

Abstract
Loss of the retinoblastoma protein, pRB, leads to apoptosis, and several results have suggested that this is dependent on the E2F transcription factors. However, so far, the ability of the different E2F family members to contribute to apoptosis is controversial. Here, we show that ectopic expression of E2F3 results in apoptosis in both primary mouse fibroblasts and transgenic mice. Apoptosis induced by E2F3 is associated with the accumulation of E2F1 and, strikingly, we found that E2F3-induced apoptosis is dependent on E2F1. On the basis of these results, we propose that the accumulation of crucial levels of E2F1 activity, and not total E2F activity, is essential for the induction of apoptosis in response to a deregulated pRB pathway. These results are consistent with previous findings that E2F1, but not other E2Fs, can have tumour-suppressing activities.
Keywords: E2Fs, pRB, apoptosis, transgenic mouse
Introduction
Deregulation of the retinoblastoma protein (pRB) pathway is one of the hallmarks of human cancers (Weinberg, 1995). Tumours harbouring genetic alterations in the pRB pathway often have additional mutations affecting the apoptotic response (Weinberg, 1995; Sherr, 1996). As these data suggest that apoptosis has a crucial role in tumour suppression, it is of considerable scientific interest to understand the molecular mechanisms triggering apoptotis. The hypophosphorylated form of pRB binds to, and negatively regulates, the ‘activating' E2Fs (E2F1–3; Attwooll et al, 2004). Thus, loss of functional pRB leads to deregulation of the activating E2Fs and increased expression of E2F-regulated genes. The finding that loss of either E2f1 or E2f3 can rescue the apoptosis observed in pRb-deficient mouse embryos (Tsai et al, 1998; Ziebold et al, 2001) suggests that deregulation of E2F transcription factors is a key step for apoptosis induction after loss of pRB. E2F1 overexpression, both in vivo and in tissue culture, results in apoptosis (Qin et al, 1994; Shan & Lee, 1994; Holmberg et al, 1998; Pierce et al, 1998a, 1998b, 1999). Still controversial, however, is whether other activating E2Fs can trigger apoptosis. On the basis of the observation that loss of both E2f1 and E2f3 rescues apoptosis in pRb-null embryos, Trimarchi & Lees (2002) proposed that apoptosis is triggered in cells when deregulated E2F activity reaches a threshold level. In contrast, tissue-culture-based experiments have supported the notion that induction of apoptosis is a specific function of E2F1 (DeGregori et al, 1997). Hallstrom & Nevins (2003) recently reported that the E2F1 marked box domain has a unique proapoptotic activity among E2Fs.
Contrary to previous reports, we have analysed the ability of E2F3 to trigger apoptosis in vivo. We show here that E2F3 deregulation triggers apoptosis and, interestingly, that E2F3-induced apoptosis is dependent on E2F1 and is associated with increased E2F1 levels. Taken together, our data suggest that loss of pRB, here exemplified by E2F3 overexpression, causes deregulation of the activating E2Fs. This deregulation results in the increased expression of E2F1, ultimately leading to apoptosis.
Results
Deregulated E2F3 activity induces apoptosis in vivo
We have recently described the generation of a mouse model that expresses E2F3 fused to a modified version of the ligand-binding domain of the oestrogen receptor (ER–E2F3) in the intermediate lobe of the pituitary gland (ILP; Denchi et al, 2005). ER–E2F3 is expressed under the control of the tissuespecific promoter POMC and the fusion protein is inactive in the absence of the inducer 4-hydroxy tamoxifen (OHT). Activation of E2F3 leads to increased expression of E2F target genes and proliferation of the otherwise quiescent melanotrophs of the ILP (Fig 1A; Denchi et al, 2005). Sustained E2F3 activation results in hyperplasia of the ILP, but not in tumour formation, suggesting that a tumour suppression mechanism is capable of restraining proliferation induced by E2F3 deregulation (Denchi et al, 2005). Therefore, we analysed whether E2F3 deregulation leads to apoptosis and, consequently, whether apoptosis acts as a principal tumour suppression mechanism. Overexpression of E2F1 in tissue culture cells and in transgenic mice results in apoptosis (Qin et al, 1994; Holmberg et al, 1998; Pierce et al, 1998a, 1998b, 1999) and suppression of this apoptosis can result in tumour formation (Pierce et al, 1998a, 1998b). To assess whether deregulated E2F3 activity can result in apoptosis, a group of transgenic mice and a group of wild-type mice (five mice/genotype) were treated with tamoxifen for 3 days. As mentioned above, E2F3 activation results in S-phase entry (Fig 1A); we also observed that E2F3 induces apoptosis, as measured by active caspase-3 staining and TdT-mediated dUTP nick end-labelling (TUNEL) assay (Fig 1B). Importantly, no apoptosis was observed in control mice, either tamoxifen-treated wild type or nontreated transgenic (Fig 1B; data not shown). Notably, the observed difference in apoptosis between transgenic induced mice and controls is statistically significant (both assays P<0.005; Fig 1B). Similar levels of apoptosis were also detected in animals treated for longer periods of time (data not shown). Therefore, we conclude that deregulation of E2F3 activity results in apoptosis in vivo.
Figure 1.

E2F3 activation triggers apoptosis in transgenic mice. (A) 5-Bromodeoxyuridine (BrdU) incorporation assay (3 h pulse). Shown are melanotrophs of the intermediate lobe of the pituitary gland (ILP) derived from transgenic mice treated with tamoxifen (Tg+TAM) and controls, wild type treated with tamoxifen (WT+TAM) and transgenic nontreated (Tg). Right panel: quantification of percentage of BrdU-positive cells. Results represent standard deviation of the mean as described in the Methods. (B) TdT-mediated dUTP nick end-labelling assay (TUNEL; top panels) and immunohistochemical (IHC) analysis for activated caspase 3 (Ac. Casp3; bottom panels). Panels show ILP sections derived from wild-type (WT+TAM) and transgenic (Tg+TAM) mice treated with tamoxifen for 3 days. Right panels: quantification of TUNEL- and active caspase-3-positive cells in transgenic mice treated with tamoxifen (dashed bars) and in controls, wild type treated with tamoxifen (white bars). (C) IHC analysis for activated caspase 3 (Ac. Casp3, bottom panels) and TUNEL assay (top panels). Panels show ILP melanotrophs derived from wild-type (p53+/+), transgenic (p53+/+, Tg) and compound p53−/−; transgenic mice (p53−/−, Tg) treated with tamoxifen for 3 days. Graphs on the right: quantification of TUNEL- and activated caspase-3-positive cells of wild-type (white box, p53+/+), transgenic (black box, p53+/+, Tg) and compound p53−/−; transgenic mice (dashed box, p53−/−, Tg).
E2F3 can induce p53-independent apoptosis
Next, we assessed whether E2F3-induced apoptosis in the ILP is mediated by p53. Strikingly, we found that loss of p53 had no effect on the ability of E2F3 to induce apoptosis, as measured by both TUNEL assay (P=0.47) and activation of caspase 3 (P=0.29; Fig 1C). These results are in agreement with previous published data showing that loss of pRB and ectopic expression of E2F1 can trigger p53-dependent and p53-independent apoptosis (Morgenbesser et al, 1994; Macleod et al, 1995; Holmberg et al, 1998). We therefore conclude that deregulated E2F3 activity induces apoptosis independently of p53 in the mouse pituitary gland.
E2F3 induces E2F1-dependent apoptosis in MEFs
We investigated alternative mechanisms by which E2F3 could induce apoptosis, and tested whether E2F1 could be involved. Among the E2F family members, E2F1 is the best characterized for its ability to induce apoptosis and, interestingly, it is an E2F-responsive gene, which is transactivated by the activating E2Fs (Hsiao et al, 1994; Johnson et al, 1994). We, therefore, speculated that activation of E2F3 could lead to an increase in E2F1 levels that could subsequently lead to apoptosis. We tested this hypothesis by expressing ER–E2F3 in mouse embryo fibroblasts (MEFs). Wild-type and E2f1−/− MEFs were infected and, after selection, the viability of cells was assessed by Trypan blue exclusion in the presence or absence of OHT. As previously reported, the viability and growth potential of wild-type and E2f1−/− infected MEFs are similar (Fig 2A). On activation of E2F3, the growth of wild-type cells is severely impaired and cells undergo apoptosis (Fig 2A). In contrast, E2F3 activation leads to an insignificant amount of apoptosis in MEFs lacking E2F1 and a consequential increase in viable cells (Fig 2A). Importantly, however, E2F3 activation still leads to a decrease in proliferation rate, which is probably caused by E2F3-induced senescence (Denchi et al, 2005). The observed difference in apoptosis is not because of the different expression levels of ER–E2F3 (Fig 2B) and, moreover, the functionality of ER–E2F3 is not impaired in E2f1−/− cells, as E2F target genes, such as Cyclin E1 and PCNA (proliferating cell nuclear antigen), are induced to similar levels in the two cell lines (Fig 2B,F). We also excluded possible effects arising from either OHT treatment or ER expression, because HAER-expressing (ER fused to an HA tag) cells do not show any growth defects in the absence or presence of OHT (data not shown; Moroni et al, 2001). As an independent marker of apoptosis, we measured the levels of active caspase 3 in wild-type and E2f1−/− cells. As shown in Fig 2C, activation of E2F3 leads to a significant increase in activated caspase 3 in wild-type but not E2f1−/− MEFs (Fig 2C). Interestingly, we found that increased E2F3 activity led to a strong upregulation of E2f1 expression in wild-type MEFs (Fig 2C). As E2f1 is a known E2F target gene, our results therefore suggest that activation of E2F3 leads to E2F1-dependent apoptosis through direct transactivation of the E2f1 gene.
Figure 2.
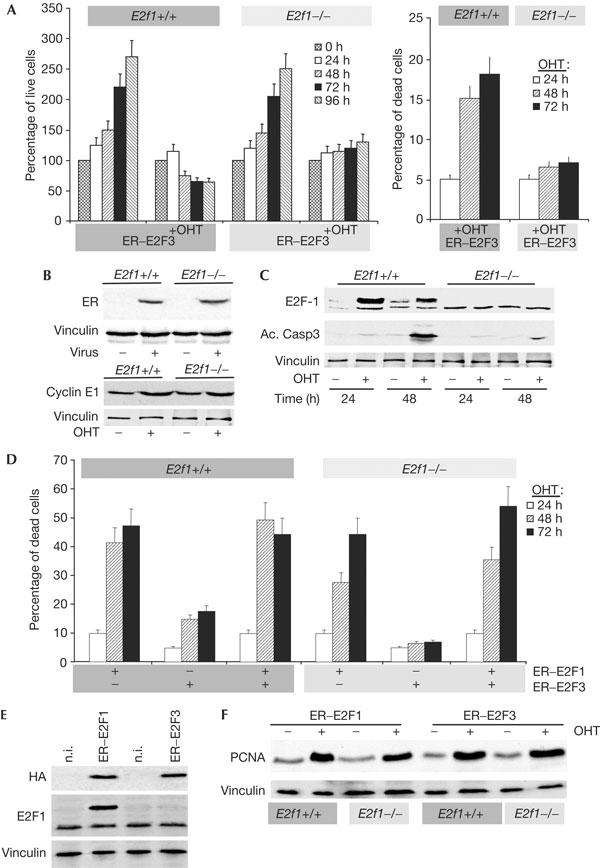
E2F3 results in E2F1-dependent apoptosis in mouse embryo fibroblasts (MEFs). (A) Early-passage MEFs derived from wild-type (E2f1+/+) or E2f1−/− littermate embryos were infected with pBabe–HAER–E2F3 (ER–E2F3). After selection, cells were either not treated or treated with 4-hydroxy tamoxifen (+OHT). Live cells (left panel) and apoptotic cells (right panel) were measured by Trypan blue exclusion at the indicated durations. (B) Top panel: expression of ER–E2F3 in E2f1−/− and E2f1+/+ cells as detected with an anti-ER antibody. Bottom panel: ER–E2F3 fusion protein is functional in both E2f1−/− and E2f1+/+ MEFs as shown by Cyclin E1 induction. As a loading control, blots were incubated with an anti-vinculin antibody. (C) Lysates were generated from MEFs collected at the indicated durations. Western blots were probed with anti-E2F1, anti-activated caspase-3 and vinculin antibodies. (D) MEFs from wild-type (E2f1+/+) or E2f1−/− littermate embryos were infected with HAER–E2F1, HAER–E2F3 or both. After selection, cells were induced with OHT and cell death was measured by Trypan blue exclusion at the indicated durations. (E) Top panel: expression of ER–E2F1 and ER–E2F3 in E2f1−/− and E2f1+/+ cells, as detected with an HA (haemagglutinin) antibody. Bottom panel: ER–E2F1 expression relative to endogenous E2f1 expression. As loading control, blots were incubated with an anti-vinculin antibody. n.i., not infected. (F) ER–E2F1 and ER–E2F3 proteins are equally functional in both E2f1−/− and E2f1+/+ MEFs, as shown by PCNA (proliferating cell nuclear antigen) induction. As a loading control, blots were incubated with an anti-vinculin antibody.
Next, we infected wild-type and E2f1−/− MEFs with a retrovirus expressing ER–E2F1, ER–E2F3 or both. In agreement with previous reports (e.g. Qin et al, 1994), E2F1 activation results in robust induction of apoptosis in wild-type and E2f1−/− MEFs despite a slight difference in the kinetics of the response (Fig 2D). These data show that E2f1−/− MEFs do not have a general defect in the apoptotic machinery. In agreement with previous reports, we found that E2F1 triggers apoptosis more efficiently than E2F3, despite being expressed at similar levels (Fig 2E). Moreover, we analysed the effect of the concomitant induction of ER–E2F1 and ER–E2F3 in wild-type and E2f1−/− MEFs. In both cell types, we found that E2F3 does not significantly increase apoptosis to a level higher than when only E2F1 is activated. These results support the hypothesis that E2F3-induced apoptosis is dependent on E2F1, and, mechanistically, that E2F3 activation leads to E2F1 accumulation, which results in apoptosis. This hypothesis is supported further by the fact that loss of p53 in MEFs, to a similar extent, reduces E2F1- and E2F3-induced apoptosis (supplementary Fig 1 online). To investigate whether a reciprocal dependency exists between E2F1 and E2F3 in inducing apoptosis, we infected MEFs containing floxed E2f3 alleles (Wu et al, 2001). As shown in supplementary Fig 1 online, deletion of E2f3 did not lead to a significant reduction of apoptosis. Taken together with the fact that overexpression of E2F1 induces apoptosis in E2f1−/− MEFs, whereas E2F3 overexpression does not, these data suggest that the E2F1 levels rather than the overall levels of E2F activity are crucial for determining whether a cell will undergo apoptosis.
E2F3 induces E2F1-dependent apoptosis in vivo
Induction of E2F3 in the ILP results in an increase of E2F1 levels, suggesting that E2F1 could also have a role in E2F3-induced apoptosis in vivo (Fig 3A). Therefore, we generated transgenic mice expressing ER–E2F3 in an E2f1−/− genetic background. Significantly, we found that E2F3 activation in the pituitary gland failed to induce apoptosis in E2f1−/− mice, whereas wild-type siblings showed significant apoptosis, as measured by active caspase-3 staining and by TUNEL assay (both assays P<0.005; Fig 3B). Significantly, loss of E2f1 does not impair E2F3-induced activation of E2F target genes, as shown by PCNA staining (Fig 3B). Similarly, E2F3 activation resulted in unscheduled proliferation in both wild-type and E2f1−/− mice, as shown by 5-bromodeoxyuridine (BrdU) incorporation and PCNA staining (Fig 3B). These data show that loss of E2f1 does not impair E2F3 functionality and, moreover, that induction of apoptosis and induction of proliferation are distinct functions of E2F3. Thus, we conclude that activation of E2F3 triggers E2F1-dependent apoptosis in vivo.
Figure 3.

E2F3 induces E2F1-dependent apoptosis in transgenic mice. (A) Immunohistochemical (IHC) analysis for E2f1 in melanotrophs of the intermediate lobe of the pituitary gland derived from wild-type (WT+TAM) and transgenic mice (Tg+TAM) induced with tamoxifen. (B) IHC analysis for activated caspase 3 (Ac. Casp3, top panels), TdT-mediated dUTP nick end-labelling assay (TUNEL, second panels from the top) and IHC analysis for PCNA (proliferating cell nuclear antigen; second panels from the bottom) and 5-bromodeoxyuridine (BrdU) (bottom panels). Panels show melanotrophs derived from wild-type (E2f1+/+), transgenic (E2f1+/+, Tg) and compound E2f1−/−; transgenic mice (E2f1−/−, Tg) treated with tamoxifen for 3 days. Panels on the left: quantification of activated caspase-3-, TUNEL-, PCNA- and BrdU-positive melanotrophs of wild-type (white box, E2f1+/+), transgenic (black box, E2f1+/+, Tg) and compound E2f1−/−; transgenic mice (dashed box, E2f1−/−, Tg).
E2F3-induced proliferation is enhanced in E2f1−/− mice
As the activation of E2F3 in E2f1−/− mice can induce unscheduled proliferation but not apoptosis, we tested the effect of sustained E2F3 activity in the absence of apoptosis. Mice (ten mice/genotype/treatment) were injected daily with tamoxifen, and pituitary homeostasis was monitored throughout a period of 6 months. As recently described (Denchi et al, 2005), increased E2F3 activity in the pituitary gland leads to hyperplasia. Significantly, this hyperplasia is enhanced in E2f1−/− mice when compared with E2f1+/+ mice (Fig 4A,B). Interestingly, however, we did not detect the formation of tumours in any of the mice monitored during the course of the experiments. On the basis of these results, we conclude that E2F1-mediated apoptosis contributes to pituitary homeostasis after E2F3 deregulation, but its suppression is not sufficient to result in tumour formation. These results are consistent with data showing that long-term activation of E2F3 in transgenic mice leads to senescent-like features, characterized by lack of proliferation, senescent-associated heterochromatin foci and increased expression of p19Arf and p16Ink4a (Denchi et al, 2005).
Figure 4.

E2F1 loss enhances E2F3-induced hyperplasia. (A) Haematoxylin and eosin staining of pituitaries derived from transgenic mice (Tg, E2f1+/+) and compound E2f1−/−; transgenic (E2f1−/−, Tg) mice treated with tamoxifen for 3 months and pituitaries derived from nontreated E2f1−/− mice. (B) Thickness of the intermediate lobe of the pituitary gland (ILP) relative to wild-type controls at the indicated time of tamoxifen treatment in E2f1−/−, transgenic mice wild type for E2f1 (E2f1+/+, POMC–ER–E2F3) and transgenic mice null for E2f1 (E2f1−/−, POMC–ER–E2F3). Bars indicate the relative thickness of the ILP expressed as percentage relative to wild-type controls. (C) Model for E2F-induced apoptosis. The retinoblastoma protein represses the activity of the activating E2Fs, which regulate the expression of several genes involved in proliferation. E2F1 is required for E2F-induced apoptosis, and it regulates the expression of a number of genes and the activity of proteins involved in apoptosis.
Discussion
Here, we have shown that E2F3, like E2F1, can trigger apoptosis both in vivo and in MEFs. Our results show that E2F3-induced apoptosis is dependent on E2F1, and that the molecular mechanism involves increased expression of E2F1. Interestingly, loss of E2F1 affects the apoptotic response to E2F3 deregulation without affecting the ability of E2F3 to induce E2F target genes and unscheduled proliferation of melanotrophs. On the basis of this, we propose that it is E2F1 that directly triggers apoptosis, rather than a checkpoint activated by abnormal proliferation. Moreover, as primary cells lacking E2F1 undergo apoptosis like wild-type cells after activation of ectopic E2F1, but not E2F3, our data suggest that it is E2F1 activity rather than the total amount of E2F activity that triggers apoptosis. Thus, we propose that deregulation of the pRB pathway, here exemplified by E2F3 expression, leads to accumulation of E2F1, which specifically leads to apoptosis (Fig 4C). On the basis of previous reports, we envision that increased E2F1 activity leads to apoptosis through an ARF–p53-dependent pathway as well as p53-independent pathways, which include the activation of E2F1 target genes such as APAF1 and p73 or of CHK2 (Irwin et al, 2000; Moroni et al, 2001; Kang et al, 2002; Rogoff et al, 2004). Importantly, our data reconcile previous published observations by demonstrating that the activating E2Fs can all induce apoptosis, but that this is dependent on E2F1, which specifically regulates the activation of proapoptotic genes.
Methods
Mice and tamoxifen administration. The generation of the POMC–ER–E2F3 mice has been described previously (Denchi et al, 2005). POMC–ER–E2F3 transgenic mice, E2f1 mutant mice (Yamasaki et al, 1998), p53−/− mice (Jacks et al, 1994) and compound mice were all bred in a mixed genetic background (129 sv//C57 Bl/6). Tamoxifen (Sigma, St Louis, MO, USA) was resuspended in sunflower oil (Sigma) and injected daily (1 mg dose).
Western blotting and antibodies. Proteins were extracted using E1A lysis buffer (50 mM Hepes pH 7.0, 250 mM NaCl, 0.1% NP-40) buffer. Protein lysates were separated on an SDS–polyacrylamide gel electrophoresis gel and transferred to Immobilon-P (Millipore, Billerica, MA, USA) by electroblotting. Primary antibodies used were anti-PCNA (Pharmingen, Franklin Lakes, NJ, USA), anti-BrdU (Becton Dickinson, Franklin Lakes, NJ, USA), anti-E2F1 (C20; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-HA (haemagglutinin, HA.11; Babco, Denver, PA, USA), anti-activated caspase 3 (ASP175; Cell Signaling Technology, Beverly, MA, USA), anti-ER (MC-20; Santa Cruz) and anti-Cyclin E1 (M-20, Santa Cruz).
Immunohistochemistry. For all our analyses, at least five mice/genotype/treatment were analysed, two independent sections/animal were stained and sequentially different fields were counted to evaluate the fraction of positive cells/total. A total of six to ten independent counts (300 cells each) were performed. A two-tailed t-test was performed to evaluate statistical significance. All antibodies were used on paraffin-embedded sections. For detection, we used the EnvisionTM kit (DAKO, Glostrup, Denmark). TUNEL assay was performed using the Apoptag® Peroxidase Plus Kit (Chemicon, Termecula, CA, USA, S7101).
Virus-mediated gene transfer and cell counts. The pBabe–ER–E2F constructs have been described (Vigo et al, 1999; Müller et al, 2001). The retrovirus-mediated gene transfer was conducted, as described (Moroni et al, 2001). To activate ER–E2F3, OHT was added to the medium to a final concentration of 300 nM. The number of live cells is expressed as percentage relative to the number of cells at time 0. The number of dead cells is expressed as percentage relative to the number of live cells at the indicated time.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported by grants from AIRC (Associazione Italiana Ricerca sul Cancro), The Italian Health Ministry, the Danish Research Ministry and the Danish Medical Research Council. E.L.D. was supported by a fellowship from FIRC (Fondazione Italiana Ricerca sul Cancro). We thank L. Yamasaki and T. Jacks for providing E2f1 and p53 mutant mice, G. Leone for providing TKO MEFs and C. Attwooll for critical reading of the manuscript.
References
- Attwooll C, Denchi EL, Helin K (2004) The E2F family: specific functions and overlapping interests. EMBO J 23: 4709–4716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR (1997) Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci USA 94: 7245–7250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, Attwooll C, Pasini D, Helin K (2005) Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol 25: 2660–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom TC, Nevins JR (2003) Specificity in the activation and control of transcription factor E2F-dependent apoptosis. Proc Natl Acad Sci USA 100: 10848–10853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg C, Helin K, Sehested M, Karlström O (1998) E2F-1 induced p53-independent apoptosis in transgenic mice. Oncogene 17: 143–155 [DOI] [PubMed] [Google Scholar]
- Hsiao KM, McMahon SL, Farnham PJ (1994) Multiple DNA elements are required for the growth regulation of the mouse E2F1 promoter. Genes Dev 8: 1526–1537 [DOI] [PubMed] [Google Scholar]
- Irwin M et al. (2000) Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 407: 645–648 [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol 4: 1–7 [DOI] [PubMed] [Google Scholar]
- Johnson DG, Ohtani K, Nevins JR (1994) Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev 8: 1514–1525 [DOI] [PubMed] [Google Scholar]
- Kang KH, Lee JH, Kim KC, Ham SW, Kim MY, Choi KH (2002) Induction of p73beta by a naphthoquinone analog is mediated by E2F-1 and triggers apoptosis in HeLa cells. FEBS Lett 522: 161–167 [DOI] [PubMed] [Google Scholar]
- Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T (1995) p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev 9: 935–944 [DOI] [PubMed] [Google Scholar]
- Morgenbesser SD, Williams BO, Jacks T, DePinho RA (1994) p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature 371: 72–74 [DOI] [PubMed] [Google Scholar]
- Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H, Helin K (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol 3: 552–558 [DOI] [PubMed] [Google Scholar]
- Müller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, Helin K (2001) E2Fs regulate the expression of genes involved in differentiation, development, proliferation and apoptosis. Genes Dev 15: 267–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AM, Fisher SM, Conti CJ, Johnson DG (1998a) Deregulated expression of E2F1 induces hyperplasia and cooperates with ras in skin tumor development. Oncogene 16: 1267–1276 [DOI] [PubMed] [Google Scholar]
- Pierce AM, Gimenez-Conti IB, Schneider-Broussard R, Martinez LA, Conti CJ, Johnson DG (1998b) Increased E2F1 activity induces skin tumors in mice heterozygous and nullizygous for p53. Proc Natl Acad Sci USA 95: 8858–8863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AM, Schneider-Broussard R, Gimenez-Conti IB, Russell JL, Conti CJ, Johnson DG (1999) E2F1 has both oncogenic and tumorsuppressive properties in a transgenic model. Mol Cell Biol 19: 6408–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X-Q, Livingston DM, Kaelin WG, Adams P (1994) Deregulated E2F1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA 91: 10918–10922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogoff HA, Pickering MT, Frame FM, Debatis ME, Sanchez Y, Jones S, Kowalik TF (2004) Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol 24: 2968–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Lee W-H (1994) Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol Cell Biol 14: 8166–8173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ (1996) Cancer cell cycles. Science 274: 1672–1677 [DOI] [PubMed] [Google Scholar]
- Trimarchi JM, Lees JA (2002) Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 3: 11–20 [DOI] [PubMed] [Google Scholar]
- Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T (1998) Mutation of E2F1 supresses apoptosis and inappropriate S-phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell 2: 293–304 [DOI] [PubMed] [Google Scholar]
- Vigo E, Muller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, Helin K (1999) CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol 19: 6379–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA (1995) The retinoblastoma protein and cell cycle control. Cell 81: 323–330 [DOI] [PubMed] [Google Scholar]
- Wu L et al. (2001) The E2F1-3 transcription factors are essential for cellular proliferation. Nature 414: 457–462 [DOI] [PubMed] [Google Scholar]
- Yamasaki L, Bronson R, Williams BO, Dyson NJ, Harlow E, Jacks T (1998) Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1(+/−) mice. Nat Genet 18: 360–364 [DOI] [PubMed] [Google Scholar]
- Ziebold U, Reza T, Caron A, Lees J (2001) E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev 15: 386–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information