

Abstract
Understanding substrate binding and product release in cytochrome P450 (CYP) enzymes is important for explaining their key role in drug metabolism, toxicity, xenobiotic degradation and biosynthesis. Here, molecular simulations of substrate and product exit from the buried active site of a mammalian P450, the microsomal CYP2C5, identified a dominant exit channel, termed pathway (pw) 2c. Previous simulations with soluble bacterial P450s showed a different dominant egress channel, pw2a. Combining these, we propose two mechanisms in CYP2C5: (i) a one-way route by which lipophilic substrates access the enzyme from the membrane by pw2a and hydroxylated products egress along pw2c; and (ii) a two-way route for access and egress, along pw2c, for soluble compounds. The proposed differences in substrate access and product egress routes between membrane-bound mammalian P450s and soluble bacterial P450s highlight the adaptability of the P450 fold to the requirements of differing cellular locations and substrate specificity profiles.
Keywords: molecular dynamics simulation, cytochrome P450, drug metabolism, protein dynamics, substrate selectivity, product release
Introduction
Cytochrome P450 (CYP) enzymes form a ubiquitous monooxygenase haem protein family. They have an important role in the synthesis and degradation of many physiologically important compounds and in the detoxification of xenobiotics in many species of microorganisms, plants and animals (Graham-Lorence & Peterson, 1996). Thus, this family is important from medical, agrochemical and pharmaceutical perspectives. The P450 proteins are categorized into families and subfamilies according to their sequence similarities. Prokaryotic P450s are soluble proteins; most eukaryotic P450s are membrane-bound enzymes and are attached to either the endoplasmic reticulum or mitochondrial membranes. All structures of cytochromes P450 known so far show the same tertiary fold. The active site of the enzyme is next to the haem cofactor, buried in the core of the protein. It is usually isolated from the surrounding solvent (Wade et al, 2004).
This raises the question of how reactants enter or exit the active site of the enzyme. The crystal structures determined so far show channels accessible to a water-molecule-sized probe at pathway (pw) 1, 2a, 2c, 2e and a so-called solvent channel (Fig 1; Wade et al, 2004). The numbering of the pws corresponds to the three pw classes (1, 2 and 3) found by thermal motion pathway analysis (Lüdemann et al, 1997). The pws are defined by the secondary structure elements lining them, and pw2 has been further subdivided according to random expulsion molecular dynamics (REMD) ligand egress trajectories (Lüdemann et al, 2000a, 2001). All subclasses of pw2 share proximity with the BC loop/B′ helix. Pw2a shows a channel opening between the FG loop and the B′ helix/BC loop. In microsomal P450s, the protein at the outer entrance of pw2a (around the G′ helix or FG loop region) is rather hydrophobic and is expected to dip into the lipid membrane (Fig 2). Pw2c is located between the B′ helix/BC loop, G and I helices. Pw2e reaches the protein surface through the BC loop. The solvent channel (Haines et al, 2001) shows an opening between the F, E and I helices.
Figure 1.

CYP2C5 crystal structure (1N6B; Wester et al, 2003) used in the simulations, showing a solvent-accessible surface and the location of three active site channels. A substrate molecule, DMZ, is shown in the active site above the haem. Selected helices are labelled. pw2a, pw2e and the solvent channel are shown. Pw2c would point out of the page towards the reader to the left of the B′ helix, but it is closed in this structure; the opening of pw2c is shown in Fig 3. The figure was made using the PyMOL software (http://pymol.sourceforge.net).
Figure 2.
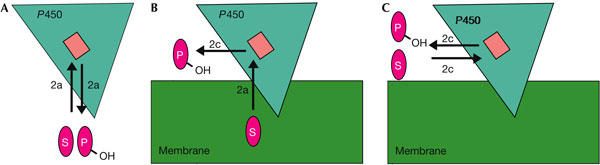
Schematic representation of putative P450 reaction cycles underlining the differences in substrate access and product egress routes in bacterial and mammalian P450s. (A) pw2a serves as substrate access and product egress channel in soluble P450s. (B) One-way route through mammalian membrane-bound P450: lipophilic substrates access the active site through pw2a and products exit through pw2c. (C) Soluble substrates access the active site and products exit through pw2c. Substrate (S) and product (P) are depicted in magenta, the membrane in green, the haem cofactor (active site) in light pink and P450 in turquoise. Mammalian P450s are anchored to the membrane through the N-terminal transmembrane helix (not shown) and by interactions with the region around the FG loop.
Experimental results, as well as theoretical approaches, indicate one route for substrate access, pw2a, that is common to P450s. Pw2a may also serve as a product egress route in some P450s (Gerber, 1994; Lüdemann et al, 2000a, 2000b; Prasad et al, 2000; Mueller et al, 2003), such as soluble bacterial proteins. However, for membrane-bound P450s, the pws for product egress and substrate access are likely to differ. A hydrophobic substrate is likely to enter the active site of the enzyme from the membrane, along pw2a; a hydroxylated watersoluble product is likely to be released into the cytoplasm and requires a different pathway (Fig 2).
The number of crystal structures of mammalian P450s available is limited and the enzymatic mechanism of membrane-bound P450s is poorly understood. Recently, the first crystal structure of a mammalian membrane-associated P450 (CYP2C5) in complex with a substrate, 4-methyl-N-methyl-N-(2-phenyl-2H-pyrazol-3-yl) benzenesulphonamide (DMZ), was solved (PDB-id: 1N6B; Wester et al, 2003). The solved structure is a chimera because, to assist expression and crystallization, the amino-terminal membrane-anchor peptide was removed and an internal hydrophobic sequence in the FG loop/G′ helix region was substituted with a more hydrophilic sequence from a related enzyme. This chimera is catalytically active in the absence as well as in the presence of lipid (Cosme & Johnson, 2000). In this crystal structure, pw2a, pw2e and the solvent channel are accessible to a water moleculesized probe (Wade et al, 2004; Fig 1). A previously determined structure of the substrate-free CYP2C5 (PDB-id: 1DT6; Williams et al, 2000) shows four active site channels along pw 2a, 2c, 2e and the solvent channel.
We address the question of how compounds exit from the active site of the CYP2C5 enzyme by applying standard molecular dynamics (MD) simulations and the REMD simulation method (Lüdemann et al, 2000a; Winn et al, 2002). Simulating ligand egress from the P450 active site is computationally challenging because the timescale of ligand egress is orders of magnitude greater than the times (typically ∼1–10 ns) for which the protein dynamics can be simulated using standard MD simulation techniques. The REMD method overcomes this problem by application of a small, artificial, randomly orientated force to the centre of mass of the ligand to enhance the probability of spontaneous exit of the ligand in a time range suitable for molecular dynamic simulations. REMD thus provides an objective way to identify ligand egress routes from protein interiors. It should be borne in mind, however, that REMD only partially accounts for the reorganization of protein and solvent on ligand egress as the timescale for ligand egress is necessarily shortened by the simulation methodology.
Here, we describe the egress routes of three compounds: progesterone (PROG), 21-hydroxy-progesterone (PROG-OH) and DMZ. CYP2C5 catalyses the 21-hydroxylation of PROG in rabbit (Dieter et al, 1982a, 1982b). DMZ is a substrate of CYP2C5 and four human CYP2C enzymes (CYP2C8, 2C9, 2C18 and 2C19).
This article describes the identification of pw2c as the predominant egress route in the CYP2C5 chimera, and the mechanism of substrate/product egress along this pathway. This pw leads from the active site to the solvent and is different from the predominant pw seen in simulations of bacterial P450s (pw2a), which in CYP2C5 is expected to connect the active site to the membrane. These observations allow us to propose two alternative mechanisms by which substrates and products are guided through the enzyme, with their usage dependent on the cellular location of the substrate.
Results and Discussion
pw2c is the predominant egress pathway
Details of all REMD simulations resulting in ligand expulsion from the protein are given in Table 1. The ligand exit trajectories were clustered into pws based on the secondary structure elements lining them, as described in the Introduction. There is one predominant pw, pw2c, that is common to all simulated compounds and was observed in 26 out of 33 ligand egress trajectories, including all the ligand egress trajectories for which the magnitude of the acceleration due to the random force was less than 0.25 kcal/g/Å. This pw is not observed to be open in the high-resolution crystal structure used for the simulation (PDB: 1N6B), but is open to a water probe in the lower resolution structure of CYP2C5 (PDB: 1DT6; Fig 1).
Table 1.
Parameters used and routes observed in the REMD simulations resulting in ligand expulsion for the complexes of PROG, PROG-OH and DMZ with CYP2C5
| Compound | No. of trajectories | Pathway | A (kcal/g/Å)a | Na | rmin (Å)a | Trajectory length (ps) |
|---|---|---|---|---|---|---|
| PROG | 12 | 2c | 0.10–0.25 | 10–100 | 0.001–0.010 | 3–189 |
| 1 | 3 | 0.25 | 80 | 0.001 | 5 | |
| 1 | Solvent channel | 0.25 | 100 | 0.010 | 19.5 | |
| |
2 |
Otherb |
0.25 |
20-100 |
0.001 |
6.5-18.5 |
| PROG-OH | 8 | 2c | 0.10–0.25 | 20–100 | 0.005–0.010 | 9.5–39.5 |
| |
1 |
Solvent channel |
0.25 |
100 |
0.010 |
44 |
| ADMZc | 3 | 2c | 0.25 | 40–100 | 0.001–0.005 | 34–206.5 |
| |
1 |
2e |
0.25 |
40 |
0.001 |
19 |
| BDMZc | 3 | 2c | 0.25 | 40–100 | 0.001–0.005 | 20–223.5 |
| 1 | 2e | 0.25 | 100 | 0.005 | 42 |
aA is the magnitude of the acceleration resulting from the random expulsion force acting on the centre of mass of the ligand. The direction of the force is kept for N time steps of 2 fs. If, during this time period, a specified distance, rmin, is covered by the substrate molecule, the direction of the force is maintained, otherwise a new direction is chosen randomly. Details of the REMD method are given in Lüdemann et al (2000a). These parameters were chosen on the basis of preliminary simulations of CYP2C5, so that artefactual distortion of the secondary structure was minimal while still permitting ligand egress to be observed within a reasonable simulation time. Some trajectories simulated (not listed) did not result in ligand egress within 400 ps, with the parameters given.
bOther routes not previously identified in crystal structures or simulations as potential active site channels for substrate or product passage.
cSimulations were started from two different configurations for DMZ in the CYP2C5 crystal structure (1N6B) (see the Methods).
Egress along pw2c involves hydrogen-bond breakage
The breakage of a hydrogen-bond between the side chain of K241 and the carbonyl oxygen of V106 lining the ligand release pw seems to be crucial for pw2c channel opening (Fig 3 and movies in supplementary information online). This hydrogen-bond breakage, which leads to an enhanced flexibility of the B′ helix/BC loop, is seen in both REMD and MD simulations (see Fig 4 and text below). A role for this hydrogen bond in channel opening was suggested by Wester et al (2003), based on the observation that, in the absence of the hydrogen bond between the side chain of K241 and the carbonyl oxygen of V106, the crystal structure 1DT6 showed an opening at pw2c, whereas, in the presence of this hydrogen bond in the crystal structure 1N6B, no such channel opening was observed. Interestingly, the crystal structure of CYP2C8 (Schoch et al, 2004) shows an arginine substituting for K241 and making a hydrogen bond to a backbone carbonyl oxygen in the B′ helix. This observation, together with recent mutagenesis data (Melet et al, 2004), suggests a similar opening mechanism in CYP2C8. A related mechanism of channel opening by dynamic hydrogen bond exchange to an arginine has also been described for pw2a in P450eryF (Winn et al, 2002).
Figure 3.
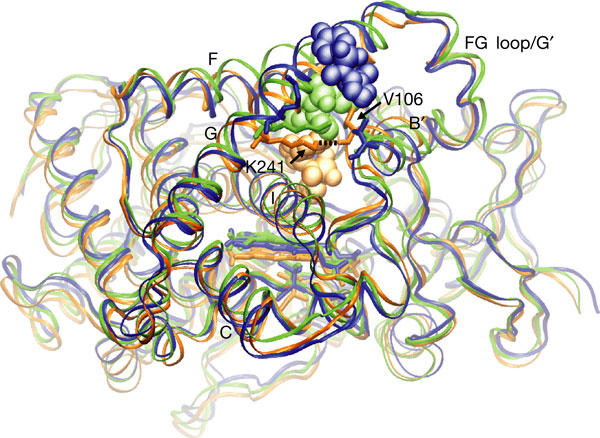
Three snapshots from one trajectory in which the substrate DMZ egresses from CYP2C5 through pw2c. The three snapshots along the exit trajectory are colour coded: copper (starting configuration), green (intermediate state) and blue (DMZ has left the protein). The protein backbone is represented by a ribbon, the haem by sticks and the ligand DMZ by CPK spheres. Selected helices are labelled. The initial orientation of DMZ in the active site is the ADMZ orientation seen in the crystal structure (1N6B) and discussed in Methods. The exit of the ligand is accompanied by a hydrogen-bond breakage (black dashed line) between K241 and the carbonyl oxygen of V106 (shown in liquorice stick representation). The view in this figure is rotated ca. 60° about the vertical axis with respect to the view in Fig 1.
Figure 4.
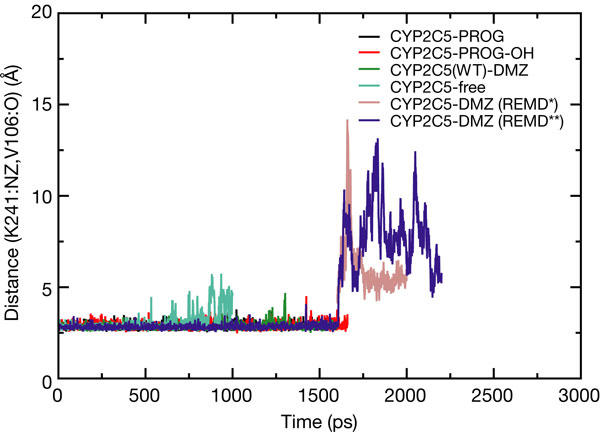
Variation of the K241–V106 hydrogen-bond length (K241:NZ-V106:O distance in Å) as a function of time is shown for standard MD simulations of CYP2C5 with PROG, PROG-OH, DMZ (1,600ps) and without ligand (free; 1,000ps), and for two REMD simulations with DMZ (*: A=0.25, N=80, rmin=0.005; **: A=0.25, N=40, rmin=0.005) started from the coordinates obtained after 1,600 ps of the standard simulation (whose trajectories are shown in movies in supplementary information online).
REMD identifies routes not observed in standard MD
A 1,600 ps standard MD simulation for each of the substrates/products in complex with CYP2C5 did not show any channel opening. In addition, a 1,000 ps simulation of a substrate-free structure (generated by replacing the substrate in the active site with water molecules) did not result in channel opening, although it showed transient breakage of the K241–V106 hydrogen bond (Fig 4). Furthermore, the reversion of the chimeric watersoluble CYP2C5 sequence back to the more hydrophobic wild-type sequence of CYP2C5, affecting residues close to pw2a, did not result in any channel opening in 1,600 ps. Essential dynamics analysis (Amadei et al, 1993) showed that the major motions were at the N and carboxyl termini and in the loop regions (notably, the FG loop, the HI loop and the EF loop), where there is also variation between the different crystal structures of CYP2C5. In contrast, all secondary structure elements were well preserved during the MD simulations and did not show much movement around pw2c.
These results are consistent with the expectation that channel opening occurs on much longer timescales than the 1.6 ns of the MD simulations, but the observation of dynamics in the formation of the K241–V106 hydrogen bond in the ligand-free structure points to the initial protein motions necessary to permit substrate binding along pw2c.
Further evidence for ligand passage along pw2c
The epitope of an inhibitory monoclonal antibody of CYP2C5 (Kronbach & Johnson, 1991) includes S115 and K118, N-terminal to the C helix close to pw2c and pw2e, and this might explain why the antibody seems to interfere with substrate binding or product exit. Site-directed mutagenesis data for CYP2B1 also provide evidence for an access channel in the region of pw2c (Scott et al, 2004a). CYP2B1 is a high Km progesterone 16α-hydroxylase, whereas CYP2C5 is a low Km progesterone 21-hydroxylase. CYP2B1 and CYP2C5 have 51% sequence identity and a few active site mutations can confer progesterone 21-hydroxylase activity on CYP2B1 (Kumar et al, 2003). Substitutions in the N-terminal portion of the I helix of CYP2B1 were able to cause changes of two- to fivefold in kcat and Km values for several substrates (Scott et al, 2004a). The I290F mutation had the greatest effect on catalysis and binding. The side chain of the equivalent residue in CYP2C5, I286, packs against the K241–V106 hydrogen bond that is perturbed on ligand egress by pw2c, and, together with V285, which packs against K241, may help to stabilize this interaction between the G helix and the B′C loop.
Summary and implications
The alternative scenarios for ligand passage to and from the P450 active site suggested by this and previous work are shown in Fig 2. Previous REMD simulations (Winn et al, 2002), supported by steered MD simulations (Lüdemann et al, 2000a, 2000b) and diverse experimental data, indicate pw2a as the most likely route for substrate access and product egress in the soluble bacterial P450s, although other channels may be used as indicated by several crystal structures of prokaryotic P450s (Wade et al, 2004), most notably Mycobacterium tuberculosis CYP51 (Podust et al, 2004). Conversely, the present results for a membrane-bound mammalian P450, CYP2C5, point to pw2c being the product egress route. This route, which would release products close to the membrane, would provide a convenient way to channel products to other membrane-bound enzymes, such as other P450s, for which they are substrates, in either degradative or biosynthetic pws. Pw2c could be part of a one-way route by which substrates from the membrane enter by pw2a and are transformed into products that leave by pw2c. Alternatively, it may be a two-way route allowing soluble substrates to enter and their products to leave the active site by pw2c. The relative extent to which pw2a and pw2c are used will thus depend on the interactions with the membrane, which are expected to affect the opening of pw2a, and the nature of the substrate.
To what extent our results for CYP2C5 are transferable to other mammalian P450s can only be speculated at present. Crystal structures of a number of recently published mammalian P450s have shown a diversity of protein conformations despite retention of the same P450 tertiary fold. The ability of this fold to undergo large conformational changes is demonstrated by the crystal structure of the mammalian CYP2B4 in which the active site is so wide open (at pw2a) that a second CYP2B4 molecule in the crystal reaches into the active site and coordinates the haem of the first (Scott et al, 2004b). Our results indicate that the use of pw2c is dependent on the type of interactions between the BC loop and the G helix, as well as the length and flexibility of the BC loop. Here, we find that a hydrogen bond between these secondary structure elements (at K241) is broken by ligand egress and may have a tethering role influencing ligand passage along pw2c, similar to that for pw2a in P450eryF (Winn et al, 2002). A similar hydrogen-bond arrangement is seen in the structure of CYP2C8. The data on CYP2B1 and CYP2C8 mutations, as well as monoclonal antibody binding to a CYP2C4 double mutant (T115S, N118K; Kronbach & Johnson, 1991), also circumstantially support their use of pw2c. Future simulations of other P450s, as well as simulations in the presence of a lipid bilayer and other proteins, such as the P450 reductase, will undoubtedly help to pinpoint the pws used by the enzyme reactants and the mechanisms of ligand passage. As such, our results provide a framework for the investigation of alternate substrate access and product egress routes in P450s, and their implications for substrate specificity.
Methods
The simulations of the CYP2C5 chimera were based on its crystal structure with DMZ bound (resolution 2.3 Å, PDB-id: 1N6B; Wester et al, 2003). In this crystal structure, two binding modes were assigned to DMZ. These have opposite orientations of the long axis of the substrate, and were named ADMZ (with the benzylic methyl group 4.4 Å from the haem Fe) and BDMZ (with the benzylic methyl group directed away from the haem).
PROG and PROG-OH were docked into the active site of CYP2C5 (with DMZ removed) using the program Autodock (Morris et al, 1998). The docked configuration with the most favourable energy score was used for subsequent simulations.
The enzyme complexes, as well as the free enzyme, were solvated with crystallographic water molecules, and then with water molecules, as well as four Na+ ions to neutralize the system, placed in energetically favourable locations using the GRID program (Goodford, 1985). This partially solvated system was then immersed in a periodic box of TIP3P water molecules. The AMBER94 force field (Cornell et al, 1995), with PME treatment of forces (cutoff=8.5 Å), and the AMBER program (Version 7, http://amber.scripps.edu) were used. The systems were energy-minimized for 200 steps and then subjected to MD simulation at 300 K and 1 bar for 1,600 ps (1,000 ps in the case of the ligand-free structure). The simulations were analysed for channel opening by visual inspection and by essential dynamics to identify low-frequency motions (Amadei et al, 1993).
The egress pws of the compounds were simulated by REMD (Lüdemann et al, 2000a; Winn et al, 2002), implemented by modification of AMBER7. The REMD simulations were started from the coordinates obtained after the 1,600 ps standard MD simulations, when the systems were well equilibrated as indicated by the root-meansquare deviation (r.m.s.d.) shown in Fig 5, among other criteria.
Figure 5.

R.m.s.d. (Å), of the ligand carbon atoms and the backbone (Cα, C, N) atoms of CYP2C5, is shown as a function of time during two standard MD simulations of CYP2C5 complexed with PROG and PROG-OH.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Supplementary Movie1
Supplementary Movie2
Supplementary Movie3
Acknowledgments
We thank Professor E. Johnson and Dr D. McRee for valuable discussions about CYP2C5 crystal structures, Dr D. Harris for the provision of haem parameters, and Dr A. Banerjee for critical comments. This work was supported in part by a German Academic Exchange Service (DAAD) fellowship to Sudarko, National Institutes of Health (USPHS) grant GM59467, Human Frontier Science Program grant RG0234/2000-M and the Klaus Tschira Foundation.
References
- Amadei A, Linssen AB, Berendsen HJ (1993) Essential dynamics of proteins. Proteins 17: 412–425 [DOI] [PubMed] [Google Scholar]
- Cornell WD, Cieplak P, Payly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA (1995) A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J Am Chem Soc 117: 5179–5197 [Google Scholar]
- Cosme J, Johnson EF (2000) Engineering microsomal cytochrome P4502C5 to be a soluble, monomeric enzyme. Mutations that alter aggregation, phospholipid dependence of catalysis, and membrane binding. J Biol Chem 275: 2545–2553 [DOI] [PubMed] [Google Scholar]
- Dieter HH, Muller-Eberhard U, Johnson EF (1982a) Identification of rabbit microsomal cytochrome P-450 isozyme, form 1, as a hepatic progesterone 21-hydroxylase. Biochem Biophys Res Commun 105: 515–520 [DOI] [PubMed] [Google Scholar]
- Dieter HH, Muller-Eberhard U, Johnson EF (1982b) Rabbit hepatic progesterone 21-hydroxylase exhibits a bimodal distribution of activity. Science 217: 741–743 [DOI] [PubMed] [Google Scholar]
- Gerber NC (1994), PhD Thesis, University of Illinois, Urbana, USA
- Goodford PJ (1985) A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem 28: 849–857 [DOI] [PubMed] [Google Scholar]
- Graham-Lorence S, Peterson JA (1996) P450s: structural similarities and functional differences. FASEB J 10: 206–214 [DOI] [PubMed] [Google Scholar]
- Haines DC, Tomchick DR, Machius M, Peterson JA (2001) Pivotal role of water in the mechanism of P450BM-3. Biochemistry 40: 13456–13465 [DOI] [PubMed] [Google Scholar]
- Kronbach T, Johnson EF (1991) An inhibitory monoclonal antibody binds in close proximity to a determinant for substrate binding in cytochrome P450IIC5. J Biol Chem 266: 6215–6220 [PubMed] [Google Scholar]
- Kumar S, Scott EE, Liu H, Halpert JR (2003) A rational approach to Re-engineer cytochrome P450 2B1 regioselectivity based on the crystal structure of cytochrome P450 2C5. J Biol Chem 278: 17178–17184 [DOI] [PubMed] [Google Scholar]
- Lüdemann SK, Carugo O, Wade RC (1997) Substrate access to cytochrome P450cam: a comparison of a thermal motion pathway analysis with molecular dynamics simulation data. J Mol Model 3: 1–5 [Google Scholar]
- Lüdemann SK, Lounnas V, Wade RC (2000a) How do substrates enter and products exit the buried active site of cytochrome P450cam? 1. Random expulsion molecular dynamics investigation of ligand access channels and mechanisms. J Mol Biol 303: 797–811 [DOI] [PubMed] [Google Scholar]
- Lüdemann SK, Lounnas V, Wade RC (2000b) How do substrates enter and products exit the buried active site of cytochrome P450cam? 2. Steered molecular dynamics and adiabatic mapping of substrate pathways. J Mol Biol 303: 813–830 [DOI] [PubMed] [Google Scholar]
- Lüdemann SK, Gabdoulline RR, Lounnas V, Wade RC (2001) Substrate access to cytochrome P450cam investigated by molecular dynamics simulations: an interactive look at the underlying mechanisms. Internet J Chem 4: 6 [Google Scholar]
- Melet A, Marquessoares C, Schoch GA, Macherey AC, Jaouen M, Dansette PM, Sari M-A, Johnson EF, Mansuy D (2004) Analysis of human cytochrome P450 2C8 substrate specificity using a substrate pharmacophore and site-directed mutants. Biochemistry 43: 15379–15392 [DOI] [PubMed] [Google Scholar]
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ (1998) Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19: 1639–1662 [Google Scholar]
- Mueller EJ, Loida PJ, Sligar SG (2003) in Cytochrome P450: Structure, Mechanism, and Biochemistry, Ortiz de Montellano PR (ed) pp 83–124. New York and London: Plenum Press [Google Scholar]
- Podust LM, Yermalitskaya LV, Lepesheva GI, Podust VN, Dalmasso EA, Waterman MR (2004) Estriol bound and ligand-free structures of sterol 14α-demethylase. Structure 12: 1937–1945 [DOI] [PubMed] [Google Scholar]
- Prasad S, Mazumdar S, Mitra S (2000) Binding of camphor to Pseudomonas putida cytochrome p450(cam): steadystate and picosecond time-resolved fluorescence studies. FEBS Lett 477: 157–160 [DOI] [PubMed] [Google Scholar]
- Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF (2004) Structure of human microsomal cytochrome P450 2C8. Evidence for a peripheral fatty acid binding site. J Biol Chem 279: 9497–9503 [DOI] [PubMed] [Google Scholar]
- Scott EE, Liu H, Qun He Y, Li W, Halpert JR (2004a) Mutagenesis and molecular dynamics suggest structural and functional roles for residues in the N-terminal portion of the cytochrome P450 2B1 I helix. Arch Biochem Biophys 423: 266–276 [DOI] [PubMed] [Google Scholar]
- Scott EE, White MA, He YA, Johnson EF, Stout CD, Halpert JR (2004b) Structure of mammalian cytochrome P450 2B4 complexed with 4-(4-chlorophenyl)imidazole at 1.9-Å resolution: insight into the range of P450 conformations and the coordination of redox partner binding. J Biol Chem 279: 27294–27301 [DOI] [PubMed] [Google Scholar]
- Wade RC, Winn PJ, Schlichting I, Sudarko (2004) A survey of active site access channels in cytochromes P450. J Inorg Biochem 98: 1175–1182 [DOI] [PubMed] [Google Scholar]
- Wester MR, Johnson EF, Marquessoares C, Dansette PM, Mansuy D, Stout CD (2003) Structure of a substrate complex of mammalian cytochrome P450 2C5 at 2.3 Å resolution: evidence for multiple substrate binding modes. Biochemistry 42: 6370–6379 [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE (2000) Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cells 5: 121–131 [DOI] [PubMed] [Google Scholar]
- Winn PJ, Lüdemann SK, Gauges R, Lounnas V, Wade RC (2002) Comparison of the dynamics of substrate access channels in three cytochrome P450s reveals different opening mechanisms and a novel functional role for a buried arginine. Proc Natl Acad Sci USA 99: 5361–5366 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Movie1
Supplementary Movie2
Supplementary Movie3