

Abstract
EMSY is a large nuclear protein that binds to the transactivation domain of BRCA2. EMSY contains an ∼100-residue segment at the amino terminus called the ENT (EMSY N-terminal) domain. Plant proteins containing ENT domains also contain members of the royal family of chromatin-remodelling domains. It has been proposed that EMSY may have a role in chromatin-related processes. This is supported by the observation that a number of chromatin-regulator proteins, including HP1β and BS69, bind directly to EMSY by means of a conserved motif adjacent to the ENT domain. Here, we report the crystal structure of residues 1–108 of EMSY at 2.0 Å resolution. The structure contains both the ENT domain and the HP1β/BS69-binding motif. This binding motif forms an extended peptide-like conformation that adopts distinct orientations in each subunit of the dimer. Biophysical and nuclear magnetic resonance analyses show that the main complex formed by EMSY and the chromoshadow domain of HP1 (HP1-CSD) consists of one EMSY dimer sandwiched between two HP1-CSD dimers. The HP1β-binding motif is necessary and sufficient for EMSY to bind to the chromoshadow domain of HP1β.
Keywords: EMSY, HP1β, chromoshadow domain, heterochromatin, X-ray crystallography
Introduction
The recent discovery of EMSY represents an important link between BRCA2 and the development of sporadic breast and ovarian cancer (Hughes-Davies et al, 2003). Although little is known about the cellular role of EMSY, indirect evidence suggests that this nuclear protein is involved in DNA-damage response and chromatin remodelling, in partnership with HP1β, BS69 and BRCA2. EMSY contains an unusual domain called ENT that directly associates with BRCA2. Analysis of the human genome showed that the ENT domain is found only in EMSY, suggesting that this conserved protein module has a specific cellular role (Hughes-Davies et al, 2003; Maurer-Stroh et al, 2003). The first 98 residues of EMSY comprise the ENT domain, which is composed of a unique arrangement of five α-helices that fold into a helical bundle formation (Chavali et al, 2005). The ENT domain forms a homodimer by the antiparallel packing of the long amino-terminal α-helix from each subunit. It is stabilized mainly by hydrophobic residues at the dimer interface (Chavali et al, 2005).
EMSY interacts with the chromoshadow domain of HP1β (HP1-CSD; Hughes-Davies et al, 2003). Deletion mapping narrowed the interaction site within EMSY to a region between residues 80 and 140, showing an HP1-binding consensus site adjacent to the ENT domain (Fig 1A). Mutagenesis of residues within the VxL motif of EMSY abolished the interaction with HP1-CSD (Hughes-Davies et al, 2003). In addition, a binding motif for BS69 (PxLxP) was found at amino acids 104–108 of EMSY (Fig 1A; Hughes-Davies et al, 2003). Mutation of the central leucine residue of the motif ablated the interaction with BS69 (Hughes-Davies et al, 2003). This region shares significant sequence similarity with the HP1β-binding site of the chromatin-assembly factor-1 (CAF-1) protein (Fig 1A).
Figure 1.
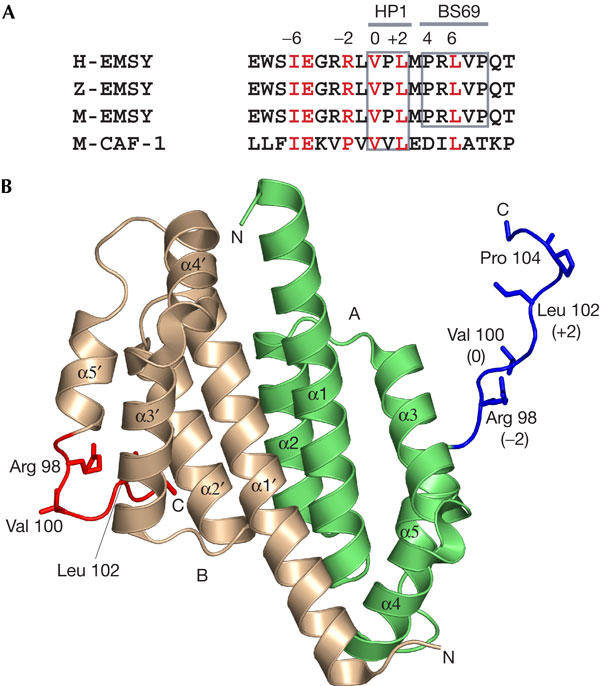
Structure of the HP1β- and BS69-binding region of EMSY. (A) Sequence alignment of the HP1- and BS69-binding platform of human EMSY (H) with the homologous region of zebra fish (Z) and mouse (M) EMSY proteins. The HP1β-binding region of mouse CAF-1 is also aligned. (B) Structure of EMSY1–108 showing the extended region identified in blue and red for chains A and B, respectively. Chain A is shown in green and chain B in the colour of wheat. Residues 98, 100, 102 and 104 are shown in a stick representation.
To understand the architecture of the HP1β/BS69-binding (HB) platform, we determined the crystal structure of EMSY1–108. This N-terminal fragment contains the ENT domain (residues 1–97) and the HB platform (residues 98–108), a region that has been implicated in binding the chromoshadow domain of HP1β (Brasher et al, 2000) and the Mynd domain of BS69 (Ansieau & Leutz, 2002). In addition, we present biophysical and nuclear magnetic resonance (NMR) studies on the interaction between EMSY and HP1β and show the relationship between the dimerization domain of EMSY and its HP1β interaction partner. These findings have important implications in understanding the network of protein complexes that regulate chromatin structure.
Results and Discussion
Structure of the HP1-binding platform of EMSY
The crystal structure of an N-terminal fragment of EMSY comprising residues 1–108 (EMSY1–108) was determined to 2.0 Å resolution. The architecture of a dimer of EMSY1–108 is shown in Fig 1B. The overall structure of EMSY1–108 is similar to the structure of the ENT domain (Chavali et al, 2005) and consists of a homodimer. There is one dimer in the asymmetric unit. The N termini of the two structures show some differences. The first ten residues of EMSY1–108 were disordered and could not be modelled into the two chains. Residues 101–106 could be modelled into chain A of EMSY1–108 and only 101–104 could be identified from the electron density map of chain B.
The HP1/BS69-binding platform of EMSY starts at Arg 97, which is located at the end of helix α5 of the ENT domain, adopting a random coil conformation twisting into a β-turn at residue Pro 104 (Fig 1B). The HB platform adopts two distinct orientations in the EMSY1–108 homodimer structure. In one molecule of the dimer, the platform extends away from the carboxyl terminus of the ENT domain into the surrounding solvent, making the HP1/BS69-binding site fully accessible to incoming proteins (Fig 1B). In the other molecule, the HB platform interacts directly with the core of the ENT domain. The remainder of the structure is identical in conformation to EMSY1–100 and could be superimposed to an r.m.s.d. of ∼0.7 Å. The two monomers in EMSY1–108 superposed with an r.m.s.d. of 0.5 Å.
A sequence adjacent to the ENT domain binds HP1
The interaction between EMSY and the dimeric HP1-CSD was investigated using biophysical methods. Analytical gel filtration was performed with EMSY1–108 and HP1-CSD at various concentrations. In addition to the elution peaks corresponding to the free dimers of EMSY1–108 and HP1-CSD, two higher-molecular-mass peaks were observed (Fig 2A). Analytical ultra-centrifugation (AUC) experiments were performed under four different conditions—1.5 and 15 μM EMSY1–108, and an equimolar concentration or a twofold excess of HP1-CSD. HP1-CSD alone sedimented with an apparent molecular mass of 15,000 Da (data not shown), which is slightly less than the calculated mass of the dimer (16,200 Da), suggesting that it has a dissociation constant Kd in the submicromolar range. This is consistent with the Kd that was measured previously (Brasher et al, 2000). The apparent mass of the mixture of EMSY and HP1-CSD proteins versus total mass concentration is shown in Fig 2B. We were not able to deconvolute the individual contributions of EMSY and HP1-CSD to the sedimentation profile. The global fitting of the data to the sum of the two components yielded the observed masses of 22,000 and 56,000 Da, which is consistent with the dimer of EMSY (25,000 Da) and a 2:4 complex of EMSY:HP1-CSD (57,000 Da). However, because of the complexity of the system (both EMSY and HP1-CSD are dimers and may dissociate under these conditions), we cannot exclude the presence of the 2:2 complex. In summary, the analytical gel filtration and AUC results indicate that there are two complexes present in solution, with 2:2 and 2:4 stoichiometries of EMSY1–108 and HP1-CSD.
Figure 2.
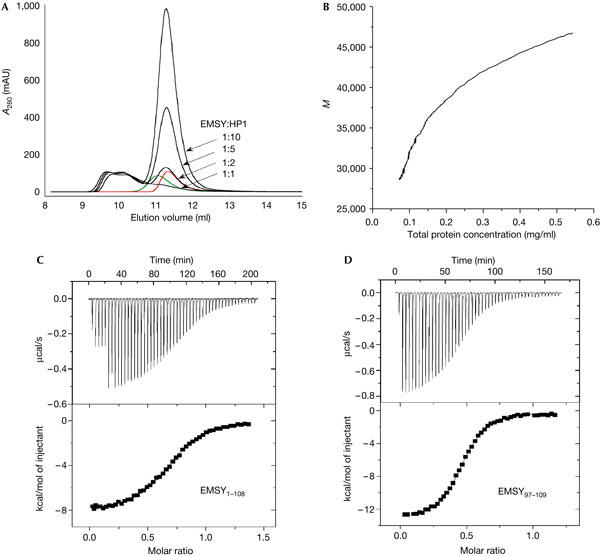
Stoichiometry of the EMSY:HP1-CSD complex. (A) Analytical gel filtration of EMSY and HP1-CSD. Dimers of EMSY1–108 (green) and HP1-CSD (red) eluted at 11.1 and 11.3 ml, respectively. The protein concentration was 50 μM. EMSY1–108 (50 μM) was mixed with HP1-CSD (50–500 μM) to give the ratios 1:1, 1:2, 1:5 and 1:10 (black) and incubated at 22°C for 2 h before being injected onto the column. Two complexes between EMSY1–108 and HP1-CSD are observed (eluting at 9.8 and 10.2 ml, respectively) and the equilibrium between them is not disrupted on addition of an excess of HP1-CSD. (B) Analytical ultracentrifugation. The apparent molecular mass (M) of the mixture of EMSY1–108 and HP1-CSD plotted versus the total mass concentration. (C,D) ITC analysis of the interaction between EMSY and HP1-CSD. The upper panels show the exothermic heat pulse of each injection and the lower panels show the integrated heat data fit to a single set of identical sites binding model. (a) EMSY1–108 (328 μM) titrated into HP1-CSD (54 μM). n=0.70±0.01 and Kd=1.8±0.1 μM. (b) EMSY97–109 (280 μM) titrated into HP1-CSD (54 μM). n=0.47±0.01 and Kd=1.0±0.1 μM. The data were collected at 20°C.
The binding affinity was determined by isothermal titration calorimetry (ITC), and the same value was obtained whether EMSY1–108 was titrated into HP1-CSD or vice versa and also using different concentrations of the two proteins. The isotherm fitted well to a model in which there is a single set of identical binding sites and gave a Kd of 1.8±0.1 μM (Fig 2C). When EMSY1–108 was titrated into HP1-CSD, the stoichiometry n was 0.70±0.01, which might reflect the presence of two complexes of 2:2 and 2:4 stoichiometries of EMSY1–108:HP1-CSD. Similarly, a value of 1.30±0.01 was obtained when HP1-CSD was titrated into EMSY1–108. No dissociation of the EMSY1–108 dimer or HP1-CSD dimer was detected in control experiments in which the same concentration of protein was titrated into buffer alone. Occupancy of only one of the two binding sites of the EMSY1–108 dimer was expected in both the AUC and gel filtration experiments, as they were carried out at protein concentrations around Kd. The ITC, however, was carried out under saturating conditions. The stoichiometry obtained by ITC could be explained by asymmetry in the binding platform of the two subunits of the dimer, as is indicated in the crystal structure. Alternatively, the intermediate stoichiometry may be the result of a proportion of misfolded protein. ITC experiments were also carried out on a longer EMSY fragment consisting of residues 1–140, and the value of n obtained was closer to 0.5, as expected for a stoichiometry of one EMSY dimer to two HP1-CSD dimers (data not shown).
Previously, residues Val 100 and Leu 102 of EMSY were shown to be important for binding the chromoshadow domain of HP1 (Hughes-Davies et al, 2003). Our studies show that the sequence adjacent to the ENT domain, containing these residues, is necessary and sufficient for high-affinity binding. First, a synthetic peptide corresponding to residues 97–109 of EMSY (Fig 1A) bound to HP1-CSD with the same affinity, measured by ITC, as did EMSY1–108 (Fig 2D). Second, no binding could be detected between HP1-CSD and the shorter EMSY1–100 construct that contains only the ENT domain (data not shown). The stoichiometry n of the EMSY97–109:HP1-CSD complex was 0.47±0.01, that is, one EMSY97–109 peptide binds to one HP1-CSD dimer. Finally, most of the cross-peaks in the two-dimensional (2D) 1H–15N heteronuclear single-quantum (HSQC) NMR spectrum of 15N-labelled EMSY1–108 were unchanged in the presence of HP1-CSD (data not shown). This suggests that binding to the adjacent HB platform does not induce any substantial change in the ENT domain structure.
Mapping the binding site of EMSY on HP1
Previously, it was reported that a mutation (I161E) in HP1-CSD, which disrupts the dimer interface to produce monomeric HP1-CSD, does not bind to a peptide derived from CAF-1 and other proteins containing a PxVxL motif (Brasher et al, 2000). The recent structure of a CAF-1 peptide binding across the symmetry axis of the HP1-CSD dimer explains why this mutant does not bind to PxVxL peptides (Thiru et al, 2004). If EMSY also recognizes HP1 across the dimer interface, then the I161E mutation should also negate the interaction. We produced the I161E mutant and measured its binding affinity for EMSY by analytical gel filtration and ITC. HP1-CSD I161E elutes as a monomer and no binding is observed for the mixture of the two proteins (Fig 3). ITC was performed with a mixture of EMSY1–108 and EMSY97–109 peptides by titrating them into HP1-CSD I161E under the same conditions as previously used for wild-type HP1-CSD. Neither of them bound to the recombinant HP1-CSD mutant (data not shown). These results confirm that EMSY binding, like CAF-1, also requires the formation of dimeric HP1-CSD and this supports the proposed model of the EMSY–HP1 complex.
Figure 3.
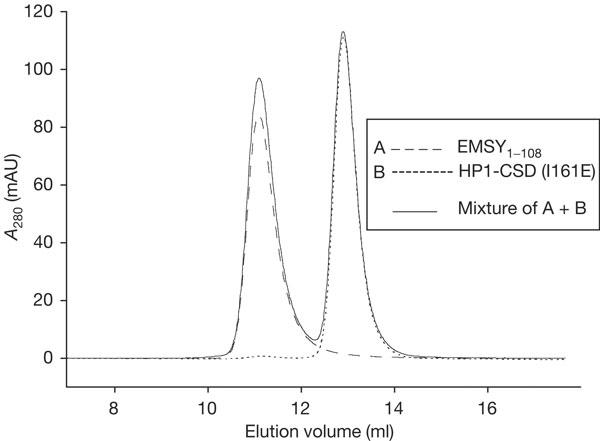
EMSY binds only to dimeric HP1β. Analytical gel filtration of EMSY and HP1-CSD I161E. The dimer of EMSY1–108 (broken line) eluted at 11.1 ml and the monomer of HP1-CSD I161E (dotted line) eluted at 12.9 ml. No shifts in the elution volumes were seen for the mixture of the two (solid line) at a ratio of 1:1, confirming that dimeric HP1-CSD is required for binding to EMSY.
To identify the residues on HP1 that interact with EMSY, we investigated the changes in the 2D 1H–15N HSQC NMR spectrum of 15N-labelled HP1-CSD in the presence of the EMSY97–109 peptide. Many cross-peaks were significantly shifted, and a subset gave two HSQC cross-peaks of almost equal intensity, with both shifted relative to the single cross-peak in the free protein (supplementary Fig 1A online). Similar behaviour was also observed previously for the interaction of HP1 with the CAF-1 peptide (Brasher et al, 2000). This is consistent with peptide binding changing HP1 from a symmetric dimer to an asymmetric dimer, as would be expected for the stoichiometry of one HP1 dimer binding to a single peptide. Using the NMR assignments of free HP1-CSD (Brasher et al, 2000; E. Laue & N. Murzina, personal communication), we were able to define the EMSY-binding site of HP1-CSD as the same region that interacts with the CAF-1 peptide (Thiru et al, 2004). Out of the 64 non-proline residues of HP1-CSD, 35 residues show split or shifted cross-peaks and the most affected residues locate mainly to the C terminus and the region that binds to the CAF-1 peptide (supplementary Fig 1B online). A total of 23 residues were not affected by binding the EMSY97–109 peptide, and these residues locate mainly to the N terminus of HP1-CSD (supplementary Fig 1C online). Two residues cannot be identified and another four cannot be classified without re-assigning the cross-peaks for the HP1-CSD complex. For ten of the affected residues, only one of the split peaks is significantly shifted, which confirms asymmetric binding of the peptide across the HP1-CSD dimer. Residues in HP1-CSD that directly interact with EMSY97–109 cannot be discriminated from those that are affected by conformational changes. However, the HP1-CSD residues that were defined by NOE restraints to interact with CAF-1 are a subset of those affected on binding to EMSY97–109, and therefore we conclude that both peptides interact with HP1-CSD in a similar manner.
Model of the EMSY–HP1β complex
To gain structural insights into the architecture of the EMSY–HP1β complex, the VxL motif of the EMSY structure was directly superposed onto the side chains of the equivalent residues of the CAF-1 peptide in the HP1–CAF-1 complex (Thiru et al, 2004). Fig 4 depicts an energy minimized model of the EMSY–HP1 complex constructed based on the crystal structure of the HP1–CAF-1 complex. The model shows that the HP1-CSD dimer probably binds to the HB platform on one side of the EMSY dimer and predicts that the EMSY homodimer can bind to another HP1 homodimer on the other HB site. However, in the structure of the EMSY1–108 homodimer, only one of the HB platforms is accessible for binding, whereas the equivalent VxL motif on the adjacent molecule interacts directly with the ENT domain. This may represent a mode of regulating the binding affinity of EMSY for HP1. It is likely that the binding of HP1 and that of BS69 to the HB platform of the same EMSY molecule are mutually exclusive, as the binding sites are in close proximity. However, this does not exclude the possibility that the EMSY homodimer could form a heterotrimeric complex, binding BS69 on one molecule and HP1 on the other molecule.
Figure 4.
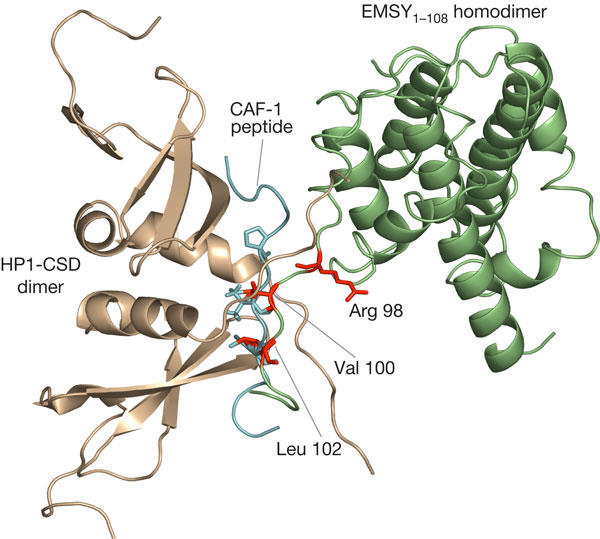
Model of the EMSY–HP1β complex. Ribbon diagram of an energy-minimized model of HP1–EMSY constructed by superposition of residues from the VxL motif of EMSY onto equivalent residues of the CAF-1 peptide. The model shows EMSY in green, HP1-CSD in brown and the VxL motif residues of EMSY in red. The CAF-1 peptide from the HP1–CAF-1 complex is truncated and depicted in cyan to provide an understanding of the binding region.
In the HP1–CAF-1 complex, the CAF-1 peptide forms an intermolecular β-sheet with residues from the C terminus of each subunit of the HP1 dimer (Thiru et al, 2004). This model, supported by our NMR and mutagenesis data, suggests that an analogous type of interaction will occur in the HP1–EMSY complex but with some minor distinctions. Many of the key residues involved in HP1 recognition are conserved in EMSY. These include residues at positions +2, 0 and −6, which have all been implicated in HP1 binding (Fig 1A). An obvious exception is the proline residue at the −2 position of CAF-1, which contributes significantly to HP1β binding (Thiru et al, 2004). In EMSY, this residue is replaced by arginine, and in our model, the side chain of Arg 98 is positioned away from the HP1-binding site, suggesting that it is unlikely to participate directly in binding (Fig 4). However, it is possible that modification of this arginine residue, for example, by methylation, may regulate the affinity of EMSY for HP1.
HP1 has a chromoshadow domain that interacts with numerous proteins by means of their PxVxL motif and a chromo domain that binds to Lys 9-methylated histone H3. Thus, HP1 seems to function as an adapter that recruits various complexes to regions of chromatin methylated at Lys 9 of histone H3. While it is thought that the chromo-domain interaction is important for localization of HP1 to heterochromatin and that the chromoshadow-domain interaction is important for stable binding and retention (Thiru et al, 2004), HP1 dimerization by means of the chromoshadow domain seems to be essential also for efficient localization to heterochromatin. This suggests that both chromo domains within the HP1 dimer must interact simultaneously with two separate H3 molecules. The finding that EMSY dimerizes through its ENT domain, thereby providing two potential interaction sites for HP1 and other proteins, adds another layer of complexity to the protein interaction network that coordinates chromatin remodelling.
Methods
EMSY97–109 (sequence RRLVPLMPRLVPQ) was prepared by G. Bloomberg (University of Bristol, UK).
Gene cloning and subcloning. EMSY (1–108) was PCR amplified using oligonucleotides with BamHI and EcoRI restriction sites at the 5′ and 3′ ends, respectively. The PCR product was subcloned into the polylinker region of mini-pRSET(A). His-tagged MOD1C (chromoshadow domain of mouse HP1β, residues 104–171 of HP1-CSD) in a pET16b vector was kindly provided by Dr N. Murzina. Site-directed mutagenesis of HP1-CSD (I161E) was performed using QuickChange.
Protein expression and purification. EMSY and HP1-CSD were overexpressed in Escherichia coli strains C41(DE3) and BL21(DE3), respectively, and purified according to the supplementary information online.
Crystallization and structure solution of EMSY1–108. Crystals were grown at 14°C in 200 mM NaCl and 20% PEG3350 (pH 6.9), and belong to the monoclinic space group P21 (see the supplementary results online). The structure of EMSY1–108 was determined by molecular replacement. Data were obtained to a resolution of 2.0 Å, and processing and refinement were carried out as described in the supplementary results online. The statistics of the final round of refinement are summarized in supplementary Table 1 online. The accession code for EMSY1–108 is 1UTU.
Analytical size-exclusion chromatography. Single protein samples were prepared at 50 μM and mixtures at 50:50, 50:100, 50:250 and 50:500 μM of EMSY1–108 and HP1-CSD, respectively. For the mutant HP1-CSD I161E, the protein concentration was 50 μM, and for the mixture with EMSY1–108, it was 50:50 μM. Analysis was performed on a Superdex 75 HR10/30 analytical gel-filtration column (Amersham Biosciences, UK) at ∼22°C. The column equilibrated in 50 mM Tris–HCl, pH 7.5, 100 mM NaCl and 1 mM dithiothreitol (DTT). Isocratic elution was performed at a flow rate of 0.5 ml/min.
Analytical ultracentrifugation. Analytical ultracentrifugation was used to determine the stoichiometry of the interaction between EMSY1–108 and HP1-CSD. Samples were dialysed into 50 mM Tris–HCl, pH 7.5, 100 mM NaCl and 1 mM DTT at 4°C. Mixtures of EMSY1–108 and HP1-CSD were used at concentrations of 15:15, 15:30, 1.5:1.5 and 1.5:3.0 μM. The experiments were performed in a Beckman Optima XL-I ultracentrifuge equipped with a 60Ti rotor using interference and absorbance at 280 and 230 nm, respectively. Samples were loaded into six-sector 12-mm path-length cells. Samples were spun at 30,000, 37,000 and 47,000 r.p.m. for the single proteins and at 18,000, 24,000 and 30,000 r.p.m. for the complex, until they reached equilibrium as judged by the changes in the subsequent scans. Runs were overspun at 47,000 r.p.m. for 1 h for determination of baselines. Samples were checked for degradation after the run by SDS–polyacrylamide gel electrophoresis. Data were analysed using the UltraSpin software (http://www.mrc-cpe.cam.ac.uk/).
Isothermal titration calorimetry. EMSY1–108 and HP1-CSD were dialysed into 50 mM Tris–HCl (pH 7.5) and 100 mM NaCl at 4°C. EMSY97–109 was dissolved in the same buffer (no change in pH was observed). The samples were concentrated as appropriate and centrifuged at 14,000 r.p.m. at 4°C for 15 min. Protein concentrations were determined spectrophotometrically by measuring absorbance at 280 nm. The peptide concentration of EMSY97–109 was determined by amino-acid analysis. HP1-CSD was applied to the reaction cell and EMSY1–108 or EMSY97–109 was applied at concentrations that gave a two-baseline binding isotherm. ITC experiments were performed on a VP-ITC MicroCalorimeter (MicroCal, USA) at 20°C, with a reference power of 10 μcal/s and an initial delay of 200 s. The stirring speed in the reaction cell was set to 305 r.p.m. Titration was performed in 3 μl (first injection/s) or 6 μl aliquots at 0.5 μl/s and the spacing between injections was 200 s. Heat of ligand dilution was determined in an independent control experiment by diluting EMSY into the buffer. A range of protein concentrations was tested but, typically, a concentration of EMSY at ∼300 μM in the syringe and HP1-CSD at ∼50 μM in the cell gave an isotherm with both baselines defined. As controls, the experiments were also performed by titrating HP1-CSD into EMSY1–108 and into EMSY97–109, and heat of ligand dilution was determined by diluting HP1-CSD into buffer. The data were analysed using the Origin software (Microcal) and fitted as a single set of identical sites.
NMR spectroscopy. 15N-labelled HP1 was prepared in the absence or presence of the EMSY97–109 peptide (concentration ratio 1:1.1) and dialysed into 10 mM sodium phosphate (pH 8.0) in the same vessel at 4°C overnight using Spectra/Por Float-A-Lyzer with a molecular mass cutoff of 500 Da. The samples were prepared for NMR in 5% D2O at a protein concentration of 300 μM HP1. HSQC spectra were recorded on a Bruker DRX 800 MHz spectrometer at 30°C.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
A.J.D. is a Royal Society University Research Fellow, and work in the A.J.D. laboratory is supported by grants from Cancer Research UK, BBSRC, Association for International Cancer Research and the Royal Society. L.S.I., S.M.V.F. and D.V. are supported by the MRC. C.M.S.E. is supported by a Cambridge Gates Studentship. T.K. is supported by grants from Cancer Research UK and L.H-D. is the recipient of a Cancer Research UK Clinical Scientist award. We thank S. Breward for the help with molecular biology. We also thank Dr N. Murzina and Professor E. Laue for providing us with NMR assignment data and for useful discussions.
References
- Ansieau S, Leutz A (2002) The conserved Mynd domain of BS69 binds cellular and oncoviral proteins through a common PXLXP motif. J Biol Chem 277: 4906–4910 [DOI] [PubMed] [Google Scholar]
- Brasher SV, Smith BO, Fog RH, Nietlispach D, Thiru A, Nielsen PR, Broadhurst RW, Ball LJ, Murzina NV, Laue ED (2000) The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J 19: 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavali GB, Ekblad CM, Basu BP, Freund SMV, Deprintsev D, Hughes-Davies L, Kouzarides T, Itzhaki LS, Doherty AJ (2005) Crystal structure of the ENT domain of human EMSY. J Mol Biol, in press [DOI] [PubMed] [Google Scholar]
- Hughes-Davies L et al. (2003) EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell 115: 523–535 [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP (2003) The Tudor domain ‘Royal Family': Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sc 28: 69–74 [DOI] [PubMed] [Google Scholar]
- Thiru A, Nietlispach D, Mott HR, Okuwaki M, Lyon D, Nielsen PR, Hirshberg M, Verreault A, Murzina NV, Laue ED (2004) Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J 23: 489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information