

Abstract
Histone acetylation regulates many chromosome functions, such as gene expression and chromosome segregation. Histone deacetylase inhibitors (HDACIs) induce growth arrest, differentiation and apoptosis of cancer cells ex vivo, as well as in vivo in tumour-bearing animal models, and are now undergoing clinical trials as anti-tumour agents. However, little attention has been paid to how HDACIs function in these biological settings and why different cells respond in different ways. Here, we discuss the consequences of inhibiting histone deacetylases in cycling versus non-cycling cells, in light of the dynamics of histone acetylation patterns with a specific emphasis on heterochromatic regions of the genome.
Keywords: chromatin, histone deacetylase inhibitors, nuclear organization, pericentric heterochromatin, anticancer therapy
Introduction
It is well established that histone modifications influence chromatin state and genome functions. These modifications, which include acetylation, phosphorylation, methylation and ubiquitination, can generate synergistic or antagonistic interaction affinities for chromatin-associated proteins (Jenuwein & Allis, 2001). Two of the most studied modifications are the acetylation and deacetylation of lysines in the core histones, which are controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. The essential role of histone acetylation and deacetylation in normal cell growth is supported by the frequent alterations in the structure or expression of HATs and HDACs in many cancers. HDAC inhibitors (HDACIs) are thus undergoing clinical trials as anticancer agents (McLaughlin & La Thangue, 2004). Initially, a model was proposed in which, in euchromatin, HDACI-mediated chromatin changes at gene promoters activate a specific gene, or set of genes, responsible for cell-cycle alteration, progression or apoptosis. However, transcriptome analysis showed that only a small number (2%–5%) of genes are affected (Glaser et al, 2003; Van Lint et al, 1996), raising the possibility of non-histone (Drummond et al, 2005; Marks et al, 2004), or non-euchromatic, targets for these agents, which we discuss here. In particular, the histone acetylation state of constitutive heterochromatin—the site of satellite repeats in the mammalian genome—is important for chromosome segregation. Pericentric heterochromatin is marked by distinctive histone modifications, such as histone hypoacetylation (Fig 1; Maison & Almouzni, 2004).
Figure 1.
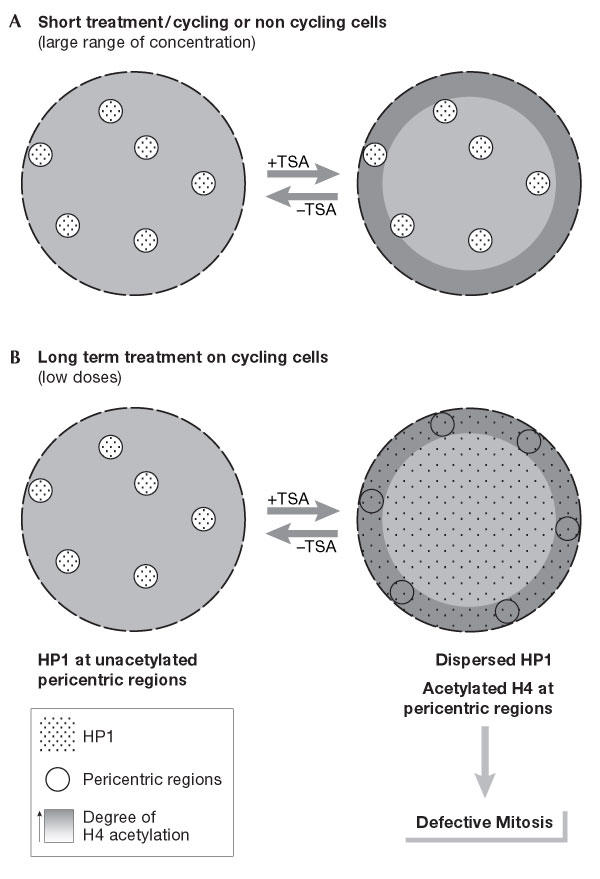
Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. (A) A very strong and rapid (within 90 min) enhancement of acetylated forms of H4 at the nuclear periphery is observed with a wide range of doses of histone deacetylase inhibitors (HDACIs) applied on a variety of cell types (Taddei et al, 1999). (B) Pericentric heterochromatin in cycling cells (immortalized human HeLa and U2OS, as well as mouse L929 cells) is specifically responsive to prolonged treatment with deacetylase inhibitors. At low doses, these defined regions relocate at the nuclear periphery and lose their properties of retaining heterochromatin protein 1 (HP1) proteins, which are spread throughout the nucleoplasm. Subsequent defects in chromosome segregation arise in mitosis. All these changes can reverse rapidly after drug removal (Taddei et al, 2001).
The effect of different HDACIs on pericentric heterochromatin structure and mitotic function in fission yeast, mouse and human transformed cells leads to chromosome segregation defects in mitosis (Ekwall et al, 1997; Taddei et al, 2001). Pericentric heterochromatin in proliferating mouse cells relocates at the nuclear periphery, and loses its concentration of heterochromatin protein 1 (HP1) on prolonged exposure (several days) to low doses of HDACIs (Fig 1B). Intriguingly, such an effect on centromere nuclear localization is not observed when the HDACI trichostatin A (TSA) is used for a short time or at high doses on non-proliferating cells (Gilchrist et al, 2004).
In this review, we consider the basis for such differences in sensitivity to HDACIs, especially now that these drugs are in clinical trials. In particular, we consider the importance of the proliferation state of cells and the integrity of their checkpoint status. These issues are discussed in the context of our current knowledge of the dynamics of histone acetylation patterns during the cell cycle at heterochromatic regions.
HDAC and HDAC inhibitors
The classification of mammalian HDACs is based on structural homologies with the three distinct yeast HDACs (de Ruijter et al, 2003): reduced potassium dependency 3 (Rpd3; class I), histone deacetylase 1 (Hda1; class II) and silencing information regulator 2 (Sir2)/homologue of Sir2 (Hst; class III). Class III HDACs have a unique enzymatic mechanism that depends on the cofactor NAD+, and are virtually unaffected by the HDACIs that are now in clinical trials. Here, we focus on class I and class II HDACs.
The most potent of the known inhibitors is TSA, which belongs to the group of hydroxamic acids and is active at nanomolar concentrations in vitro. TSA has a similar Ki for all HDAC isoforms that have been examined. Except for trapoxin and depudesin, which irreversibly inhibit the enzyme through covalent binding, all HDACIs act reversibly; this may explain their transient effect in clinical applications.
Not all HDACs are equally sensitive to the different HDACIs (Drummond et al, 2005). Thus, not all HDACIs should be considered alike, and we should be cautious when interpreting their effects in vivo as these depend on the cell type and the organism in question. The fact that their activity could also involve non-histone targets should not be ignored. Thus, given their lack of overall specificity towards HDACs, the continued identification of isoformspecific inhibitors that are targeted to histones will undoubtedly remain a major emphasis of HDACI development.
Histone dynamics and chromatin modifications
Histones in chromatin can be modified in at least two ways: (i) histones in a nucleosomal form can be modified in situ, or (ii) histones in their soluble form can be modified before their incorporation into chromatin or concomitantly with their deposition. Both of these methods must be taken into account when interpreting the results of TSA treatment. First, the acetylation state of a chromatin locus can result from the antagonist activities of HATs and HDACs on pre-existing nucleosomes. This occurs independently of replication and of the proliferative state of the cell. Alternatively, de novo modification of free histones and their direct incorporation should be considered. A replication-dependent pathway of histone acetylation is particularly crucial for newly synthesized histone H4 acetylated at lysines 5 and 12 (Fig 2; Sobel et al, 1995). Fluorescence recovery after photobleaching (FRAP) and biochemical analysis at a global level show that core histones H3 and H4 have a limited turnover outside replication time (Kimura & Cook, 2001). During replication, newly synthesized DNA is rapidly wrapped into nucleosomes that consist of a mixture of parental and newly synthesized histones (Sogo & Laskey, 1995). The newly synthesized histone H4 is easily identified by its characteristic diacetylated state (Fig 2B). In addition, the incorporation of acetylated histone may occur outside S phase using the histone variant H3.3 instead of the replicative histone variants H3.1 and H3.2, which are incorporated only during S phase (Fig 2C; Kamakaka & Biggins, 2005). Intriguingly, H3.3 has been reported as being enriched in modifications that are associated with transcriptionally active chromatin, such as acetylation, as shown on extracted H3.3 in Drosophila cells (McKittrick et al, 2004). This offers another means by which the pattern of chromatin modifications could be switched.
Figure 2.
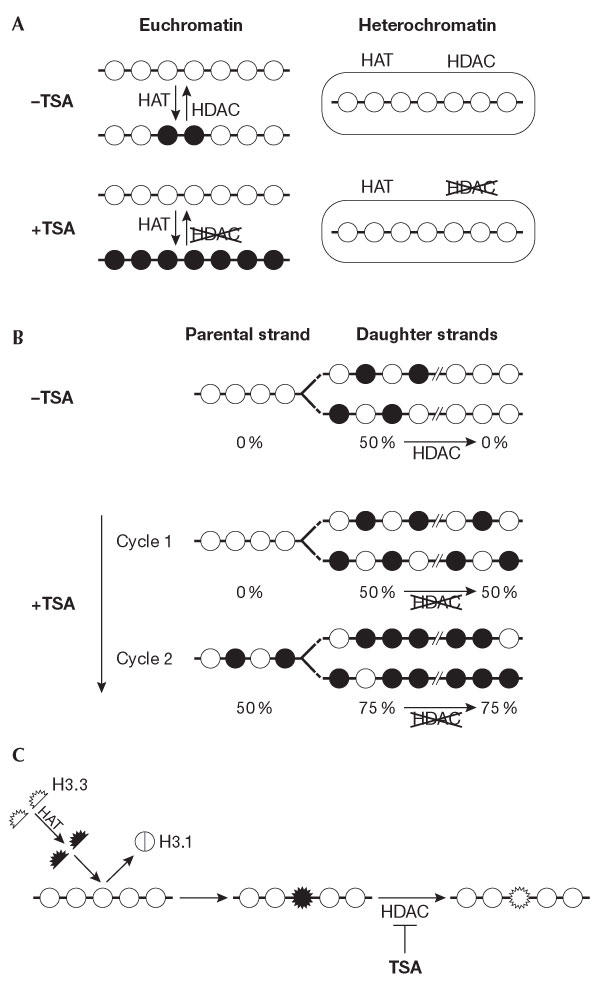
Different dynamics of histone acetylation. (A) Replication-independent dynamics. The acetylation state of a chromatin locus results from the antagonist activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs) on pre-existing nucleosomes. These two activities are locally regulated. Indeed, inhibiting deacetylases by trichostatin A (TSA) stabilizes the acetylation state of histone H4 in euchromatin regions with a very strong efficiency at the nuclear periphery, whereas it has no effect on heterochromatin regions (Taddei et al, 2001). (B) Replication-dependent dynamics of histone acetylation. After DNA replication, DNA daughter strands are rapidly wrapped into nucleosomes that contain a mixture of parental and newly synthesized histones. The bulk of newly synthesized histone H4 is acetylated at lysines 5 and 12 by a cytoplasmic HAT and is incorporated into new chromatin in this form. During late S phase, the acetates at H4 lysines 5 and 12 are rapidly lost in heterochromatin regions, probably owing to the presence of an HDAC at the site of chromatin assembly that can be inhibited by TSA treatment. This inhibition will lead to the maintenance of the acetylated state of newly incorporated histones, which dilutes by twofold the non-acetylated parental histones at each round of replication in the presence of TSA (Taddei et al, 1999). (C) Dynamics of histone acetylation by histone exchange. The variant H3.3 (expressed throughout the cell cycle) can be deposited into chromatin at any time. It has been shown that H3.3 is enriched in modifications that are associated with transcriptional activity. We propose that it can be acetylated before incorporation, thus providing another means to change chromatin patterns. The action of HDACIs that is envisaged in this context is depicted.
Effects of HDACIs on pericentric heterochromatin
On treatment with HDACIs such as TSA, a zone of histone hyperacetylation becomes visible at the nuclear periphery, which is distinct from heterochromatin. A wide range of concentrations triggers a rapid response that leads to a visible enhancement at the nuclear periphery of acetylation on H3 and H4 for a large variety of cell types, including primary fibroblasts, telomerase-immortalized fibroblasts, HT1080 fibrosarcoma cells, human HeLa cells, mouse L929 cells, and Xenopus A6 cells and nuclei from embryos after gastrulation (Fig 1A; Gilchrist et al, 2004; Taddei et al, 1999). Although these effects are similar across many cell types for a wide range of TSA concentrations, the exact mechanism that underlies this phenomenon remains to be elucidated. Notably, constitutive heterochromatic regions, as defined by their late replication timing and HP1 enrichment, are excluded from this hyperacetylated domain (Fig 1B). This suggests that brief TSA treatment does not allow the stabilization of any of the acetylated forms of histone H4, and that most HATs do not normally have access to heterochromatin regions.
TSA can, however, influence histone acetylation at pericentric heterochromatin regions. This is observed when using low doses (160 nM for HeLa cells and 80 nM for L929 mouse cells) and requires several cell cycles. Indeed, one cell cycle in the presence of TSA was not sufficient to detect any visible effect on a heterochromatin marker such as HP1 or centromere localization (Taddei et al, 2001). However, after two or three cell cycles in the presence of TSA, increased levels of histone acetylation were observed at centromeric regions by chromatin immunoprecipitation (ChIP; Maison et al, 2002), HP1 spreading, centromere relocalization and destabilization of the higher-order structure of pericentric heterochromatin (Fig 1B).
The gradual response of heterochromatin to the effects of low-dose TSA treatment can be explained by the different dynamics of histone H4 acetylation, which are restricted to the time of DNA replication for heterochromatin (Taddei et al, 1999). The presence of TSA at the time of heterochromatin replication stabilizes the diacetylated form of newly synthesized histone H4 that is incorporated there (Fig 2B; Taddei et al, 1999). But it should be noted that only half of the H4 histones in heterochromatin will be diacetylated following TSA treatment after the first round of replication, because newly formed nucleosomes are made of parental as well as newly synthesized histones (Fig 2B). The nature of the HDAC that is targeted at heterochromatin during late replication is unknown. It is interesting to note that the inactivation of mouse suppressor of defective silencing 3 (mSds3), which is an essential component of the switch independent 3 (mSin3)/HDAC co-repressor complex, results in the alteration of pericentric heterochromatinspecific modifications and HP1 protein association, the formation of micronuclei and aberrant cell-cycle profiles in primary mouse embryonic fibroblast cells (David et al, 2003). Importantly, Sin3 is concentrated specifically at active kinetochores of mouse and human chromosomes (Craig et al, 2003).
We therefore propose that the concentration of parental histones must be diluted to generate a visible effect on the nuclear organization of pericentric heterochromatin regions. The use of HDACIs at high concentration (320 nM) prevents this by inducing immediate cell-cycle arrest on proliferating cells, and is not observed at all on quiescent cells (Gilchrist et al, 2004). Similar observations were reported in fission yeast (Ekwall et al, 1997), in which several cell cycles in the presence of TSA were required to observe an effect on chromosome segregation and localization of the HP1 homologue Swi6 in centromeric regions. Defective mitosis is also observed immediately after treatment of human lung diploid fibroblast cells with a high dose of TSA (1,600 nM), but in this case cells are blocked in mitosis, probably due to checkpoint activation, and cannot undergo several cell cycles (Cimini et al, 2003).
In addition to the effect of HDACIs in constitutive heterochromatin, as observed in centromeric regions, it is also important to consider the effect in facultative heterochromatin, such as the inactive X chromosome in female mammals. This may be interesting to explore given the fact that the breast and ovarian tumour suppressor BRCA1 (breast cancer early onset 1) has been implicated in the inactivation of the X chromosome (Ganesan et al, 2002). The effect of HDACI treatment on mouse female embryonic stem cells is difficult to interpret because the absence of normal X inactivation could be due to a defect in differentiation, an arrest in G1 or even cell death in the treated population (O'Neill et al, 1999). TSA treatment of differentiated mouse embryonic fibroblast cells did not seem to have any effect on an inactive X-linked green fluorescent protein (GFP) reporter gene, although a synergistic effect was reported when TSA was combined with the DNA methyl transferase inhibitor 5-azadC and the deletion of Xist, an RNA that is encoded on the X chromosome and has a key role in its inactivation (Csankovszki et al, 2001). This latter case illustrates potential differences in the response to TSA in different types of heterochromatin. Thus, in the context of constitutive pericentric heterochromatin, the capacity to proliferate is an important parameter to consider, but a general extrapolation to all types of heterochromatin is not necessarily valid.
Implications for HDACIs as therapeutic agents
The different responses of cycling and non-cycling cells to HDACI treatment is of major interest because these proteins are considered to be a promising new class of chemotherapeutic drug and are now in early-phase clinical trials. Cell types respond differently to varying amounts of HDACIs (Taddei et al, 2001) and several inhibitors including TSA have been shown to induce cell-cycle arrest and cell death at high doses (Yoshida et al, 1995). In addition, inhibiting HDACs in a developing organism has different consequences depending on the stage of development, with the early gastrulation stage appearing to be a specifically sensitive time period in both starfish and Xenopus development (Almouzni et al, 1994; Ikegami et al, 1993). Interestingly, this time period corresponds to a transition in cell-cycle and checkpoint properties. Thus, it becomes increasingly clear that in addition to the proliferative status of cells, other parameters are crucial for determining sensitivity to HDACIs.
One reason for the tumour-selective cytotoxicity of HDACIs when used at high doses (500 nM TSA or equivalent) is thought to be the disruption of two cell-cycle checkpoints (Warrener et al, 2003). The first is the G2-phase checkpoint, which allows cells to enter an aberrant mitosis even if the G2 phase is not properly completed. On treatment with TSA, cells that are likely to go through an aberrant G2 phase with defective chromatin will thus die. This checkpoint is independent of the ataxia telangiectasia mutated (ATM) DNA damage checkpoint and involves the p38 mitogen-activated protein kinase (MAPK) checkpoint, which is also triggered by topoisomerase II inhibitors (Mikhailov et al, 2004). The second checkpoint is the mitotic spindle checkpoint, which normally detects aberrant mitosis and blocks mitotic exit until the defect is rectified. The disruption of both checkpoints results in the premature exit of cells from an abortive mitosis, followed by apoptosis. Recently, it was reported that HDACI sensitivity in a specific type of leukaemia is a property of fully transformed cells and is mediated by a specific death pathway that involves the tumour necrosis factor-related apoptosis-inducing ligand (TRAIL; Insinga et al, 2005; Nebbioso et al, 2005). The mechanism responsible for the cancer selectivity of the TRAIL response to HDACIs and how these drugs function in other cancer cells must be explored. Interestingly, the initiation of neuroblastoma caspase-dependent cell death by HDACI is mediated by the acetylation of ku70—an auto-antigen that is involved in DNA repair—which leads to the activation of the mitochondria-centred apotoptic response (Subramanian et al, 2005). This shows that different mechanisms can account for HDACI-induced cell death in different cell types. Most importantly, from a clinical point of view, it will be crucial to establish predictive markers that can determine which tumours are responsive to a specific drug in order to optimize the use of that drug. In this respect, the most obvious marker is the acetylation status of chromatin in the tumour cells.
Indeed, the tumour selectivity of HDACIs could be related to global chromatin modifications that are associated with full oncogenic transformation. In support of this hypothesis, a recent report shows that tumour cells undergo an overall loss of monoacetylation on histone H4 at lysine 16 and a trimethylation on histone H4 at lysine 20, which is predominantly associated with repetitive sequences such as pericentric heterochromatin repeats (Fraga et al, 2005). This global change in chromatin structure could render some function of the genome more sensitive to HDACIs, which in turn could affect the activation of key genes and also the integrity of heterochromatin. We speculate that mechanisms of mitosis or of genome surveillance, such as DNA repair or checkpoint, may monitor the proper heterochromatic state of the cells. When the heterochromatin state is altered, this mechanism would allow cells to go through mitotic catastrophe or other types of cell death.
We are still far from understanding the exact mechanisms by which HDACIs can affect cancer cells. Here, we draw attention to possible non-transcriptionally mediated effects that should be considered potential HDACI targets. A variety of cellular mechanisms that lead to cell death can be exploited by treating these targets with HDACIs. The outcome of the treatment will depend on the exact defects of the cancer cell itself, which can be a combination of genetic and epigenetic changes. Indeed, epigenetic control includes histone modifications and DNA methylation. These epigenetic marks are established during differentiation but some are lost during cell transformation, which could explain why undifferentiated cells as well as transformed cells are specifically sensitive to HDACIs.
Acknowledgments
We thank E. Heard for critical reading of the manuscript. G.A. and W.A.B. are supported by the Network of Excellence Epigenome (LSHG-CT-2004-503433). G.A.'s team is supported by La Ligue Nationale Contre le Cancer (Equipe labellisée 2004), the Commissariat à l'Energie Atomique (LRC no.26), European Contract RTN (HPRN-CT-2002-00238) and Collaborative Programme between the Curie Institute and the Commisariat à l'Energie Atomique (PIC Paramètres Epigénétiques).
References
- Almouzni G, Khochbin S, Dimitrov S, Wolffe AP (1994) Histone acetylation influences both gene expression and development of Xenopus laevis. Dev Biol 165: 654–669 [DOI] [PubMed] [Google Scholar]
- Cimini D, Mattiuzzo M, Torosantucci L, Degrassi F (2003) Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol Biol Cell 14: 3821–3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig JM, Earle E, Canham P, Wong LH, Anderson M, Choo KH (2003) Analysis of mammalian proteins involved in chromatin modification reveals new metaphase centromeric proteins and distinct chromosomal distribution patterns. Hum Mol Genet 12: 3109–3121 [DOI] [PubMed] [Google Scholar]
- Csankovszki G, Nagy A, Jaenisch R (2001) Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol 153: 773–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Turner GM, Yao Y, Protopopov A, DePinho RA (2003) mSin3-associated protein, mSds3, is essential for pericentric heterochromatin formation and chromosome segregation in mammalian cells. Genes Dev 17: 2396–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370: 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC (2005) Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol 45: 495–528 [DOI] [PubMed] [Google Scholar]
- Ekwall K, Olsson T, Turner BM, Cranston G, Allshire RC (1997) Transient inhibition of histone deacetylation alters the structural and functional imprint at fission yeast centromeres. Cell 91: 1021–1032 [DOI] [PubMed] [Google Scholar]
- Fraga MF et al. (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37: 391–400 [DOI] [PubMed] [Google Scholar]
- Ganesan S et al. (2002) BRCA1 supports XIST RNA concentration on the inactive X chromosome. Cell 111: 393–405 [DOI] [PubMed] [Google Scholar]
- Gilchrist S, Gilbert N, Perry P, Bickmore WA (2004) Nuclear organization of centromeric domains is not perturbed by inhibition of histone deacetylases. Chromosome Res 12: 505–516 [DOI] [PubMed] [Google Scholar]
- Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK (2003) Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther 2: 151–163 [PubMed] [Google Scholar]
- Ikegami S, Ooe Y, Shimizu T, Kasahara T, Tsuruta T, Kijima M, Yoshida M, Beppu T (1993) Accumulation of multiacetylated forms of histones by trichostatin A and its developmental consequences in early starfish embryos. Rouxs Arch Dev Biol 202: 144–151 [DOI] [PubMed] [Google Scholar]
- Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, Altucci L, Nervi C, Minucci S, Pelicci PG (2005) Inhibitors of histone deacetylases induce tumorselective apoptosis through activation of the death receptor pathway. Nat Med 11: 71–76 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Kamakaka RT, Biggins S (2005) Histone variants: deviants? Genes Dev 19: 295–310 [DOI] [PubMed] [Google Scholar]
- Kimura H, Cook PR (2001) Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J Cell Biol 153: 1341–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison C, Almouzni G (2004) HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol 5: 296–304 [DOI] [PubMed] [Google Scholar]
- Maison C, Bailly D, Peters AH, Quivy JP, Roche D, Taddei A, Lachner M, Jenuwein T, Almouzni G (2002) Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet 30: 329–334 [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Miller T, Kelly WK (2004) Histone deacetylase inhibitors. Adv Cancer Res 91: 137–168 [DOI] [PubMed] [Google Scholar]
- McKittrick E, Gafken PR, Ahmad K, Henikoff S (2004) Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc Natl Acad Sci USA 101: 1525–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin F, La Thangue NB (2004) Histone deacetylase inhibitors open new doors in cancer therapy. Biochem Pharmacol 68: 1139–1144 [DOI] [PubMed] [Google Scholar]
- Mikhailov A, Shinohara M, Rieder CL (2004) Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J Cell Biol 166: 517–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebbioso A et al. (2005) Tumorselective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med 11: 77–84 [DOI] [PubMed] [Google Scholar]
- O'Neill LP, Keohane AM, Lavender JS, McCabe V, Heard E, Avner P, Brockdorff N, Turner BM (1999) A developmental switch in H4 acetylation upstream of Xist plays a role in X chromosome inactivation. EMBO J 18: 2897–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel RE, Cook RG, Perry CA, Annunziato AT, Allis CD (1995) Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc Natl Acad Sci USA 92: 1237–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo JM, Laskey RA (1995) Chromatin replication and assembly. In Elgin SCR (ed), Chromatin Structure and Gene Expression pp 49–71. New York, NY, USA: IRL [Google Scholar]
- Subramanian C, Opipari AW Jr, Bian X, Castle VP, Kwok RP (2005) Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA 102: 4842–4847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Roche D, Sibarita JB, Turner BM, Almouzni G (1999) Duplication and maintenance of heterochromatin domains. J Cell Biol 147: 1153–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Maison C, Roche D, Almouzni G (2001) Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat Cell Biol 3: 114–120 [DOI] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Verdin E (1996) The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr 5: 245–253 [PMC free article] [PubMed] [Google Scholar]
- Warrener R, Beamish H, Burgess A, Waterhouse NJ, Giles N, Fairlie D, Gabrielli B (2003) Tumor cellselective cytotoxicity by targeting cell cycle checkpoints. FASEB J 17: 1550–1552 [DOI] [PubMed] [Google Scholar]
- Yoshida M, Horinouchi S, Beppu T (1995) Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays 17: 423–430 [DOI] [PubMed] [Google Scholar]