

Abstract
Thymosin α1 (Tα1) is noted for its immunomodulatory activities and therapeutic potential in treatment of infectious diseases and cancer. However, the molecular mechanism of its effectiveness is not completely understood. Here, we report that Tα1 induces interleukin (IL)-6 expression through the IκB kinase (IKK) and nuclear factor-κB (NF-κB) pathway. Using IKKβ-deficient bone-marrow-derived macrophages and mouse embryo fibroblasts (MEFs), we show that IKKβ is essential for IKK and NF-κB activation as well as efficient IL-6 induction. Further analysis using tumour necrosis factor receptor-associated factor 6 (TRAF6)-deficient MEFs shows that TRAF6 is crucial for activation of IKK and induction of IL-6 by Tα1. Intriguingly, Tα1 triggers protein kinase C (PKC)ι/ζ activation, which is TRAF6 dependent and involves IKK. In addition, Tα1 induces the formation of a signalsome composed of TRAF6, p62 and PKCι/ζ as well as IKK. Thus, our study identifies Tα1 as a unique activator of the TRAF6 signal pathway and provides a cohesive interpretation of the molecular basis of the therapeutic utility of Tα1.
Keywords: thymosin α1, TRAF6, PKCι/ζ, IKK
Introduction
Thymosin α1 (Tα1), a 28-amino-acid peptide, was originally isolated from bovine thymus as a thymic hormone and is now obtained by synthetic preparation (Low et al, 1979; Wang et al, 1980). Tα1, known for its ability as an immune adjuvant for vaccines, is in clinical trials worldwide for the treatment of hepatitis B virus, hepatitis C virus and cancer (Billich, 2002). Studies on the molecular basis of its action have suggested that Tα1 can induce T-cell and dendritic cell (DC) maturation as well as interleukin (IL)-12 expression (Knutsen et al, 1999; Romani et al, 2004). In addition, a recent study indicated that Tα1 upregulates expression of Toll-like receptor (TLR) 2, 5, 8 and 9, and protects mice from challenge by invasive aspergillosis in a myeloid differentiate factor 88 (MyD88)-dependent way (Romani et al, 2004).
The canonical TLR signalling pathway includes several crucial intermediates such as MyD88, IL-1 receptor-associated kinase activator (IRAK) and tumour necrosis factor (TNF) receptor-associated factor 6 (TRAF6; Akira & Takeda, 2004). It has been established that TLRs, on activation by their agonists, recruit MyD88, followed by bringing IRAK1 and IRAK4 to the intracellular domains of the transmembrane receptors. IRAK1 is phosphorylated by IRAK4 and then interacts with TRAF6, which is essential for nuclear factor-κB (NF-κB) activation. Typical NF-κB activation involves the IκB kinase (IKK) complex, which consists of two catalytic subunits, IKKα and IKKβ, as well as the regulatory subunit IKKγ. Activated IKK phosphorylates IκB, which immediately undergoes degradation and subsequently liberates NF-κB. Several studies have suggested that activation of NF-κB by TRAF6 involves atypical protein kinase C (aPKC; PKCι and PKCζ) as well as the adaptor protein p62 (Sanz et al, 2000; Leitges et al, 2001; Duran et al, 2003). As aPKC associates with IKK (Sanz et al, 1999), it is likely that, on IL-1 stimulation, TRAF6 forms a complex with p62 and aPKC, which in turn activates IKK. This has been shown by the overexpression of dominant negative (dn) mutants of either PKCι or PKCζ, which block IKK and NF-κB activation (Lallena et al, 1999). Moreover, genetic evidence shows that activation of IKK and NF-κB by IL-1, lipopolysaccharide (LPS) and TNFα is largely impaired in PKCζ-deficient lung cells (Leitges et al, 2001). Interestingly, in PKCζ-deficient mouse embryo fibroblasts (MEFs), activation of NF-κB, but not IKK, by IL-1 is inhibited (Leitges et al, 2001), suggesting a redundant role of PKCι in this pathway.
We investigated signalling pathways responsive to Tα1 that lead to induction of IL-6. We obtained evidence showing that an essential pathway that triggers NF-κB activation by Tα1 is dependent on IKKβ, which requires TRAF6 and aPKC. We showed that, on Tα1 stimulation, TRAF6 is activated and forms a signalsome that contains TRAF6, p62 and PKCι/ζ. Therefore, our study shows that IKKβ and TRAF6 are decisive components in the Tα1-induced immune response.
Results
Tα1 induces production of IL-6 in vitro and in vivo
As Tα1 has been shown to induce IL-12 expression (Romani et al, 2004), we investigated whether Tα1 directly activates macrophages to produce IL-6. Bone-marrow-derived macrophages (BMDMs) were treated with different amounts of Tα1 for 24 h and supernatant samples were assessed. IL-6 secretion was induced by Tα1 at a concentration of 25 ng/ml and reached higher levels at 100 ng/ml (Fig 1A). However, Tα1 at a much higher concentration (e.g. 4 μg/ml) did not further increase, but instead inhibited IL-6 secretion (data not shown). Tα1 also stimulated BMDMs to produce IL-12 (Fig 1C). In contrast, scrambled Tα1 (sTα1; same amino acids as Tα1, but in an altered primary sequence) failed to induce IL-6 and IL-12 in BMDMs (Fig 1B,C).
Figure 1.
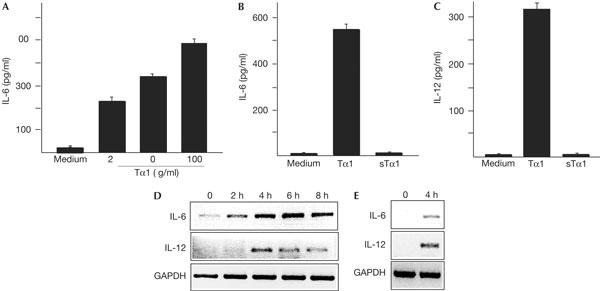
Tα1 induces expression of IL-6 and IL-12 in vitro and in vivo. (A) Bone-marrow-derived macrophages (BMDMs) were treated with indicated amounts of Tα1 for 24 h and the level of IL-6 was assessed by enzyme-linked immunosorbent assay. Levels of IL-6 (B) and IL-12 (C) in BMDMs treated with Tα1 (100 ng/ml) or sTα1 (100 ng/ml) were determined. (D) Levels of IL-6 and IL-12 mRNA in BMDMs treated with Tα1 (100 ng/ml) for indicated durations were assessed by RT–PCR. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (E) Mice were injected intravenously with Tα1 (10 μg per mouse). After 3 h, mice were killed and total RNA was isolated using Trizo (Invitrogen). The levels of IL-6 and IL-12 mRNA in the liver were determined by RT–PCR.
The induction of IL-6 and IL-12 by Tα1 is associated with increases in messenger RNA. BMDMs were treated with Tα1 and levels of IL-6 and IL-12 mRNA were examined by reverse transcription–PCR (RT–PCR) analysis. IL-6 and IL-12 mRNA levels were detected after a 2 h incubation with Tα1 and decayed after 6 h (Fig 1D). Furthermore, mice were injected with Tα1 or phosphate-buffered saline (PBS), and the levels of IL-6 and IL-12 mRNA in the liver were analysed. IL-6 and IL-12 mRNA levels increased 4 h after injection in Tα1- but not PBS-treated livers (Fig 1E; data not shown). Thus, Tα1 can induce IL-6 and IL-12 production in both BMDMs and mice.
IKKβ is essential for cytokine induction by Tα1
Tα1 triggers NF-κB activation (Romani et al, 2004), although the pathway of activation has not yet been explained. Therefore, we investigated whether Tα1 activates IKK, which is known to be essential for activation of NF-κB. Incubation of BMDMs with Tα1 resulted in strong activation of IKK (Fig 2A). As Tα1 is derived from pro-thymosin α1 (Sarandeses et al, 2003) and is found to be circulating in the blood (Welch et al, 1988), we reasoned that effectors for Tα1 activity should not be limited to immune cells. Therefore, we examined the responses of several different types of cell to Tα1. As we expected, Tα1 activated IKK in MEFs (Fig 2A, right panel), embryonic stem (ES) cells, NIH-3T3 cells and human T-lymphocyte Motl4 cells (data not shown). In contrast, sTα1 induced minimal activation of IKK in MEFs (Fig 2A, right panel).
Figure 2.
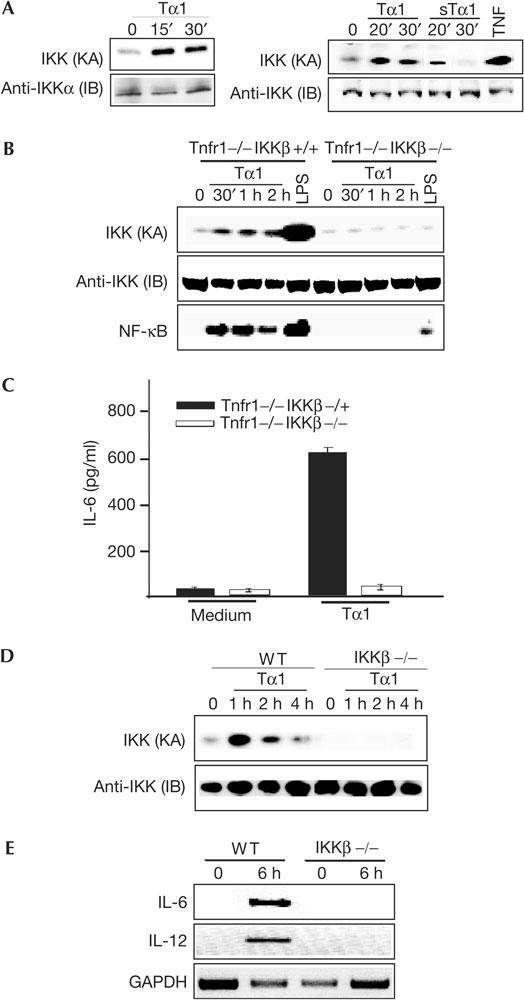
IKKβ is required for activation of IKK and NF-κB and induction of IL-6 and IL-12 by Tα1. (A) Left panel: Bone-marrow-derived macrophages (BMDMs) were treated with Tα1 (100 ng/ml) for 15 or 30 min and cell lysates were prepared. A 100 μg portion of cell lysates was used to assess IKK kinase activity (KA) by a kinase assay using GST–IκBα(1–54) as a substrate. Recovery of IKK was determined by an immunoblotting (IB) analysis with an anti-IKKα antibody. Right panel: Mouse embryo fibroblasts (MEFs) were treated with Tα1 (100 ng/ml) or sTα1 (100 ng/ml) for indicated durations, cell lysates were prepared and then IKK KA was determined. (B) BMDMs from Tnfr1−/− and Ikkβ−/−Tnfr1−/− mice were treated with Tα1 (400 ng/ml) or lipopolysaccharide (LPS; 30 min, 1 μg/ml) for indicated durations and then lysed. IKK KA was determined and NF-κB DNA-binding activity was measured by an electrophoretic mobility shift assay. (C) Levels of IL-6 in Tα1-treated BMDMs from Ikkβ-/+Tnfr1−/− and Ikkβ−/−Tnfr1−/− mice were determined. (D) Wild-type (WT) and IKKβ-deficient MEFs were treated with Tα1 (100 ng/ml) for indicated durations and IKK KA was determined. (E) Levels of IL-6 and IL-12 mRNA in WT and IKKβ-deficient MEFs treated with Tα1 (100 ng/ml) were assessed by RT–PCR. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Our previous studies suggested that IKKβ is crucial for polynucleotide-triggered activation of IKK and NF-κB and subsequent immune responses (Chu et al, 1999, 2000). Thus, we proposed that IKKβ also has a pivotal role in the activation of IKK and NF-κB, as well as induction of cytokines, by Tα1. To test this possibility, we decided to use Tnfr1−/−Ikk+/− (referred to as ‘IKKβ heterozygous') mice to generate Tnfr1−/−Ikkβ−/− (referred to as ‘IKKβ-deficient') mice (Chu et al, 2000; Senftleben et al, 2001). BMDMs from Tnfr1−/− and IKKβ-deficient mice were then isolated to determine the role of IKKβ in the Tα1-driven NF-κB activation. Incubation of Tnfr1−/− BMDMs with Tα1 resulted in strong activation of IKK and NF-κB (Fig 2B). In contrast, the loss of IKKβ severely impaired activation of IKK by Tα1 and, as a result, Tα1 stimulation of NF-κB activity was almost abolished in IKKβ-deficient BMDMs (Fig 2B). In line with a previous report (Chu et al, 2000), activation of IKK and NF-κB by LPS was largely reduced in IKKβ-deficient cells (Fig 2B). As a control, we measured p38 kinase activity in Tnfr1−/− and IKKβ-deficient BMDMs, and found that both were equally responsive to Tα1 in activating p38 (data not shown).
We next determined whether IKKβ is crucial for IL-6 and IL-12 induction by Tα1. Heterozygous and IKKβ-deficient BMDMs were incubated with Tα1 for 24 h and the levels of IL-6 and IL-12 were determined. The loss of IKKβ almost abolished induction of IL-6 by Tα1 compared with wild-type (WT) controls (Fig 2C). However, IL-12 was not detected in either heterozygous or IKKβ-deficient BMDMs under our experimental conditions (data not shown). As it is difficult to generate enough IKKβ-deficient mice for experiments, and Tα1 is able to activate MEFs (Fig 2A), we examined activation of IKK by Tα1 in IKKβ-deficient MEFs. As we expected, activation of IKK in IKKβ-deficient MEFs was largely impaired (Fig 2D). To examine further whether induction of IL-6 and IL-12 by Tα1 occurs at the level of transcription, we performed RT–PCR. As shown in Fig 2E, induction of IL-6 and IL-12 mRNA was not observed in IKKβ-deficient MEFs. Taken together, our data suggest that IKKβ is essential for the activation of IKK and NF-κB, and for induction of IL-6 and IL-12 by Tα1.
Activation of IKK involves aPKC
A previous study indicated that Tα1 activates PKC in thymocytes (Baumann et al, 1997). However, it is not clear which class of PKCs is activated and what role PKC has in the Tα1 pathway. To resolve these issues, we first investigated whether Tα1 activates aPKC in MEFs. As shown in Fig 3A, aPKC activation was induced by Tα1 after a 7.5 min incubation and its activity reached a maximal level around 1 h (Fig 3A). Next, we explored the possibility of involvement of aPKC in the activation of IKK by Tα1 using a specific aPKC inhibitor, Go6983. At a concentration of 120 nM, Go6983 inhibited activation of IKK by Tα1, but not by TNFα (Fig 3B). At the same concentration, Go6983 had no effect on activation of either c-Jun N-terminal kinase or extracellular signal-regulated kinase 1/2 by Tα1 (data not shown). To verify the involvement of PKCζ in activation of IKK, we transfected PKCζ short interfering RNA (siRNA) and mock siRNA into 3T3-like MEFs. As shown in Fig 3C, lower levels of phosphorylation of PKCζ and IKKα/β by Tα1 were observed in cells transfected with PKCζ siRNA compared with mock transfectants. As a result, activation of IKK by Tα1 was impaired in PKCζ siRNA transfectants (Fig 3C). Taken together, these experimental results indicate that PKCζ activation by Tα1 takes precedence over that of IKK.
Figure 3.
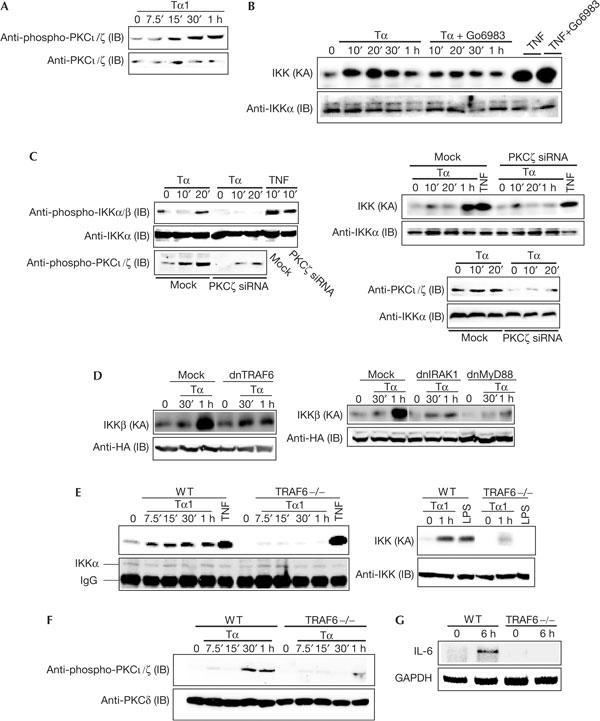
TRAF6 is involved in the activation of aPKC and IKK by Tα1. (A) Mouse embryo fibroblasts (MEFs) were treated with Tα1 (100 ng/ml) for indicated durations. The phosphorylation level of PKCι/ζ was determined by immunoblotting (IB) using an anti-phospho-PKCι/ζ antibody. (B) MEFs were treated with Tα1 (100 ng/ml) in the presence or absence of Go6983 (120 nM) and IKK kinase activity (KA) was determined. (C) 3T3-like MEFs were transfected with PKCζ siRNA or its control siRNA. After 48 h, cells were split and starved overnight. Cells were treated with Tα1 (400 ng/ml) for indicated durations. Phosphorylation of IKKα/β and PKCζ (left panel) was assessed by IB analyses using anti-phospho-antibodies against IKKα/β or PKCζ, and IKK KA (right panel) was determined. Levels of PKCι/ζ in control or PKCζ siRNA transfectants (lower right panel) were assessed by IB analysis using anti-PKCι/ζ antibodies. (D) MEFs were transfected with expression vectors HA–IKKβ (0.2 μg) together with control vector, dnIRAK1 (0.4 μg), dnMyD88 (0.2 μg) or dnTRAF6 (0.4 μg). After 24 h, cells were treated with Tα1 (100 ng/ml) for 1 h. Cell lysates were prepared and the IKKβ kinase complex was immunoprecipitated using an anti-HA antibody followed by a kinase assay to measure IKK KA. (E) Wild-type (WT) and TRAF6-deficient cells were treated with Tα1 (100 ng/ml), TNFα (10 min, 20 ng/ml) or lipopolysaccharide (LPS; 30 min, 10 μg/ml) for indicated durations. A 100 μg portion of cell lysates was used to assess IKK KA, followed by IB analysis. (F) WT and TRAF6-deficient MEFs were treated with Tα1 for 6 h. Total RNA was isolated and then subjected to RT–PCR analysis. (G) WT and TRAF6-deficient MEFs were treated with Tα1 (100 ng/ml) and then phosphorylation of PKCι/ζ was assessed. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Tα1 activates IKK and aPKC through TRAF6
A recent study suggested that MyD88 is involved in the Tα1 pathway (Romani et al, 2004). As IRAK1 and TRAF6 act downstream to MyD88 in the TLR signalling pathway, we investigated whether MyD88, IRAK1 or TRAF6 has a role in activation of IKK by Tα1. First, we performed a transient transfection assay using dnIRAK1, dnMyD88 and dnTRAF6 expression vectors. Results in Fig 3C show that all double-negative mutants, dnIRAK1, dnMyD88 and dnTRAF6, blocked IKK activation by Tα1. Next, we evaluated IKK activation in TRAF6-deficient MEFs. Incubation of WT cells with Tα1 strongly induced IKK activation, whereas the loss of TRAF6 severely impaired activation of IKK by Tα1 (Fig 3D). As controls, both WT and deficient cells were similarly responsive to TNFα in activating IKK (Fig 3D); activation of IKK by LPS was almost abrogated in TRAF6-deficient cells (Fig 3D). Indeed, TRAF6 is involved in activation of IKK by Tα1, as introduction of WT TRAF6 restored activation of IKK by Tα1 in TRAF6-deficient cells (data not shown). In addition, we determined whether TRAF6 contributes to the expression of IL-6 by Tα1. As shown in Fig 3E, induction of IL-6 was largely impaired in TRAF6-deficient MEFs.
As Tα1 activates both IKK and PKCι/ζ, we tested whether TRAF6 acts upstream of aPKC. As shown in Fig 3F, activation of PKCι/ζ by Tα1 was inhibited in TRAF6-deficient cells. Taken together, our results show that MyD88 and IRAK1 are involved in the activation of IKK, and that TRAF6 is crucial for the activation of IKK and PKCι/ζ by Tα1.
Tα1 induces formation of a TRAF6–p62–aPKC complex
We sought to determine whether TRAF6 and aPKC interact in response to Tα1. WT MEFs were treated with Tα1 and cell lysates were prepared. PKCι/ζ were immunoprecipitated, and co-immunoprecipitation of TRAF6 was detected by an immunoblotting (IB) analysis. Results in Fig 4A show that Tα1 triggered association of PKCι/ζ with TRAF6. Next, we investigated whether Tα1 stimulation promotes TRAF6 and p62 interaction, and whether association of PKCι/ζ with TRAF6 is functional. Cells were treated with Tα1 and immunoprecipitations were performed with either anti-TRAF6 or anti-PKCι/ζ antibodies, followed by IB analyses. Tα1 induced the interaction of TRAF6 with p62 (Fig 4B). Phosphorylated PKCι/ζ was observed to associate with TRAF6 on Tα1 stimulation (Fig 4B). Finally, we investigated whether Tα1 initiates IKK association with PKCι/ζ. Interestingly, interaction of PKCι/ζ with IKK was observed in untreated cells, and was not further potentiated by Tα1 treatment (Fig 4C). Taken together, our data suggest that Tα1 induces formation of a signalsome, consisting of TRAF6, p62, PKCι/ζ and IKK.
Figure 4.
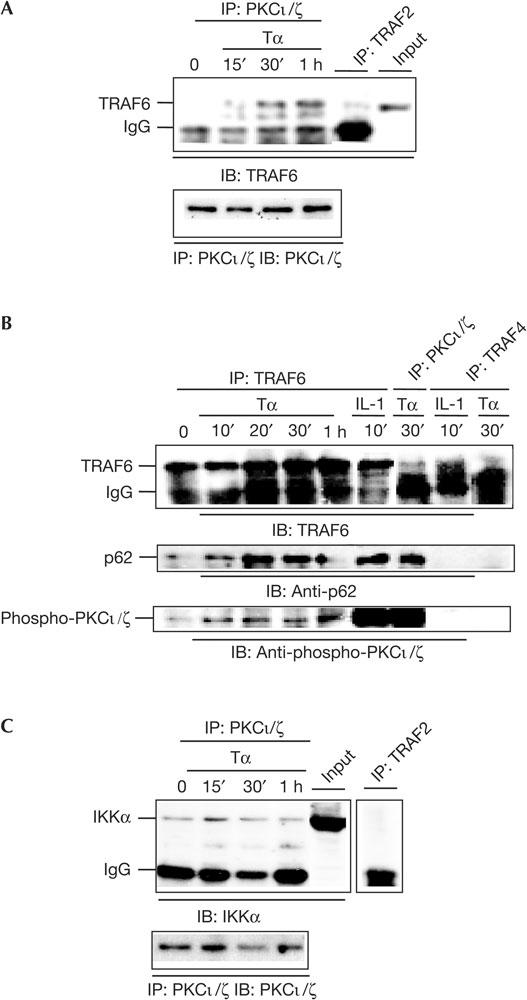
Tα1 induces formation of a TRAF6–p62–PKCζ complex. (A) Mouse embryo fibroblasts (MEFs) were treated with Tα1 (100 ng/ml) for the indicated time points and then lysed. A 200 μg portion of lysates was incubated overnight with anti-PKCι/ζ or anti-TRAF2 antibodies together with 20 μl of protein A/G beads. The beads were washed four times with lysis buffer (0.2 M NaCl) and then co-immunoprecipitation of TRAF6 was assessed by immunoblotting (IB) analysis using an anti-TRAF6 antibody. IP, immunoprecipitation. (B) MEFs were treated with Tα1 (400 ng/ml) or IL-1 (20 ng/ml) for indicated durations. A 200 μg portion of cell lysates was incubated overnight with anti-PKCι/ζ, anti-TRAF6 or anti-TRAF4 antibodies together with 20 μl of protein A/G beads. The beads were washed four times with lysis buffer (0.2 M NaCl) and co-immunoprecipitation of p62, phospho-PKCι/ζ or TRAF6 was determined by IB analyses using anti-p62, anti-phospho-PKCι/ζ or anti-TRAF6 antibodies. (C) MEFs were treated with Tα1 (100 ng/ml), and 200 μg of cell lysates was incubated with an anti-PKCι/ζ antibody. Co-immunoprecipitation of IKK was detected by IB analysis using an anti-IKKα antibody.
Discussion
Tα1 has been shown to induce expression of various cytokines by peripheral blood lymphocytes, including colony-stimulating factors, interferon-γ, IL-2 and IL-7, and to stimulate maturation of thymocyte cells (reviewed by Billich, 2002). The effects of Tα1 seem to be dependent on the presence of other mitogens or interferon, leading to the conclusion that Tα1 is not able to function as a classic primary signal in the induction of cytokines, but is used instead as an adjuvant to amplify signals related to activation of the host immune system. However, our macrophage data presented here, together with recent evidence showing that Tα1 can directly induce DC maturation and secretion of IL-12, clearly show that Tα1 can act as a primary activator. Furthermore, as IL-6 induces thymocyte-cell growth and stimulates cell maturation, and IL-12 is a key immunoregulatory molecule in Th1 responses, our results can help to explain how Tα1 exerts its immunostimulatory activity.
Our study shows that the TRAF6–IKK–NF-κB signalling pathway is involved in induction of IL-6 and IL-12 by Tα1. It has been well established that NF-κB has an important role in expression of a variety of immune genes, including cytokines, chemokines and lymphokines (Ghosh & Karin, 2002). Disruption of IKKβ or NF-κB severely impairs cytokine induction in response to viral and bacterial infections and blocks differentiation and maturation of immune cells (Ghosh & Karin, 2002). Indeed, Tnfr−/−Ikkβ−/− mice are more sensitive to the environment and survive 2 weeks or less after birth, until they succumb to opportunistic infections (Senftleben et al, 2001). Thus, the finding of the physiological role of IKKβ in the Tα1 pathway conforms with previous analyses of the NF-κB signalling pathway, which is of great importance in immune-cell function. Recently, a study showed that TRAF6 has a central role in DC maturation and development (Kobayashi et al, 2003). Our results, indicating that Tα1 activates the TRAF6 pathway, will provide further insight into how Tα1 induces DC maturation.
MyD88, IRAK1 and TRAF6 are important intermediates in the TLR signalling pathway. Corroborating a recent study showing that Tα1 activates MyD88 (Romani et al, 2004), our work indicates that both MyD88 and IRAK1 are primarily involved in activation of IKK by Tα1. Moreover, our observations suggest that Tα1 triggers TRAF6-dependent activation of PKCι/ζ, which in turn activates IKK. This notion is supported by evidence that phosphorylation of IKK or PKCι/ζ and activation of IKK by Tα1 are disrupted by lowering the levels of PKCζ. Interestingly, TNFα is able to induce the association of TRAF6 and PKCι/ζ (Zhang et al, data not shown), suggesting a role for PKCι/ζ in the activation of IKK and NF-κB by TNFα. However, neither reducing nor abolishing the expression of PKCζ in MEFs has an effect on activation of IKK by TNFα (Fig 3; Leitges et al, 2001). In contrast, the loss of PKCζ in the lung severely impaired activation of IKK by TNFα and the TLR ligands IL-1 and LPS (Leitges et al, 2001). Nevertheless, although differential roles of PKCζ in the activation of IKK by TNFα in different cell types or tissues have been accredited to the levels of PKCζ expression (Leitges et al, 2001), the molecular mechanism of such a discrepancy is not well understood.
TLRs have been identified as receptors for exogenous microbial products (e.g. dsRNA, CpG-DNA and LPS) and endogenous factors (e.g. HSP60 and defensin β; Akira & Takeda, 2004). It has been reported that Tα1 induces the expression of TLR2, TLR3, TLR5 and TLR9 in DCs (Romani et al, 2004). Moreover, Tα1 augments induction of IL-12 in the response of DCs to CpG-ODN. Tα1 only stimulates IL-8 production in 293 cells overexpressing TLR9, but not TLR2 or TLR4, suggesting that TLR9 mediates the effect of Tα1 on DCs. However, TLR9 deficiency does not alter inhibition of fungal growth by Tα1. Conversely, the loss of MyD88 almost abolishes Tα1-induced protection of mice from fungal infection, indicating that MyD88 has an essential role in the Tα1 signalling pathway (Romani et al, 2004). Therefore, it is still not clear which TLR is the receptor for Tα1, and how Tα1 activates TLRs remains to be explained further.
In conclusion, Tα1 activates the TRAF6–aPKC–IKK signalling pathway that leads to the activation of NK-κB, which in turn initiates cytokine gene expression. Our study provides a rationale for the therapeutic utility of Tα1, and explains further the pathways involved in its action.
Methods
Plasmids, siRNA, Tα1, inhibitors and antibodies. Expression vectors encoding HA–IKKβ (haemagglutinin), dnIRAK1, dnMyD88 and dnTRAF6 were described previously (Chu et al, 1999; Chuang et al, 2002). PKCζ siRNA and its control were purchased from Upstate (NY, USA). Tα1 and sTα1 peptides were synthesized (sequences have been described previously; Romani et al, 2004) and their endotoxin levels were <0.03 pg/ml, determined by a standard limulus lysate assay. The lyophilized powders were reconstituted in sterile endotoxin-free PBS. All regular antibodies were purchased from Santa Cruz Biotech (CA, USA) and anti-phospho-antibodies were from Cell Signaling (MA, USA).
Animals, cell cultures, transfection, Tα1 treatment, enzyme-linked immunosorbent assays and RT–PCR. Ikkβ−/−Tnfr1−/− mice were generated as described previously (Chu et al, 2000; Senftleben et al, 2001). C57BL6 mice were from Charles River (NY, USA). BMDMs were prepared as described previously (Chu et al, 2000). Before use, BMDMs were seeded (2.5 × 105 per well in triplicate) in 96-well plates and then treated with Tα1 or sTα1 (25–100 ng/ml). After 24 h, supernatant samples were collected and assessed for IL-6 and IL-12 using enzyme-linked immunosorbent assay kits (PharMingen, CA, USA). MEFs were cultured in DMEM supplemented with 10% FBS and antibiotics. Cell-transfection experiments were performed using Lipofectamine plus reagents or Lipofectamine2000 according to the manufacturer's instructions (Invitrogen, CA, USA). RT–PCR for IL-6 and IL-12 was performed as described previously (Chu et al, 2000). PKCζ siRNA transfection was performed according to the manufacturer's instructions (Upstate, NY, USA).
Immunoprecipitation, kinase assays and immunoblotting assays. Lysates were prepared and incubated overnight with antibodies at 4°C. Immunoprecipitates were washed and used for kinase assays. Kinase assays and IB analyses were performed as described previously (Chu et al, 2000).
Acknowledgments
We thank Dr C. Biron and Dr G. Yap for critical comments. We also thank Dr P. Shank and Dr T. Mak for reagents and Dr J Wands for advice. This study was supported by National Institutes of Health (AI 54128-01 to W.M.C. and AI043477 to M.K.) and SciClone Pharmaceuticals Inc. W.M.C. is a scholar of the American Liver Foundation.
References
- Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4: 499–511 [DOI] [PubMed] [Google Scholar]
- Baumann CA, Badamchian M, Goldstein AL (1997) Thymosin α1 antagonizes dexamethasone and CD3-induced apoptosis of CD4+CD8+ thymocytes through the activation of cAMP and protein kinase C dependent second messenger pathways. Mech Age Dev 94: 85–101 [DOI] [PubMed] [Google Scholar]
- Billich A (2002) Thymosin α1 (SciClone Pharmaceuticals). Curr Opin Investig Drugs 3: 698–707 [PubMed] [Google Scholar]
- Chu WM, Ostertag D, Li ZW, Chang L, Chen Y, Hu Y, Williams B, Perrault J, Karin M (1999) JNK2 and IKKβ are required for activating the innate response to viral infection. Immunity 11: 721–731 [DOI] [PubMed] [Google Scholar]
- Chu WM et al. (2000) DNA-PKcs is required for activation of innate immunity by immunostimulatory DNA. Cell 103: 909–918 [DOI] [PubMed] [Google Scholar]
- Chuang TH, Lee J, Kline L, Mathison JC, Ulevitch RJ (2002) Toll-like receptor 9 mediates CpG-DNA signaling. J Leukoc Biol 71: 538–544 [PubMed] [Google Scholar]
- Duran A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP, Moscat J, Diaz-Meco MT (2003) The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell 6: 303–309 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M (2002) Missing pieces in the NF-κB puzzle. Cell 109: S81–S96 [DOI] [PubMed] [Google Scholar]
- Knutsen AP, Freeman JJ, Mueller KR, Roodman ST, Bouhasin JD (1999) Thymosin-α1 stimulates maturation of CD34+ stem cells into CD3+4+ cells in an in vitro thymic epithelia organ coculture model. Int J Immunopharmacol 21: 15–26 [DOI] [PubMed] [Google Scholar]
- Kobayashi T et al. (2003) TRAF6 is a critical factor for dendritic cell maturation and development. Immunity 19: 353–363 [DOI] [PubMed] [Google Scholar]
- Lallena MJ, Diaz-Meco MT, Bren G, Paya CV, Moscat J (1999) Activation of IKKβ by protein kinase C isoforms. Mol Cell Biol 19: 2180–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M, Sanz L, Martin P, Duran A, Braun U, Garcia JF, Camacho F, Diaz-Meco MT, Rennert PD, Moscat J (2001) Targeted disruption of the ζPKC gene results in the impairment of the NF-κB pathway. Mol Cell 8: 771–780 [DOI] [PubMed] [Google Scholar]
- Low TLK, Thurman GB, McAdoo M, McClure J, Rossio JL, Naylor PH, Goldstein AL (1979) Isolation, characterization, and biological activities of thymosin α1 and polypeptide β1 from calf thymus. J Biol Chem 254: 981–986 [PubMed] [Google Scholar]
- Romani L et al. (2004) Thymosin α1 activates dendritic cells for antifungal Th1 resistance through toll-like receptor signaling. Blood 103: 4232–4239 [DOI] [PubMed] [Google Scholar]
- Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J (1999) The interaction of p62 with RIP links the atypical PKCs to NF-κB activation. EMBO J 18: 3044–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Diaz-Meco MT, Nakano H, Moscat J (2000) The atypical PKC-interacting protein p62 channels NF-κB activation by the IL-1–TRAF6 pathway. EMBO J 19: 1576–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarandeses CS, Covelo G, Diaz-Jullien C, Freire M (2003) Prothymosin α is processed to thymosin α1 and thymosin α11 by a lysosomal asparaginyl endopeptidase. J Biol Chem 278: 13286–13293 [DOI] [PubMed] [Google Scholar]
- Senftleben U, Li ZW, Baud V, Karin M (2001) IKKβ is essential for protecting T cells from TNFα-induced apoptosis. Immunity 14: 217–230 [DOI] [PubMed] [Google Scholar]
- Wang SS, Makofske R, Bach A, Merrifield RB (1980) Automated solid phase synthesis of thymosin α1. Int J Peptide Protein Res 15: 1–4 [DOI] [PubMed] [Google Scholar]
- Welch RA, Lee HH, Sokol RJ, Mutchnick MG (1988) Amniotic fluid thymosin α1 levels increase during gestation. Am J Reprod Immunol Microbiol 17: 96–97 [DOI] [PubMed] [Google Scholar]