

Abstract
Aminoacylation of transfer RNAs is a key step during translation. It is catalysed by the aminoacyl-tRNA synthetases (aaRSs) and requires the specific recognition of their cognate substrates, one or several tRNAs, ATP and the amino acid. Whereas the control of certain aaRS genes is well known in prokaryotes, little is known about the regulation of eukaryotic aaRS genes. Here, it is shown that expression of AspRS is regulated in yeast by a feedback mechanism that necessitates the binding of AspRS to its messenger RNA. This regulation leads to a synchronized expression of AspRS and tRNAAsp. The correlation between AspRS expression and mRNAAspRS and tRNAAsp concentrations, as well as the presence of AspRS in the nucleus, suggests an original regulation mechanism. It is proposed that the surplus of AspRS, not sequestered by tRNAAsp, is imported into the nucleus where it binds to mRNAAspRS and thus inhibits its accumulation.
Keywords: aspartyl-tRNA synthetase, regulation, nucleus, tRNA, yeast
Introduction
For biological necessity, aminoacylation of transfer RNA has to be highly specific (Ibba et al, 2005). However, tRNA aminoacylation does not occur with absolute specificity. To reduce error levels, aminoacyl-tRNA synthetases have developed kinetic artifices that ensure best selection of their cognate tRNAs and amino acid, and destruction of the mischarged tRNAs (Ebel et al, 1973; Hendrickson & Schimmel, 2003). Because of the central role of tRNAs and aminoacyl-tRNA synthetases (aaRSs) in translation, adjustments of the rate of their synthesis can be advantageous to the cell. Such controls may prevent overexpression of aaRSs that would misacylate heterologous tRNAs, as recently shown in the case of yeast AspRS, where increased concentration of the enzyme generates aspartylated tRNAAsn and tRNAGlu in vitro (Ryckelynck et al, 2003). In vivo, such errors would lead to the synthesis of erroneous proteins.
In prokaryotes, it is known that expression of aaRS genes can be transcriptionally or post-transcriptionally controlled (Ryckelynck et al, 2005). In yeast, it was shown that AspRS interacts with the 5′ end of its own messenger RNA and lowers expression of a fused reporter protein (Frugier & Giegé, 2003). Interestingly, the affinity measured for the AspRS/mRNAAspRS complex was found to be comparable with that determined for the AspRS/tRNAAsp complex. Protein and mRNA variants allowed identification of the domains involved in this complex. In mRNAAspRS, they encompass a sequence of 248 residues extending from nucleotide −38 in the 5′ untranslated region (5′UTR) to nucleotide 210 in the coding sequence; in AspRS, it is the anticodon-binding domain and the amino-terminal extension (NTE) that contact the mRNA. This appendix, probably of helical architecture (Agou et al, 1995), protrudes from the anticodon-binding module of AspRS (Ruff et al, 1991) and contains an RNA-binding motif (29xSKxxLKKxK38) responsible for interaction with the anticodon branch of tRNAAsp (Frugier et al, 2000). Even if this interaction is not essential for aspartylation, it increases the stability of the complex and the global efficiency of aminoacylation (Ryckelynck et al, 2003). On the basis of these observations and the data reported here, we propose a new mechanism for the regulation of AspRS expression in yeast (Fig 1). In short, the model implies the import of excess cytoplasmic AspRS into the nucleus, where it binds alternatively to the newly transcribed tRNAAsp or to mRNAAspRS, which leads to the inhibition of its own expression.
Figure 1.
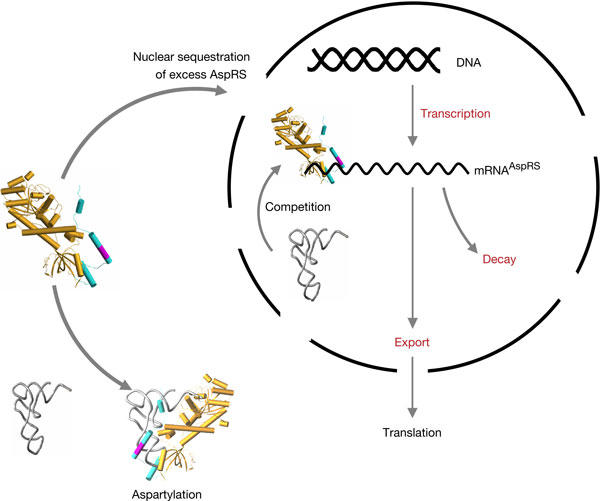
Model for yeast AspRS feedback regulation and potential effects of AspRS on the nuclear population of mRNAAsp. The three-dimensional structure of one monomer of the AspRS core (Ruff et al, 1991) is given (in light brown) with the modelled amino-terminal extension (in blue; Frugier et al, 2000) appended to the anticodon-binding domain. The location of the RNA-binding domain in the second helix of the appendix (in magenta) and its binding with tRNA are indicated (Frugier et al, 2000). The steps that are potentially affected during the regulation process are indicated in red.
Results And Discussion
The classical way to study expression regulation of a protein gene consists in increasing its copy number and then analysing the intracellular concentration of the biosynthesized protein. Thus, a yeast null strain (deleted of the endogenous AspRS gene) for AspRS was complemented with the AspRS gene cloned into a centromeric or a multicopy (2μ) plasmid. In both cases, no stimulation in AspRS expression could be detected, which suggests that AspRS expression is regulated. This regulation may involve a third molecule that would control the expression mechanism. If so, an obvious candidate is tRNAAsp, the cognate substrate of AspRS. To test this possibility, a wild-type yeast strain was transformed with a plasmid carrying the coding sequence for AspRS and two flanking sequences large enough to include all the information necessary for an endogenous transcription of the gene. This strain was subsequently co-transformed with plasmids carrying one gene copy of tRNAIle, used as a control, or tRNAAsp. As anticipated, the intracellular concentration of tRNAAsp remained unchanged in the control experiment (Fig 2A, lane 1) and increased significantly (about fivefold) in the strain transformed with the plasmid carrying the tRNAAsp gene (Fig 2A, lane 2). The variations in cellular tRNAAsp concentration are accompanied by strong intensifications (about fivefold) in the mRNAAspRS and AspRS expression patterns (Fig 2A, lanes 1,2). Indeed, expression of native AspRS is strongly enhanced when tRNAAsp is overexpressed, whereas the internal mRNA and protein controls (mRNAβ-actin and G6PDH expression) remain constant. Thus, by coexpressing AspRS and its cognate tRNAAsp, it is shown that AspRS translation is dependent on mRNAAspRS concentration and correlates with the level of cellular tRNAAsp. Because of the importance of the lysine-rich RNA-binding motif in AspRS NTE for binding its mRNA (Frugier & Giegé, 2003), an AspRS variant that lacked this motif was designed. With this mutant, variations in AspRS and mRNAAspRS expression are no longer correlated to tRNA concentrations (Fig 2A, lanes 3,4). This is indicative of a feedback regulation that necessitates an intact RNA-binding motif that enables the formation of a complex between the synthetase and its mRNA.
Figure 2.
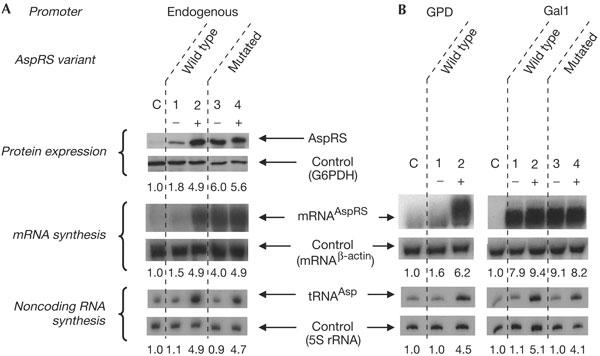
Expression levels of protein and RNAs. (A) Analysis of wild-type and mutant AspRS and mRNAAspRS expressions in yeast strains without (−) or with (+) plasmids encompassing an extra tRNAAsp gene (in the (−) strains, the tRNAAsp gene was replaced by a tRNAIle gene). Controls (c) correspond to nontransformed cells. (B) mRNAAspRS and tRNAAsp expression under the control of heterologous promoters. AspRS genes (wild type or mutated) were cloned under the control of the glyceraldehyde-3 phosphate dehydrogenase (GPD) promoter (strong constitutive promoter) or the Gal 1 promoter (strong inducible promoter) and the level of mRNAAspRS expression was determined in the presence of different tRNAAsp concentrations. No results were obtained with the mutated AspRS expressed under the GPD promoter, because the transformed yeast strains cannot grow properly. Quantitative data are given, representing mean values (about ±20%) of two independent experiments; values were calculated as a ratio of AspRS or mRNAAspRS or tRNAAsp with respect to controls and normalized towards the internal calibration controls (glucose 6-phosphate dehydrogenase (G6PDH), the mRNA encoding β-actin and 5S rRNA, respectively).
AspRS NTE was also predicted to be a nuclear localization signal (NLS; Schimmel & Wang, 1999). To bring an experimental answer to this prediction, DNA sequences corresponding to AspRS constructs deleted in their NTE (native, Δ30, Δ50 and Δ70 AspRSs) were cloned in a centromeric plasmid and transformed in a yeast strain deleted of its endogenous AspRS (null strain). Nucleus purifications from these strains allowed us to explore the synthetase compartmentalization, without being misled by the high cytoplasmic AspRS concentration. Data clearly indicate that all AspRS forms are present in the nuclear fractions (Fig 3). This finding is one of the few examples of nuclear aaRS localization (Mucha, 2002), and suggests that AspRS nuclear import requires a mechanism other than the classical NLS process (Christophe et al, 2000). Furthermore, this result sheds new light on the mechanism that accounts for AspRS regulation. Indeed, significant differences in the concentrations of AspRS variants were detected in the nucleus, whereas their cytoplasmic counterparts did not show any differences. The wild-type and truncated Δ30 AspRSs, both of which contain the RNA-binding motif, are less concentrated in the nucleus than the two other variants that do not contain this motif (Fig 3). The nuclear accumulation of Δ50 and Δ70 AspRSs could be a consequence of a loss of control consecutive to the loss of binding properties towards their mRNA and also to their reduced affinity for tRNAAsp in the cytoplasm. These data show that even when AspRS expression is no longer regulated, its concentration in the cytoplasm remains the same. Variations appear only in the nucleus, indicating that spare synthetase is segregated away from the translation site.
Figure 3.
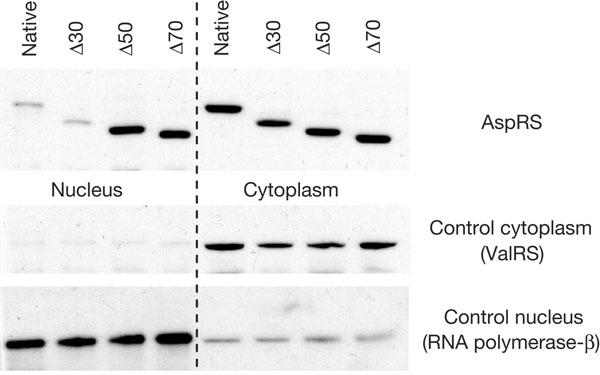
Detection of AspRS in the nucleus. Western blots on nuclear and cytoplasmic fractions using antibodies against AspRS (present in both fractions), ValRS (only cytoplasmic) and RNA polymerase II (only nuclear).
These results can be interpreted in two ways. (i) AspRS expression would be regulated by a translational mechanism that is consistent with other regulations mediated by RNA-binding proteins targeted to specific motifs in their own mRNA (McCarthy, 1998). The binding of AspRS to the 5′UTR of mRNAAspRS as well as the dependence towards tRNAAsp concentration seems reminiscent of what occurs in the well-known Escherichia coli threonine system (Romby & Springer, 2003). Here, ThrRS binds specifically to its own mRNA operator and inhibits its translation by hindering ribosome binding. ThrRS acts as a translational repressor, and the resulting inhibition is abolished by the increased concentration of tRNAThr. (ii) Nevertheless, as mRNAAspRS expression correlates directly with the profile of AspRS expression (Fig 2), it may also be that AspRS expression relies on a transcriptional control with the contribution of regulatory promoter elements (Kornberg, 1999) and/or mRNA turnover.
To check for the presence of binding sites for putative gene-specific transcriptional regulatory factors, the 5′UTR of mRNAAspRS was fused directly downstream of two foreign promoters, namely the strong constitutive promoter of glyceraldehyde-3 phosphate dehydrogenase (GPD) and the strong inducible promoter Gal 1. Despite the reduced growth rate of the corresponding yeast transformants, the level of expression of wild-type mRNAAspRS under the control of the GPD promoter is reduced when tRNAAsp concentration is low and is stimulated when it increases (Fig 2B, left panels). This means that the controlled expression of AspRS does not involve any specific regulatory promoter element and that the messenger contains all the information necessary to regulate the synthetase level in vivo. One also observes that the same construct containing the mutated AspRS does not form viable yeast transformants. On the contrary, AspRS expressed under the Gal 1 promoter does not undergo its own regulation. This can be explained by the high activation of transcription following galactose induction (Fig 2B, right panels). This feature was useful in the next experiments when testing the rate of mRNAAspRS degradation.
At this point, it is tempting to propose that the mRNAAspRS/AspRS complex is responsible for the inhibition of mRNAAspRS stabilization. Indeed, post-transcriptional control of gene expression can also be achieved by controlling mRNA decay (Gingerich et al, 2004; Wilusz & Wilusz, 2004). For example, it has been shown in E. coli that an excess of ribosomal L2 protein directs its own mRNA to a degradative pathway (Presutti et al, 1991). However, to some extent, this regulation is similar to that of AspRS, as recognition of mRNAL2 by protein L2 is essential and requires a large region in the messenger (−21 to +339). Thus, the effect of AspRS binding capacity on the stability of mRNAAspRS was tested in strains expressing wild-type or mutated AspRSs. Under galactose induction, transcription of both wild-type and mutated mRNAs is stimulated (Fig 4A). Even if the mutated mRNAAspRS seems to accumulate slightly more rapidly than the wild-type mRNA, addition of glucose in the medium blocks the Gal 1-dependent transcription, and both mRNAs show the same stability (Fig 4B). The similar rates of decay indicate that mRNAAspRS turnover is not the consequence of an accelerated degradation by AspRS binding.
Figure 4.
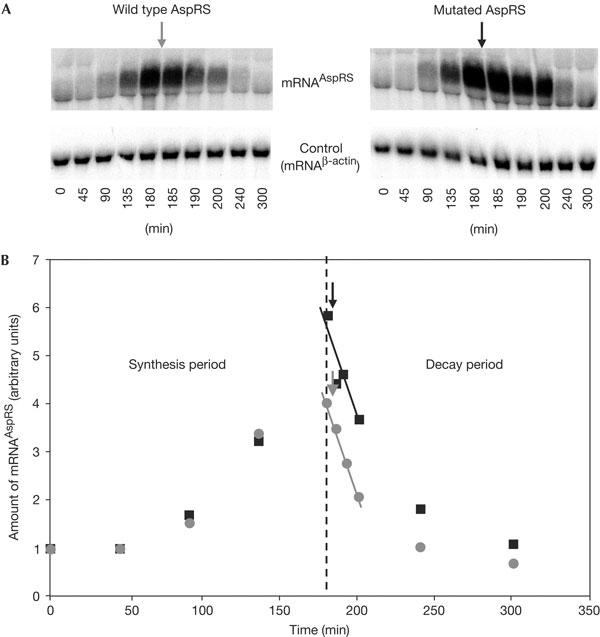
Accumulation and stability of mRNAAspRS. (A) Northern blot analysis of mRNAAspRS accumulation (45, 90, 135 and 180 min at 30°C) and degradation (185, 190, 200, 240 and 300 min at 30°C). The use of the galactose-inducible promoter allows one to overcome the regulation process of AspRS and accumulate enough mRNAAspRS and measure its degradation rate. After 3 h, glucose was substituted for galactose in the medium (arrows) to stop mRNA transcription. (B) Graphical representation of the accumulation and degradation rates corresponding to both wild-type (grey) and mutated (black) mRNAAspRS.
The above experimental evidence supports the existence of a new mechanism for the control of yeast AspRS (Fig 1) that differs from what is known in the aaRS field. One can presume that AspRS regulation is neither translational, as for E. coli ThrRS, nor linked to mRNA degradation, as for protein L2. However, the three systems share in common the fact that the crucial step in their regulation is the capacity of the protein to bind to its own mRNA. Here, the variations in mRNAAspRS concentration and the presence of the synthetase in the nucleus are in favour of an early nuclear interaction, thus determining the future fate of the transcript from the very beginning. Although all mechanistic details are not deciphered, the mechanism is based on a series of robust evidences. Key features are the synchronized synthesis of AspRS and tRNAAsp, the correlated synthesis of AspRS and mRNAAspRS, the involvement of the NTE in the control mechanism and the nuclear localization of a fraction of the synthetase. Further, the dependence of AspRS control on tRNAAsp concentration indicates that yeast maintains an adequate balance between the amounts of synthetase and its cognate tRNA. Rationalizing these facts leads to the following scenario for the regulation of yeast AspRS (Fig 1). When significant amounts of AspRS molecules are not sequestered by tRNAAsp in the cytoplasm, the synthetase in excess is imported in the nucleus. There, it binds to the 5′ end of its mRNA and thereby inhibits its own transcription. As a consequence, AspRS concentration decreases in the cytoplasm. In this model, part of yeast AspRS is imported in the nucleus, where both its substrates, namely the newly transcribed cognate tRNAAsp and the coding mRNAAspRS, are present. Thus, AspRS can either bind to the 5′ end of its mRNA and inhibit its accumulation or aminoacylate the newly synthesized tRNAAsp and further enhance its export to the cytoplasm (Azad et al, 2001; Steiner-Mosonyi & Mangroo, 2004). This nuclear localization also justifies the influence of tRNAAsp concentration on AspRS expression, as the newly transcribed tRNAAsp can compete directly with mRNAAspRS and thus release the transcriptional inhibition. As AspRS undergoes its own regulation even when expressed under the control of a different transcriptional promoter, this model, in some aspects, resembles the activation of gene expression in human immunodeficiency virus-1, where the Tat viral protein acts as an unusual transcription factor. It recognizes a stem–loop RNA structure (TAR) present at the 5′ end of the viral transcripts and this interaction promotes viral transcription by inducing chromatin modifications and by stimulating the recruitment of RNA polymerase II complexes (Marcello et al, 2001).
The interconnected control of both yeast AspRS and tRNAAsp cellular concentrations would help to significantly reduce the risk of errors during translation. This control probably occurs as shown in Fig 1, in a model that accounts best for the reported experimental data. Its originality lies in a dual character, combining features acting at the post-transcriptional level, as interaction of AspRS with its own mRNA is essential, and at the transcriptional level, leading to the arrest of mRNAAspRS transcription. At this stage of our study, we are aware that all mechanistic details of the regulation scenario are not deciphered, in particular what occurs in the nucleus and the putative participation of macromolecular partners recruited by AspRS when bound to mRNAAspRS. Also, we cannot exclude other post-transcriptional events, such as the possible inhibition of mRNA export from the nucleus or of translation, that would help the cell to keep AspRS expression under a control that is as efficient and reactive as possible. Experiments are in progress to unravel the uncertainties and also to understand, at the molecular level, how AspRS recognizes its mRNA.
Methods
Strains, plasmids and mutants. Construction of the AspRS-gene-disrupted YAL3 strain was described by Ador et al (1999). The amino-terminal-deleted AspRS genes (native, Δ30, Δ50 and Δ70) were cloned in the centromeric pRS314 (Trp+) and used to complement the YAL3 strain and study their cellular localization. YAL3 transformants were selected on a minimal medium supplemented with uracil, lysine, leucine and limiting concentrations of adenine (2 μg/ml). After 72 h incubation at 30°C, the Trp+ Sect+ colonies were isolated and screened for 5-fluoroorotic acid resistance.
For overproduction experiments, strain YBC 603 (ade2:hisG his3Δ200 leu2Δ0 lys2Δ0 met15Δ0 trp1Δ63 ura3Δ0) was transformed with plasmid pFL45S (2μ, Trp+) containing a 3.8 kb DNA fragment encoding AspRS and endogenous transcription promoter and terminator. AspRS was expressed under the control of the strong constitutive promoter of GPD on plasmid pRS426 (2μ, Ura+) and of the strong inducible promoter Gal 1 on plasmid pRS425 (2μ, Leu+). tRNA genes (tRNAAsp and tRNAIle) were PCR amplified with their own promoter and terminator regions using yeast genomic DNA as template, and introduced into pRS426 or pRS425.
The lysine-rich RNA-binding motif in the N-terminal extension of AspRS was altered by introducing a +1 frameshift (insertion) at position 83 in the coding sequence of mRNAAspRS and re-establishment of the reading frame (deletion) at position 147. Thus the wild-type sequence 30SKKALKKLQKEQEKQRKKE48 was replaced by the mutated sequence 30LVQEGLEEIAERARETEKE48.
RNA analysis. Yeast strains transformed with pRS425-Gal 1 containing the wild-type or the mutated AspRS genes were grown to an optical density (OD)600 nm of 0.3–0.4 in 2% glucose media. The cultures were induced in the presence of 2% galactose and 0.1% glucose, and aliquots were analysed by northern blotting after 45, 90, 135 and 180 min. To stop induction, galactose was replaced with 4% glucose and samples were collected after 5, 10, 20, 60 and 120 min incubation at 30°C. Other yeast strains were grown on amino-acid-supplemented minimal media for 15 h at 30°C and 180 r.p.m. Cells were collected by centrifugation, washed in PBS and total RNA was extracted as described previously (Schmitt et al, 1990). After ethanol precipitation, 15 μg (RNA stability) or 20 μg of total cellular RNA was analysed by northern blots using the NorthernMax kit (Ambion, Austin, TX, USA) and 32P-labelled probes prepared with the NonaPrimer kit (Quantum Appligene, France). Signal analysis and quantification were carried out on a Fuji Bioimager Bas2000 with Work Station Software (v1.1).
For tRNA detection, 15 μg of total RNA was resolved on a 1-mm-thick 12% polyacrylamide gel/8 M urea in Tris–borate–EDTA buffer, and transferred to a nylon membrane (Hybond-XL) and baked for 3 h at 80°C using a gel dryer. Hybridization was performed for 12 h at 60°C in 15 ml of Denhardt's solution, 5 × SSPE (saline/sodium phosphate/EDTA) and 0.5% SDS, with probes against tRNAAsp, tRNAIle and 5S RNA. Each probe corresponded to the 5′-end sequence of the given RNA and was 5′-end 32P labelled.
Cell lysis and western blotting. Cells were collected, washed, quantified (OD600 nm) and lysed in SDS loading buffer for 5 min at 90°C. Equivalent amounts of crude extract (0.01 OD) were resolved on 12% polyacrylamide SDS gels, transferred to Immobilon-P transfer membranes (Millipore, Bedford, MA, USA) and probed with anti-AspRS antibody or a 1:5,000 dilution of anti-G6PDH antibody (Sigma, St Louis, MI, USA). Primary antibody was detected by a 1:5,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit antibody (Amersham Biosciences, Buckinghamshire, UK).
Nucleus purification. Nucleus purifications were carried out according to Galy et al (2000). To obtain equivalent signals with both cytoplasmic and nuclear fractions, seven times more nuclear than cytoplasmic proteins (in equivalent number of cells) were loaded on the denaturing gel, blotted and detected using antibodies against AspRS, RNA polymerase II (αsubunit) or ValRS from yeast.
Acknowledgments
We thank M. Springer and C. Florentz for their critical reading and comments on the manuscript, G. Eriani and A. Lescure for their advice and discussion, P. Erbs and J. Boeke for yeast strains and M. Vigneron, G. Eriani and F. Fasiolo for antibodies. This work was supported by grants from the Centre National de la Recherche Scientifique (CNRS), Ministère de l'Education Nationale, de la Recherche et de la Technologie (MENRT) and Université Louis Pasteur.
References
- Ador L, Camasses A, Erbs B, Cavarelli J, Moras D, Gangloff J, Eriani G (1999) Active site mapping of yeast aspartyl-tRNA synthetase by in vivo selection of enzyme mutations lethal for cell growth. J Mol Biol 288: 231–242 [DOI] [PubMed] [Google Scholar]
- Agou F, Yang Y, Gesquière J-C, Waller J-P, Guittet E (1995) Polyanion-induced α-helical structure of a synthetic 23-residue peptide representing the lysine-rich segment of the N-terminal extension of yeast cytoplasmic aspartyl-tRNA synthetase. Biochemistry 34: 569–576 [DOI] [PubMed] [Google Scholar]
- Azad AK, Stanford DR, Sarkar S, Hopper AK (2001) Role of the nuclear pools of aminoacyl-tRNA synthetases in tRNA nuclear export. Mol Biol Cell 12: 1381–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophe D, Christophe-Hobertus C, Pichon B (2000) Nuclear targeting of proteins: how many different signals? Cell Signal 12: 337–341 [DOI] [PubMed] [Google Scholar]
- Ebel J-P, Giegé R, Bonnet J, Kern D, Bedfort N, Bollack C, Fasiolo F, Gangloff J, Dirheimer G (1973) Factors determining the specificity of the tRNA aminoacylation reaction. Biochimie 55: 547–557 [DOI] [PubMed] [Google Scholar]
- Frugier M, Giegé R (2003) Yeast aspartyl-tRNA synthetase binds specifically its own mRNA. J Mol Biol 331: 375–383 [DOI] [PubMed] [Google Scholar]
- Frugier M, Moulinier L, Giegé R (2000) A domain in the N-terminal extension of class IIb eukaryotic aminoacyl-tRNA synthetases is important for tRNA binding. EMBO J 19: 2371–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy V et al. (2000) Nuclear pore complexes in the organization of silent telomeric chromatin. Nature 403: 108–112 [DOI] [PubMed] [Google Scholar]
- Gingerich TJ, Feige JJ, LaMarre J (2004) AU-rich elements and the control of gene expression through regulated mRNA stability. Anim Health Res Rev 5: 49–63 [DOI] [PubMed] [Google Scholar]
- Hendrickson TL, Schimmel P (2003) Transfer RNA-dependent amino acid discrimination by aminoacyl-tRNA synthetases. In Translation Mechanisms, Lapointe J, Brakier-Gingras L (eds) pp 34–64. Georgetown, USA: Landes Bioscience [Google Scholar]
- Ibba M, Francklyn C, Cusack S (eds) (2005) The Aminoacyl-tRNA Synthetases. Georgetown, USA: Landes Bioscience [Google Scholar]
- Kornberg RD (1999) Eukaryotic transcriptional control. Trends Cell Biol 12: 46–49 [PubMed] [Google Scholar]
- Marcello A, Zoppe M, Giacca M (2001) Modes of transcriptional regulation by the HIV-1 Tat transactivator. IUBMB Life 51: 175–181 [DOI] [PubMed] [Google Scholar]
- McCarthy EG (1998) Posttranscriptional control of gene expression in yeast. Microbiol Mol Biol Rev 62: 1492–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha P (2002) Aminoacyl-tRNA synthetases and aminoacylation of tRNA in the nucleus. Acta Biochim Pol 49: 1–10 [PubMed] [Google Scholar]
- Presutti C, Ciafré SA, Bozzoni I (1991) The ribosomal protein-L2 in Saccharomyces cerevisiae controls the level of accumulation of its own messenger-RNA. EMBO J 10: 2215–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romby P, Springer M (2003) Bacterial translational control at atomic resolution. Trends Genet 19: 155–161 [DOI] [PubMed] [Google Scholar]
- Ruff M, Krishnaswamy S, Boeglin M, Poterzman A, Mitschler A, Podjarny A, Rees B, Thierry J-C, Moras D (1991) Class II aminoacyl transfer RNA synthetases: crystal structure of yeast aspartyl-tRNA synthetase complexed with tRNAAsp. Science 252: 1682–1689 [DOI] [PubMed] [Google Scholar]
- Ryckelynck M, Giegé R, Frugier M (2003) Yeast tRNAAsp charging accuracy is threatened by the N-terminal extension of aspartyl-tRNA synthetase. J Biol Chem 278: 9683–9690 [DOI] [PubMed] [Google Scholar]
- Ryckelynck M, Giegé R, Frugier M (2005) tRNAs and tRNA mimics as cornerstones of aminoacyl-tRNA synthetase regulations. Biochimie 11 May 2005; doi:10.1016/j.biochi.2005.02.014 [epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Schimmel P, Wang C-C (1999) Getting tRNA synthetases into the nucleus. Trends Biochem Sci 24: 127–128 [DOI] [PubMed] [Google Scholar]
- Schmitt ME, Brown TA, Trumtower BL (1990) A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res 18: 3091–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner-Mosonyi M, Mangroo D (2004) The nuclear tRNA aminoacylation-dependent pathway may be the principal route used to export tRNA from the nucleus in Saccharomyces cerevisiae. Biochem J 378: 809–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz CJ, Wilusz J (2004) Bringing the role of mRNA decay in the control of gene expression into focus. Trends Genet 20: 491–497 [DOI] [PubMed] [Google Scholar]