

Abstract
The spindle assembly checkpoint ensures accurate chromosome segregation by delaying anaphase initiation until all chromosomes are properly attached to the mitotic spindle. Here, we show that the previously reported c-Jun amino-terminal kinase (JNK) inhibitor SP600125 effectively disrupts spindle checkpoint function in a JNK-independent fashion. SP600125 potently inhibits activity of the mitotic checkpoint kinase monopolar spindle 1 (Mps1) in vitro and triggers efficient progression through a mitotic arrest imposed by spindle poisons. Importantly, expression of an Mps1 mutant protein refractory to SP600125-mediated inhibition restores spindle checkpoint function in the presence of SP600125, showing that its mitotic phenotype is induced by Mps1 inhibition in vivo. Remarkably, primary human cells are largely resistant to the checkpoint-inactivating action of SP600125, suggesting the existence of Mps1-independent checkpoint pathways that are compromised in tumour cells.
Keywords: JNK, mitosis, Mps1, spindle assembly checkpoint, SP600125
Introduction
The core of the spindle assembly checkpoint is a multiprotein complex assembled at the kinetochore, a proteinaceous structure embracing the centromeric region of each chromatid that organizes the complex microtubule–chromosome interactions during mitosis. Among the proteins that constitute the sensory machinery of the checkpoint, Mad2 and BubR1 fulfil crucial roles (Musacchio & Hardwick, 2002). In the absence of bipolar attachment, both proteins interact dynamically with kinetochores, where they are converted into an active form that can bind to and inhibit Cdc20, a regulatory subunit of an E3 ubiquitin ligase that is essential for anaphase progression, called the anaphase-promoting complex (APC; Peters, 2002). On successful bipolar attachment and/or generation of tension, Mad2 and BubR1 are displaced from the kinetochore, leading to the release of Cdc20 and APC-mediated degradation of essential mitotic targets such as Securin and Cyclin B, which allow exit from mitosis (Shah & Cleveland, 2000).
Most spindle checkpoint components are phosphorylated during mitosis and several are kinases themselves (Nigg, 2001; Musacchio & Hardwick, 2002). One of these kinases, monopolar spindle 1 (Mps1), was initially identified as a mutant that affects spindle pole duplication in Saccharomyces cerevisiae (Winey et al, 1991). Interference with the function of Mps1 causes an obvious checkpoint defect (Weiss & Winey, 1996; Abrieu et al, 2001; Stucke et al, 2002), whereas overexpression of Mps1 induces a mitotic arrest that depends on the presence of various checkpoint genes (Hardwick et al, 1996). This indicates that Mps1 acts at or near the top of the pathway and fulfils a regulatory function in spindle assembly checkpoint signalling.
Results and Discussion
SP600125 abrogates spindle checkpoint function
SP600125 was originally reported as a specific and reversible ATP-competitive inhibitor for stress- and mitogen-activated protein kinases of the c-Jun amino-terminal kinase (JNK) family, and causes human naive T cells to accumulate with a 4N DNA content (Bennett et al, 2001; Han et al, 2001). To study whether the latter effect is mediated through JNK, we analysed JNK1/2−/− double-deficient fibroblasts (Sabapathy et al, 1999), which are completely devoid of JNK activity (supplementary Fig S1 online). Interestingly, SP600125 could also induce accumulation of 4N cells in the absence of JNK (Fig 1A). Moreover, SP600125 prevented enrichment of mitotic cells in response to nocodazole, a spindle poison that triggers microtubule depolymerization and a spindle-checkpoint-dependent arrest (Fig 1B). To distinguish whether this was a result of impaired G2 progression or defective spindle checkpoint function, we added SP600125 to nocodazole-arrested JNK1/2−/− cultures. Strikingly, the percentage of phospho (p)-histone H3-positive cells that characterizes mitotic cultures decreased markedly in the presence of SP600125 (Fig 1C). Likewise, Cyclin B protein and Cyclin B-associated kinase activity, which rise in late G2 and are sustained in spindle-checkpoint-activated cells (Nigg, 2001), sharply dropped on SP600125 co-administration (Fig 1D). This indicates that these cells progressed past the spindle assembly checkpoint and activated the APC, leading to degradation of Cyclin B by the proteasome. Indeed, co-treatment with the proteasome inhibitor MG132 largely reversed these effects of SP600125 (Fig 1C,D), whereas treatment with MG132 did not alter the mitotic index of nocodazole-arrested cultures (data not shown). Together, these data show that SP600125 ablates spindle assembly checkpoint function in a JNK-independent manner and targets at least one other kinase in intact cells. This is not unlikely, as SP600125 was recently reported to inhibit several kinases in vitro in addition to JNK (Bain et al, 2003).
Figure 1.
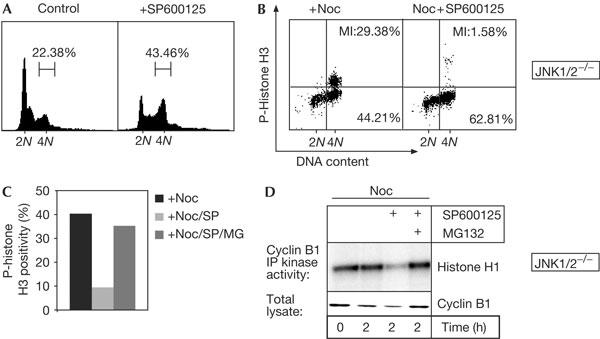
SP600125 affects spindle checkpoint function in a c-Jun amino-terminal kinase (JNK)-independent manner. (A) DNA profiles of JNK1/2−/− 3T3 fibroblasts treated with SP600125 for 10 h. Percentage of 4N cells is plotted above the DNA profiles. (B) Phospho-histone H3 (p-histone H3) positivity (y-axis) versus propidium iodide staining (x-axis) of JNK1/2−/− cells treated for 10 h with nocodazole (Noc) or Noc+SP600125. Mitotic cells (MI) are represented by the p-histone H3-positive 4N population (upper right quadrant); figures in the lower panels denote the percentage of non-mitotic 4N cells. (C,D) JNK1/2−/− cells were pretreated with Noc for 5 h and mitotic cells were obtained by shake-off. Cells were reseeded into medium containing Noc, Noc/SP600125 or Noc/SP600125/MG132, and were collected after 2 h. (C) Mitotic progression was examined by quantification of p-histone H3 positivity by fluorescence-activated cell sorting. (D) Cyclin B immunoprecipitation (IP) kinase assays using histone H1 as a substrate (upper panel), and western analysis of Cyclin B levels in total lysates (lower panel).
We next wanted to extend our findings to human cells. The addition of SP600125 to nocodazole-arrested human U2OS osteosarcoma cells induced a rapid loss of p-histone H3 positivity (Fig 2A) and cyclin B-associated kinase activity (Fig 2B), and both effects were blocked by co-treatment with MG132 (Fig 2A; data not shown). A similar effect of SP600125 was observed in taxol-arrested cultures (Fig 2B,C), and we found that the minimum concentration of SP600125 required for efficient checkpoint override ranged around 2.5 μM (Fig 2D). This concentration is well below the effective concentration for JNK inhibition in these cells (see supplementary Fig S6C online), again indicating that JNK inhibition is not required for SP600125-mediated checkpoint override. Interestingly, accumulation of 4N cells was only seen at concentrations above 10 μM in U2OS (supplementary Fig S2A online; data not shown), and time-lapse microscopy uncovered no striking mitotic aberrancies at 10 μM SP600125 (supplementary Fig S2B online). Similar results were obtained with two human breast carcinoma lines, HBL100 and T47D, in which 10 μM SP600125 was sufficient to overcome a nocodazole-mediated arrest but failed to elicit major defects in the absence of spindle damage (data not shown).
Figure 2.
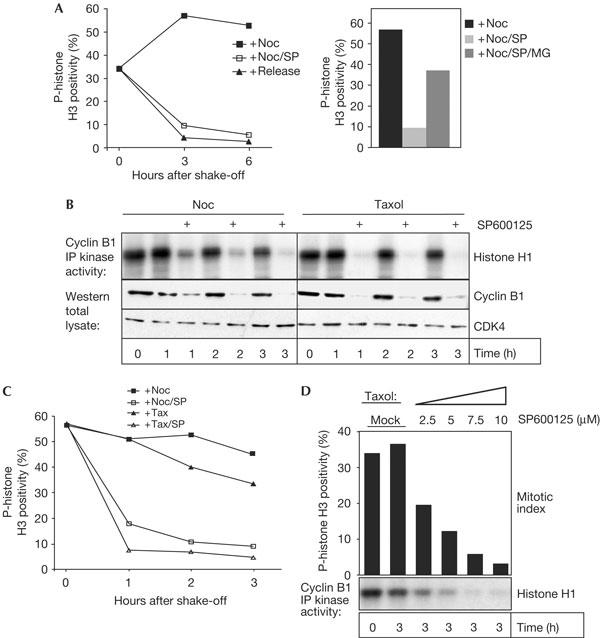
SP600125 abrogates spindle assembly checkpoint function in human cells. (A) Nocodazole (Noc)-arrested mitotic U2OS cells were collected by shake-off and reseeded into fresh medium containing Noc alone or in combination with the specified drugs. At the indicated time points, cells were collected and analysed for phospho-histone H3 (p-histone H3) positivity. The Histogram shows the relative mitotic index at 3 h after re-seeding in the presence of different drug combinations. For comparison, a fraction of the cells was released from the Noc block and analysed in parallel (release). (B,C) U2OS cells were treated as in (A), using either Noc or taxol, to obtain mitotic cells. After mitotic shake-off, cells were re-seeded into Noc or taxol. (B) Cyclin B-associated kinase activity and cyclin B protein levels were determined at the indicated time points in the specified drug combination. Cdk4 was used as a loading control. (C) Mitotic index, as determined by p-histone H3 positivity at different time points after re-seeding in the specified drug combination. (D) Cells were treated as in (B) and p-histone H3 positivity (bars) and cyclin B-associated kinase activity were determined after 3 h of co-incubation with the indicated SP600125 concentrations or an equivalent amount of solvent (mock).
SP600125 treatment leads to premature loss of BubR1
To study how SP600125 causes inactivation of the spindle checkpoint, we analysed kinetochore recruitment of two well-established spindle checkpoint proteins, Mad1 and BubR1. BubR1 is recruited to kinetochores in nocodazole- and in taxol-treated cells (Chan et al, 1999; Taylor et al, 2001; supplementary Fig S3A online), but its recruitment was clearly reduced in SP600125-treated mitotic cells (supplementary Fig S3A online). In contrast, Mad1 localization was only slightly affected by SP600125 co-treatment (supplementary Fig S3B online). Thus, SP600125 seems to specifically affect kinetochore recruitment of BubR1.
SP600125 directly inhibits kinase activity of human MPS1
We noted a certain degree of sequence similarity between the ATP-binding pocket of JNK and the human Mps1 (MPS1) kinase domain (Fig 3A). Thus, we tested whether SP600125 could inhibit Mps1 kinase activity in vitro. Endogenous MPS1 activity was inhibited more efficiently than JNK, as its activity was completely abolished at 0.5 μM SP600125 (Fig 3B). In contrast, SP600125 treatment did not significantly affect cyclin B/Cdc2 activity and only mildly inhibited BubR1 (Fig 3B) and aurora B (89% activity remaining at 10 μM SP600125, data not shown) at the maximal dose. SP600125 treatment did not interfere with kinetochore localization of Mps1, as we found abundant levels of MPS1 on kinetochores of mitotic cells in the presence of SP600125 (supplementary Fig S4A online).
Figure 3.
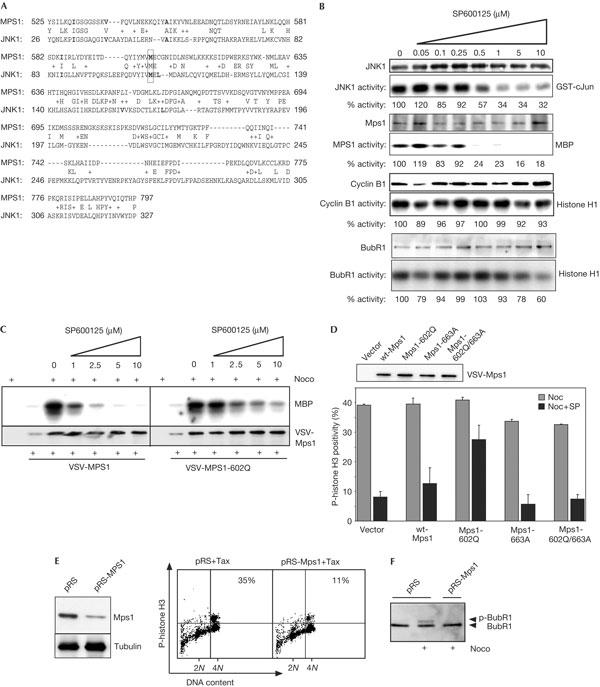
SP600125 mediates its effect on the spindle assembly checkpoint by inhibition of monopolar spindle 1 (Mps1). (A) Comparison of the amino-acid sequences encoding the kinase domains of human Mps1 (MPS1) and c-Jun amino-terminal kinase 1 (JNK1). Residues in bold denote conserved/similar amino acids in the SP600125-binding pocket of JNK1 (Heo et al, 2004); M602 in MPS1 (outlined with box) corresponding to M108 in JNK1 was chosen for mutation in the experiments shown in (C,D). (B) U2OS cells were treated with anisomycin to activate JNK1, or nocodazole (Noc) to obtain active MPS1, BubR1 and cyclin B/Cdc2. Total lysates from anisomycin-treated cells or selectively collected mitotic cells were pooled and subjected to immune complex kinase assays in the presence of 1% dimethylsulphoxide (DMSO; mock) or 1% DMSO and the indicated SP600125 concentrations. (C) Differential sensitivity of wild-type (wt) MPS1 and MPS1-M602Q to SP600125-mediated inhibition. U2OS cells were transfected with empty vector, VSV-Mps1 or VSV-Mps1-602Q and the influence of increasing SP600125 concentrations on kinase activity of the exogenous Mps1 proteins was analysed analogously to (B). Samples from thymidine-treated transfections were included as a control. Equal loading of the immunoprecipitated kinases in (B,C) was confirmed by western blot. (D) Cells transfected with empty vector, or expression vectors for the indicated VSV-tagged Mps1 proteins, were treated with Noc alone or in the presence of 10 μM SP600125 and compared for phospho-histone H3 (p-histone H3) positivity by flow cytometry. A representation of two independent experiments with the standard error is shown. A control western blot (top) confirmed similar expression of the mutants. (E) U2OS cells were co-transfected with spectrin–GFP (green fluorescent protein) and either empty pRS-puro vector (pRS) or pRS-puro-expressing small hairpin RNA against MPS1 (pRS-MPS1). After 48 h, cells were treated overnight with taxol and GFP-positive cells were analysed for p-histone H3 positivity by fluorescence-activated cell sorting (FACS; right) or transfected cells were selected by a 24 h puromycin (2 μg/ml) treatment and Mps1 knockdown levels of positively selected cells were determined by western blot (left). The percentage of mitotic cells in the 4N population is plotted in the upper right corner of the FACS profiles. (F) As in (E), but puromycinselected transfected cells were treated with Noc for 18 h before lysis. The presence of phosphorylated BubR1 in total lysates was detected by electromobility shift on western blots with a BubR1-specific antiserum.
Mutation of methionine M108 to glutamine (Q) in JNK1 renders it insensitive to SP600125-mediated inhibition (Heo et al, 2004). Interestingly, a corresponding mutation in MPS1 (M602Q) also proved significantly less sensitive to SP600125 in kinase assays (Fig 3C). Importantly, expression of this SP600125-hyposensitive mutant of MPS1 largely restored p-histone H3 positivity in the presence of SP600125, but expression of wild-type (wt) Mps1, kinase-dead Mps1 (Mps1 D663A; Stucke et al, 2002) or a kinase-dead version of MPS1-M602Q (MPS1-602Q/663A) could not rescue the SP600125-mediated checkpoint override (Fig 3D), whereas all mutants localized to kinetochores (supplementary Fig S4B online). These data clearly show that SP600125 mediates its effect on spindle checkpoint function by Mps1 inhibition.
We next used RNA interference (RNAi) on the function of MPS1. Transfection of U2OS cells with pooled expression plasmids for three individual small hairpin RNAs (shRNAs) against Mps1 (pRS-Mps1) reduced MPS1 protein levels to about 20%–30% (Fig 3E). This resulted in an approximately threefold decrease of p-histone H3 positivity in taxol or nocodazole (Fig 3E; data not shown), showing that the MPS1 protein depletion could largely abrogate a spindle-checkpoint-mediated mitotic arrest in U2OS cells. In agreement with published data (Stucke et al, 2002) and our findings with SP600125, Mps1 depletion did not induce major cell-cycle defects in the absence of spindle damage (supplementary Fig S5A online).
We then analysed BubR1 phosphorylation, which was previously shown to correlate with mitotic progression and is induced by microtubule depolymerization (Taylor et al, 2001). Mps1 depletion resulted in a clear shift of BubR1 to its hypophosphorylated form in the presence of nocodazole (Fig 3F), indicating that Mps1 depletion affects BubR1 activity. Similar to SP600125 treatment, introduction of pRS-Mps1 also resulted in a clear loss of BubR1 from kinetochores of prometaphase cells in all examined combinations (supplementary Fig S5B online).
BJ-Tert fibroblasts are refractory to SP600125
We next explored the effect of SP600125 on normal somatic cells. For this, we analysed BJ-Tert cells, primary human fibroblasts immortalized through stable expression of the catalytic component of human telomerase (Jiang et al, 1999). Surprisingly, even 10 μM of SP600125 was not sufficient to effectively overcome a taxol- or nocodazole-induced mitotic arrest in BJ-Tert cells (Fig 4A,B; data not shown). The insensitivity of BJ-Tert cells to SP600125 is not a consequence of higher Mps1 protein expression, as the level of Mps1 in BJ-Tert cells is below that in U2OS cells (supplementary Fig S6A online). Consistent with the lack of responsiveness to SP600125, BubR1 localization was largely unchanged by SP600125 treatment in BJ-Tert cells and BubR1 was retained at the kinetochores of nocodazole- or taxol-arrested prometaphase cells (supplementary Fig S6B online). However, SP600125 inhibited JNK in BJ-Tert cells in a manner comparable with that in U2OS cells (supplementary Fig S6C online). More importantly, SP600125 could prevent nocodazole- and taxol-induced BubR1 phosphorylation in both cell types (Fig 4C), showing that SP600125 was effective in BJ-Tert cells, but was insufficient to trigger a full checkpoint override.
Figure 4.
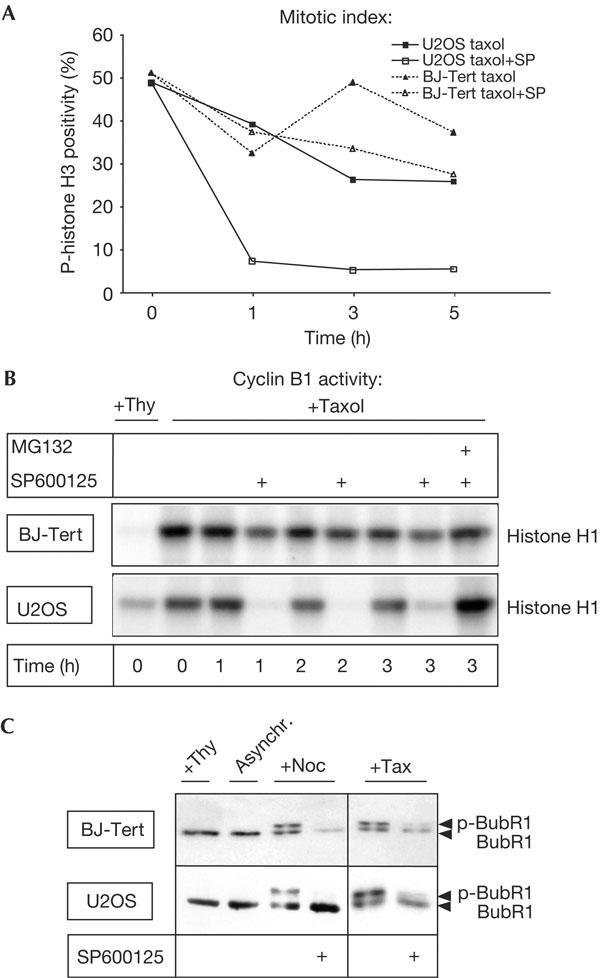
Primary BJ-Tert cells are refractory to SP600125-mediated spindle checkpoint override. (A,B) Taxol-arrested, mitotic BJ-Tert cells or U2OS cells were selectively collected by shake-off, reseeded and exposed to taxol, taxol/SP600125, or both drugs in combination with the proteasome inhibitor MG132. Cells were collected at the indicated time points and analysed for phospho-histone H3 (p-histone H3) positivity (A) or cyclin B-associated kinase activity (B). Cyclin B-associated kinase activity was compared with thymidine-arrested (S phase) cells (Thy) as control. (C) BJ-Tert and U2OS cells were treated overnight with nocodazole or taxol in the absence or presence of SP600125 and BubR1 phosphorylation was analysed by electromobility shift on western blots.
Conclusion
Our data show that SP600125 effectively disrupts mammalian spindle checkpoint function in a JNK-independent fashion. Particularly, our rescue experiments using SP600125-resistant mutants of Mps1 clearly show that SP600125 acts on Mps1 to inhibit the checkpoint. Principally, however, we cannot exclude that SP600125 also inhibits kinases other than Mps1 and JNK in vivo, especially as Bain et al (2003) previously reported inhibition of several kinases by SP600125 in vitro. However, apart from cyclin-dependent kinase (CDK) 2/Cyclin A, none of these kinases has thus far been reported to have a role in mitotic progression. It seems unlikely that CDK2/Cyclin A complexes are prominent targets of SP600125 in intact cells, as inhibition of CDK2/Cyclin A is expected to cause inhibition of entry to mitosis, and induces a G2 arrest in human cells (Furuno et al, 1999), which is not observed in our cells. Nonetheless, inhibition of other kinases cannot be ruled out, and future efforts should be directed to modifications of SP600125 that result in drugs with a higher specificity for Mps1.
Our observation that Mad1 recruitment is not affected by SP600125, at first glance, seems to be at odds with previous reports showing a crucial role of Mps1 in Mad1 localization (Abrieu et al, 2001; Martin-Lluesma et al, 2002). However, in these studies, Mps1 protein was depleted, whereas here we merely interfere with its kinase activity. Indeed, Liu et al (2003) found no apparent differences in Mad1 localization on microinjection of an inactivating anti-Mps1 antibody, whereas RNAi-mediated depletion of Mps1 produced a loss of Mad1 from the kinetochores. Thus, our data support a model in which Mad1 recruitment requires the physical presence of Mps1 but not necessarily its kinase activity.
Importantly, our data indicate the existence of multiple spindle-checkpoint-enforcing pathways in primary cells that make them resistant to SP600125. These redundant pathways seem to be lost or compromised in the cancer cell lines studied here, underlining the potential of spindle-checkpoint-interfering drugs as an attractive new approach in anticancer therapy. Obviously, more specific inhibitors for Mps1 will be essential in such efforts.
Methods
Antibodies and reagents. Antisera against CDK4 (sc-260), JNK1 (sc-474), TTK/MPS1 (sc-540, for immunoprecipitation), cyclin B1 (sc-245) and p-JNK (sc-6254) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against p-histone H3 and MPS1 (for western blots; Stucke et al, 2002) were from Upstate Biotechnologies (Lake Placid, NY, USA). Anti-VSV (P5D4) and myelin basic protein were from Sigma (St Louis, MO, USA). Anti-BubR1 was a kind gift from S. Taylor (Taylor et al, 2001) and anti-Mad1 was a generous gift from A. Musacchio. GST-c-Jun (1–135) as substrate for JNK1 was purified according to standard procedures. Histone H1 was obtained from Roche Diagnostics (Basel, Switzerland), and SP600125 was from Biomol (Plymouth Meeting, PA, USA) and was used at 10 μM unless otherwise stated. MG132, thymidine, paclitaxel (taxol) and nocodazole were all from Sigma and used at concentrations of 5 μM, 2.5 mM, 1 μM and 250 ng/ml, respectively.
Plasmids. Expression vectors for H2B–GFP (green fluorescent protein) and spectrin–GFP have been described previously (Lens et al, 2003). A pool of three targeting constructs for MPS1 (pRS-MPS1) were obtained from an shRNA kinase knockdown library (Berns et al, 2004; sequences available on request). Full-length wt-MPS1 complementary DNA (Origene) was ligated in-frame onto a VSV-tag sequence inserted into a pCR3 expression vector (Invitrogen, Carlsbad, CA, USA) to express VSV-MPS1. VSV-tagged Mps1 mutants were obtained from the latter construct by PCR-based site-directed mutagenesis (Stratagene, La Jolla, CA, USA).
Cell culture and transfections. Human BJ-Tert foreskin fibroblasts (Jiang et al, 1999) were grown in a 1:4 mixture of Medium 199/DMEM (Invitrogen) with 15% FCS and penicillin/streptomycin. Human U2OS osteosarcoma cells and murine JNK1/2−/− double-deficient embryonic fibroblasts (Sabapathy et al, 1999; kind gift from E. Wagner) were cultured in DMEM/8% FCS/penicillin/streptomycin. Transfections were performed using the standard calcium phosphate protocol. Cells were arrested in mitosis by treatment with 1 μM taxol or 250 ng/ml nocodazole for 18 h, and mitotic cells were collected by shake-off (Ditchfield et al, 2003).
Immunoblots and kinase assays Immunoblots were performed as described previously (Schmidt et al, 2000). To activate JNK1, cells were treated with anisomycin (10 μg/ml) for 20 min. To obtain active MPS1, Cyclin B/Cdc2 or BubR1, cells were incubated overnight with nocodazole (250 ng/ml) and mitotic cells were isolated by shake-off before lysis. In vitro kinase assays were performed as described previously (Schmidt et al, 2000; Stucke et al, 2002; Mao et al, 2003) using a standardized kinase buffer (Schmidt et al, 2000) in the presence of 0.25 mg/ml substrate, 50 μM ATP and 2.5 μCi [γ-32P]ATP (Amersham Biosciences Europe, Freiburg, Germany). Phosphorylated substrates were separated on SDS–polyacrylamide gels and analysed on blots by autoradiography. Equal precipitation of the kinases was confirmed by probing the blots with the appropriate antibody.
Cell-cycle analysis and immunofluorescence. Cell-cycle distribution and mitotic indices were determined by combined propidium iodide and p-histone H3 staining, as described previously (Lens et al, 2003). Kinetochore localization of BubR1, Mad1 and Mps1 was judged by analysis of individual prometaphase cells by confocal microscopy as described previously (Lens et al, 2003).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
We thank S. Taylor, A. Musacchio and H. van Dam for providing useful reagents and all other lab members of the Medema lab for continued discussion and helpful suggestions.
References
- Abrieu A, Magnaghi-Jaulin L, Kahana JA, Peter M, Castro A, Vigneron S, Lorca T, Cleveland DW, Labbe JC (2001) Mps1 is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell 106: 83–93 [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P (2003) The specificities of protein kinase inhibitors: an update. Biochem J 371: 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL et al. (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA 98: 13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns K et al. (2004) A largescale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428: 431–437 [DOI] [PubMed] [Google Scholar]
- Chan GK, Jablonski SA, Sudakin V, Hittle JC, Yen TJ (1999) Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J Cell Biol 146: 941–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS (2003) Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol 161: 267–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuno N, den Elzen N, Pines J (1999) Human cyclin A is required for mitosis until mid prophase. J Cell Biol 147: 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS (2001) c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest 108: 73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW (1996) Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science 273: 953–956 [DOI] [PubMed] [Google Scholar]
- Heo YS et al. (2004) Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J 23: 2185–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XR et al. (1999) Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet 21: 111–114 [DOI] [PubMed] [Google Scholar]
- Lens SM, Wolthuis RM, Klompmaker R, Kauw J, Agami R, Brummelkamp T, Kops G, Medema RH (2003) Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension. EMBO J 22: 2934–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui ST, Chan GK, Hittle JC, Fujii G, Lees E, Yen TJ (2003) Human MPS1 kinase is required for mitotic arrest induced by the loss of CENP-E from kinetochores. Mol Biol Cell 14: 1638–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Abrieu A, Cleveland DW (2003) Activating and silencing the mitotic checkpoint through CENP-E-dependent activation/inactivation of BubR1. Cell 114: 87–98 [DOI] [PubMed] [Google Scholar]
- Martin-Lluesma S, Stucke VM, Nigg EA (2002) Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2. Science 297: 2267–2270 [DOI] [PubMed] [Google Scholar]
- Musacchio A, Hardwick KG (2002) The spindle checkpoint: structural insights into dynamic signalling. Nat Rev Mol Cell Biol 3: 731–741 [DOI] [PubMed] [Google Scholar]
- Nigg EA (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2: 21–32 [DOI] [PubMed] [Google Scholar]
- Peters JM (2002) The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol Cell 9: 931–943 [DOI] [PubMed] [Google Scholar]
- Sabapathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF (1999) Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech Dev 89: 115–124 [DOI] [PubMed] [Google Scholar]
- Schmidt M, Goebeler M, Posern G, Feller SM, Seitz CS, Brocker EB, Rapp UR, Ludwig S (2000) Ras-independent activation of the Raf/MEK/ERK pathway upon calcium-induced differentiation of keratinocytes. J Biol Chem 275: 41011–41017 [DOI] [PubMed] [Google Scholar]
- Shah JV, Cleveland DW (2000) Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell 103: 997–1000 [DOI] [PubMed] [Google Scholar]
- Stucke VM, Sillje HH, Arnaud L, Nigg EA (2002) Human Mps1 kinase is required for the spindle assembly checkpoint but not for centrosome duplication. EMBO J 21: 1723–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ (2001) Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J Cell Sci 114: 4385–4395 [DOI] [PubMed] [Google Scholar]
- Weiss E, Winey M (1996) The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J Cell Biol 132: 111–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winey M, Goetsch L, Baum P, Byers B (1991) MPS1 and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J Cell Biol 114: 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information