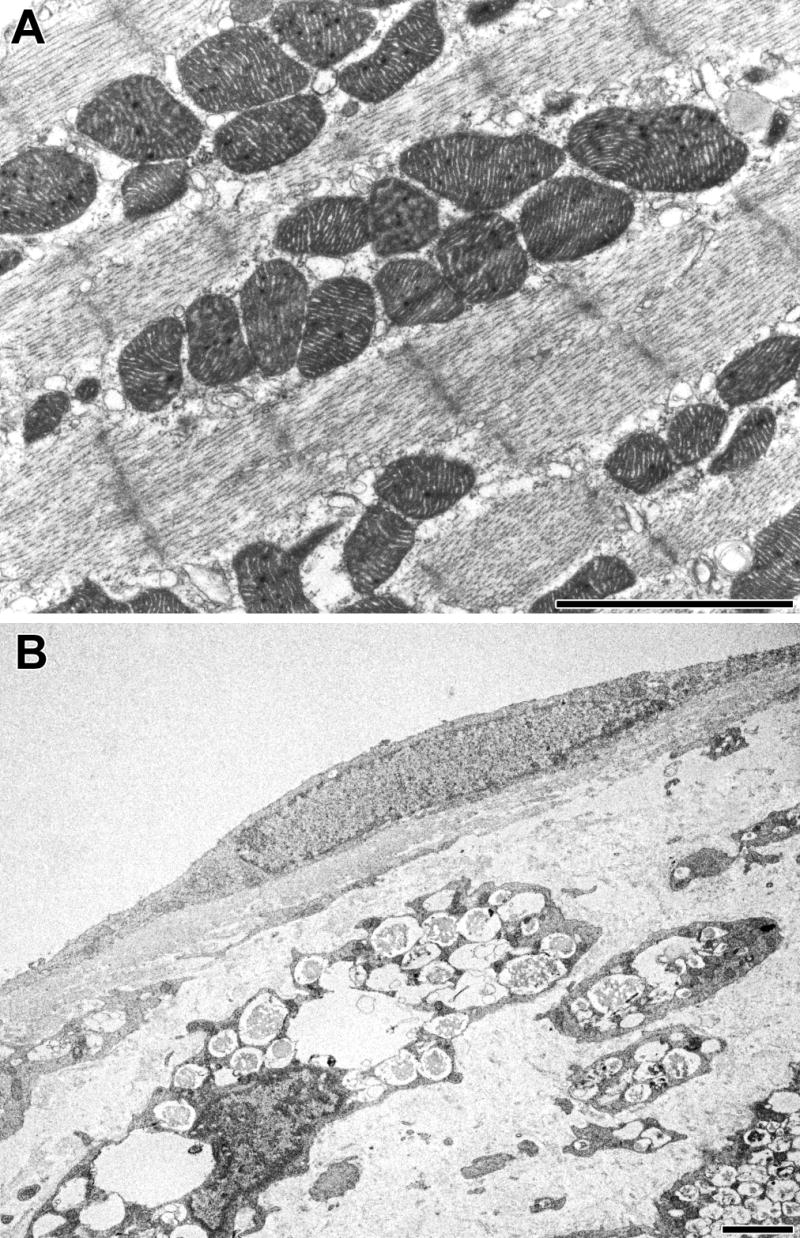
Morphologic changes in cells of MPS I heart after gene therapy. A: Myocyte of treated mouse. B: endothelial cell and underlying insterstitial cells of mitral valve of treated mouse. The scale bars are 2 μm.
Discussion
The hearts of MPS I mice are significantly enlarged and dysfunctional by 6 months of age. Unlike typical hypertrophic responses in heart, this myocardial enlargement does not involve increased myocyte mass or dilated chambers. MPS I mice have a concentric ventricular enlargement from thicker walls due primarily to the infiltration of non-myocyte cells laden with storage material. While there is some storage within the myocytes themselves, it appears to be minor compared to the storage between myocytes within the myocardial wall.
Echocardiographic and hemodynamic measurements showed significant deficits in ventricular function. The reduction in contractility of MPS I mice recorded by catheterization (dP/dT) is greater than that measured by echocardiography (Vcf). This is likely due to procedural differences. The invasive nature of catheterization requires a deeper level of anesthesia and artificial ventilation, both of which typically depress function. In addition, the larger, misshapen valves of the MPS I mice may reduce the effectiveness of the seal around the catheter and lead to additional aortic regurgitation and insufficiency during the hemodynamic evaluation.
The striking enlargement of the valves contributes to compromised ventricular function. The Doppler echocardiographic studies clearly identify mitral and aortic valve regurgitation, along with significantly longer ejection times. The large backward flows across these valves suggest incomplete closure. This would lead to lower maximum flow velocity and peak pressure gradient across the aortic valve. All these factors can contribute to less efficient pumping of blood and a larger energetic requirement for the myocardium, leading to early failure.
A unique aspect of this study is the application of radio telemetry to monitor pressure, activity, ECG and body temperature in MPS I mice without the confounding variables of anesthesia, restraint or other stressful environmental conditions. Despite the documented ventricular and valvular dysfunction of MPS I, telemetric derived pressures and heart rates were surprisingly within the normal range. Though we only examined one WT mouse with each type of transducer in this study, we had previously recorded values from dozens of C57BL/6 mice); the WT values reported here are identical to those found in the earlier published studies [20,24] and unpublished studies. But although their heart rates were similar to WT, MPS I mice had significantly reduced heart rate variability (CV%). This suggests a blunting of autonomic tone to the SA node, similar to that seen with administration of parasympathetic blockers like atropine [22]. The arterial pressure was modestly, but significantly higher in the MPS I mice; the 132 mm Hg systolic pressure could be considered borderline hypertensive. Both pulmonary and systemic hypertension have been reported in MPS patients [8–11] and attributed to vascular resistance from vessel pathology, renal influences, or chronic hypoxia from respiratory insufficiency. These factors may also apply to the MPS I mouse model, in view of their vascular pathology. Elevated systolic pressures would contribute to increased demands on the MPS I myocardium, which along with the coronary artery stenosis, would promote myocardial failure and premature mortality.
The telemetry studies demonstrated that the circadian cycle of the MPS I mice was abnormal with respect to activity, heart rate and body temperature. During the light (“sleep”) period, they were more restless, showing nearly twice the activity of WT mice. Additionally during their typical active period (dark cycle), their patterns of activity were generally more irregular than WT mice. Patients with MPS I often have disturbed sleep and daytime somnolence, attributed to obstructive sleep apnea that is caused, at least in part, by GAG deposits in tissues surrounding the upper respiratory tract [25]. The mechanism underlying the disturbed circadian rhythm in the MPS I mice is not known.
The ECG of the MPS I mice also showed striking differences in wave amplitude and widths from WT mouse ECG. The increased P wave width (duration) is probably an indicator of atrial enlargement resulting from GAG accumulation and mitral valve regurgitation. Similarly, the increased QRS-Tri wave width and reduced S wave amplitude are likely to relate to the thicker ventricular walls. Since the subendocardial tissue is disrupted by infiltrating cells, there is a discontinuity in the mechanical and electrical syncytium of the myocardium. Thus, the wave front of electrical depolarization which makes up the ECG must travel a longer, more circuitous route leading to increased ECG wave durations and multiple peaks.
There are currently two forms of treatment of patients with MPS I – bone marrow transplantation and enzyme replacement. Bone marrow transplantation has been reported to prevent cardiovascular disease both in short term [26] and long term studies [27] of Hurler patients, though the valves were not improved in the latter study. Slower progression of cardiovascular disease was reported for MPS I dogs treated by bone marrow transplantation [28]. After 1 year of enzyme replacement for MPS I patients, there was an improvement of one or two New York Heart Association functional classes [29]. Some improvement in cardiac function has been reported at various meetings (reviewed in [30]). Because enzyme replacement has become available only recently, it is too early to draw conclusions about its potential long-term effect on heart disease in MPS I.
The results from the successfully gene-treated MPS I mouse indicate that function of the cardiovascular system can be improved when the heart is provided with sufficient α-l-iduronidase. However, only myocytes and valvular endothelial cells showed correction. Interstitial cells in the valve remained highly vacuolated. These are the cells that produce the proteoglycan of the extracellular matrix; they may have to turn over so much GAG that they are not easily corrected. Interstitial cells would also have less access to circulating α-l-iduronidase that might originate from secretions of gene-modified hematopoietic cells (macrophages) in liver and spleen.
Four trials of gene therapy of the same MPS I mouse model were reported after the present study had been completed. Using an AAV vector to introduce the human IDUA cDNA, Hartung et al [31] found a very high level of expression in heart and a concominant loss of vacuolated cells. Liu et al [32] used a liver-targeted retroviral vector, and demonstrated complete normalization of heart biochemistry, structure and function at the higher dose of the vector. The same liver-targeted retroviral vector had shown excellent results in correcting the cardiovascular disease of the canine model of MPS VII (β-glucuronidase deficiency) [33]. Using a lentiviral vector, Domenico et al [34] restored only a low level of enzyme activity in heart or any tissue other than liver or spleen. On the other hand, Kobayashi et al [35], also using a lentiviral vector, found significant uptake and excellent correction of heart pathology. These studies all used an intravenous route for administration of the vector, which would be the preferred route for any eventual human application.
Acknowledgments
We thank Hui-Zhi Zhao, Helen C. Chang, James A. Jordan and Jeanne K. Kim for their technical support on this project. We thank Dr. Hong Drum for preliminary studies. This work was funded in part by the UCLA Laubisch Endowment (KPR) and NIH grant DK 38857 (EFN).
References
- 1.E.F. Neufeld, and J. Muenzer, The mucopolysaccharidoses, in: C.R. Scriver, A.L. Beaudet, W.S. Sly and D. Valle (Eds.), The Metabolic and Molecular Bases of Inherited Disease, McGraw-Hill, New York, 2001, pp. 3421–3452.
- 2.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 3.V.A. McKusick Heritable Disorders of Connective Tissue, Mosby, St Louis, 1972.
- 4.Pyeritz RE. Cardiovascular manifestations of heritable disorders of connective tissue. Prog Med Genet. 1983;5:191–302. [PubMed] [Google Scholar]
- 5.Dangel JH. Cardiovascular changes in children with mucopolysaccharide storage diseases and related disorders--clinical and echocardiographic findings in 64 patients. Eur J Pediatr. 1998;157:534–538. doi: 10.1007/s004310050872. [DOI] [PubMed] [Google Scholar]
- 6.Fischer TA, Lehr HA, Nixdorff U, Meyer J. Combined aortic and mitral stenosis in mucopolysaccharidosis type I-S (Ullrich-Scheie syndrome) Heart. 1999;81:97–99. doi: 10.1136/hrt.81.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohan UR, Hay AA, Cleary MA, Wraith JE, Patel RG. Cardiovascular changes in children with mucopolysaccharide disorders. Acta Paediatr. 2002;91:799–804. doi: 10.1080/08035250213211. [DOI] [PubMed] [Google Scholar]
- 8.Krovetz LJ, Schiebler GL. Cardiovascular manifestations of the genetic mucopolysaccharidoses. Birth Defects: Original Articles Series. 1972;8:192–196. [Google Scholar]
- 9.Renteria VG, Ferrans VJ, Roberts WC. The heart in the Hurler syndrome: gross, histologic and ultrastructural observations in five necropsy cases. Am J Cardiol. 1976;38:487–501. doi: 10.1016/0002-9149(76)90468-9. [DOI] [PubMed] [Google Scholar]
- 10.Taylor DB, Blaser SI, Burrows PE, Stringer DA, Clarke JT, Thorner P. Arteriopathy and coarctation of the abdominal aorta in children with mucopolysaccharidosis: imaging findings. AJR Am J Roentgenol. 1991;157:819–823. doi: 10.2214/ajr.157.4.1909834. [DOI] [PubMed] [Google Scholar]
- 11.Chan D, Li AM, Yam MC, Li CK, Fok TF. Hurler's syndrome with cor pulmonale secondary to obstructive sleep apnoea treated by continuous positive airway pressure. J Paediatr Child Health. 2003;39:558–559. doi: 10.1046/j.1440-1754.2003.00218.x. [DOI] [PubMed] [Google Scholar]
- 12.Shull RM, Munger RJ, Spellacy E, Hall CW, Constantopoulos G, Neufeld EF. Canine alpha-L-iduronidase deficiency. A model of mucopolysaccharidosis I. Am J Pathol. 1982;109:244–248. [PMC free article] [PubMed] [Google Scholar]
- 13.Haskins ME, Jezyk PF, Desnick RJ, McDonough SK, Patterson DF. Alpha-L-iduronidase deficiency in a cat: a model of mucopolysaccharidosis I. Pediatr Res. 1979;13:1294–1297. doi: 10.1203/00006450-197911000-00018. [DOI] [PubMed] [Google Scholar]
- 14.Clarke LA, Russell CS, Pownall S, Warrington CL, Borowski A, Dimmick JE, Toone J, Jirik FR. Murine mucopolysaccharidosis type I: targeted disruption of the murine alpha-L-iduronidase gene. Hum Mol Genet. 1997;6:503–511. doi: 10.1093/hmg/6.4.503. [DOI] [PubMed] [Google Scholar]
- 15.Russell C, Hendson G, Jevon G, Matlock T, Yu J, Aklujkar M, Ng KY, Clarke LA. Murine MPS I: insights into the pathogenesis of Hurler syndrome. Clin Genet. 1998;53:349–361. doi: 10.1111/j.1399-0004.1998.tb02745.x. [DOI] [PubMed] [Google Scholar]
- 16.Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci U S A. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng Y, Jordan MC, Rozengurt N, Roos KP, Neufeld EF. Cardiac manifestations in the mouse model of Hurler syndrome (MPSI) FASEB J. 2003;17:A528–A528. doi: 10.1016/j.ymgme.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng Y, Rozengurt N, Ryazantsev S, Kohn DB, Satake N, Neufeld EF. Treatment of the mouse model of mucopolysaccharidosis I with retrovirally transduced bone marrow. Mol Genet Metab. 2003;79:233–244. doi: 10.1016/s1096-7192(03)00116-1. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka N, Dalton N, Mao L, Rockman HA, Peterson KL, Gottshall KR, Hunter JJ, Chien KR, Ross J., Jr Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation. 1996;94:1109–1117. doi: 10.1161/01.cir.94.5.1109. [DOI] [PubMed] [Google Scholar]
- 20.Marro ML, Scremin OU, Jordan MC, Huynh L, Porro F, Roos KP, Gajovic S, Baralle FE, Muro AF. Hypertension in beta-adducin-deficient mice. Hypertension. 2000;36:449–453. doi: 10.1161/01.hyp.36.3.449. [DOI] [PubMed] [Google Scholar]
- 21.Mills PA, Huetteman DA, Brockway BP, Zwiers LM, Gelsema AJ, Schwartz RS, Kramer K. A new method for measurement of blood pressure, heart rate, and activity in the mouse by radiotelemetry. J Appl Physiol. 2000;88:1537–1544. doi: 10.1152/jappl.2000.88.5.1537. [DOI] [PubMed] [Google Scholar]
- 22.Gehrmann J, Hammer PE, Maguire CT, Wakimoto H, Triedman JK, Berul CI. Phenotypic screening for heart rate variability in the mouse. Am J Physiol Heart Circ Physiol. 2000;279:H733–740. doi: 10.1152/ajpheart.2000.279.2.H733. [DOI] [PubMed] [Google Scholar]
- 23.London B. Cardiac arrhythmias: from (transgenic) mice to men. J Cardiovasc Electrophysiol. 2001;12:1089–1091. doi: 10.1046/j.1540-8167.2001.01089.x. [DOI] [PubMed] [Google Scholar]
- 24.Huynh L, Scremin OU, Jordan MC, Roos KP. Rota-rod treadmill exercise enhance murine ventricular mass. FASEB J. 2001;15:A793. [Google Scholar]
- 25.Leighton SE, Papsin B, Vellodi A, Dinwiddie R, Lane R. Disordered breathing during sleep in patients with mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2001;58:127–138. doi: 10.1016/s0165-5876(01)00417-7. [DOI] [PubMed] [Google Scholar]
- 26.Gatzoulis MA, Vellodi A, Redington AN. Cardiac involvement in mucopolysaccharidoses: effects of allogeneic bone marrow transplantation. Arch Dis Child. 1995;73:259–260. doi: 10.1136/adc.73.3.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braunlin EA, Stauffer NR, Peters CH, Bass JL, Berry JM, Hopwood JJ, Krivit W. Usefulness of bone marrow transplantation in the Hurler syndrome. Am J Cardiol. 2003;92:882–886. doi: 10.1016/s0002-9149(03)00909-3. [DOI] [PubMed] [Google Scholar]
- 28.Gompf RE, Shull RM, Breider MA, Scott JA, Constantopoulos GC. Cardiovascular changes after bone marrow transplantation in dogs with mucopolysaccharidosis I. Am J Vet Res. 1990;51:2054–2060. [PubMed] [Google Scholar]
- 29.Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M, Izykowski B, Phillips J, Doroshow R, Walot I, Hoft R, Neufeld EF. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med. 2001;344:182–188. doi: 10.1056/NEJM200101183440304. [DOI] [PubMed] [Google Scholar]
- 30.Wraith JE. The first 5 years of clinical experience with laronidase enzyme replacement therapy for mucopolysaccharidosis I. Expert Opin Pharmacother. 2005;6:489–506. doi: 10.1517/14656566.6.3.489. [DOI] [PubMed] [Google Scholar]
- 31.Hartung SD, Frandsen JL, Pan D, Koniar BL, Graupman P, Gunther R, Low WC, Whitley CB, McIvor RS. Correction of metabolic, craniofacial, and neurologic abnormalities in MPS I mice treated at birth with adeno-associated virus vector transducing the human alpha-L-iduronidase gene. Mol Ther. 2004;9:866–875. doi: 10.1016/j.ymthe.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Xu L, Hennig AK, Kovacs A, Fu A, Chung S, Lee D, Wang B, Herati RS, Mosinger Ogilvie J. Liver-directed neonatal gene therapy prevents cardiac, bone, ear, and eye disease in mucopolysaccharidosis I mice. Molecular Therapy. 2005;11:35–47. doi: 10.1016/j.ymthe.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 33.Sleeper MM, Fornasari B, Ellinwood NM, Weil MA, Melniczek J, O'Malley TM, Sammarco CD, Xu L, Ponder KP, Haskins ME. Gene therapy ameliorates cardiovascular disease in dogs with mucopolysaccharidosis VII. Circulation. 2004;110:815–820. doi: 10.1161/01.CIR.0000138747.82487.4B. [DOI] [PubMed] [Google Scholar]
- 34.Domenico CD, Villani GR, Napoli DD, Reyero EG, Lombardo A, Naldini L, Natale PD. Gene Therapy for a Mucopolysaccharidosis Type I Murine Model with Lentiviral-IDUA Vector. Hum Gene Ther. 2005;16:81–90. doi: 10.1089/hum.2005.16.81. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi H, Carbonaro D, Pepper K, Petersen D, Ge S, Jackson H, Shimada H, Moats R, Kohn DB. Neonatal Gene Therapy of MPS I Mice by Intravenous Injection of a Lentiviral Vector. Mol Ther. 2005;11:776–789. doi: 10.1016/j.ymthe.2004.10.006. [DOI] [PubMed] [Google Scholar]