

Abstract
Antigens are able to elicit productive immune responses only when second signals are provided by adjuvant molecules. It is well established that exogenously acquired, pathogen-associated molecular patterns fulfil this adjuvant role when recognized by specific receptors on antigen-presenting cells. Recent evidence points to the existence of another class of adjuvant, which is apparently released from injured cells. Such endogenous adjuvants, referred to as 'danger' signals, could alert the immune system to situations that cause cell damage, but not necessarily those that involve infections. Endogenous adjuvants provide a good explanation for immune responses generated against tumours and autologous tissues, but it has been difficult to explain how a constant activation of the immune system is avoided, considering the frequency at which cells are injured in vivo. Here, we suggest that the efficiency with which cells reseal wounds in their plasma membrane might be an important factor in the balance between tolerance and autoimmunity. Recent observations in synaptotagmin-VII-deficient mice suggest that defective membrane repair could lead to autoimmunity in tissues that are more susceptible to mechanical injury.
Keywords: adjuvant, autoimmunity, immunity, membrane repair, synaptotagmin VII
Introduction
The generation of effective immune responses against soluble proteins requires the co-injection of molecules with adjuvant activity (Dresser, 1961, 1968). In the absence of adjuvants, injection of purified proteins can induce tolerance (Dresser, 1962). Although it has been postulated that some adjuvants enhance immune responses by retaining the antigen in a long-lived form in vivo, their main mechanism of action is thought to be the induction of costimulatory signals that can be recognized by T cells. This concept is well established in the case of adjuvants of microbial origin, such as lipopolysaccharide (LPS), due to an explosion of information in recent years. Receptors for conserved molecules from infectious organisms have been identified, and the signalling pathways by which they induce antigen-presenting cells (APCs) to express co-stimulatory molecules have been characterized (Beutler, 2004; Takeda et al, 2003). These findings have, in large part, confirmed predictions that the immune system evolved to recognize and respond to patterns that are common on infectious agents, but absent from the host (Janeway, 1989). Because these are exogenously acquired molecules, this concept has been referred to as the 'stranger' hypothesis (Rock et al, 2005).
However, there is plenty of evidence that the immune system is also able to mount effective responses in the absence of 'stranger' molecules. Examples of such situations include immunity to transplants and to tumours and autoimmunity. To explain how these responses occur, it was hypothesized that the immune system could discriminate 'danger' signals from injured host cells (Matzinger, 1994; Rock et al, 2005). This scenario is consistent with the fact that many adjuvants are tissue irritants (Dresser, 1961). This review focuses on recent studies that are beginning to provide evidence for the existence of endogenous adjuvants. Given that these molecules are released from injured cells, the regulation of their effect by plasma-membrane repair on immune responses is also discussed.
The 'danger' hypothesis
In 1994, Polly Matzinger proposed an alternative to the widely established concept that the immune system primarily discriminates self from non-self. This alternative hypothesis suggested that the immune system is mainly adapted to recognize and respond to signals generated in a 'dangerous' situation that involves cell damage (Matzinger, 1994). This model, which became known as the 'danger' hypothesis, is based on the idea that the crucial signals for the initiation of immune responses are endogenous, not exogenous (Matzinger, 2001). According to this hypothesis, the power lies with the tissues: the activation state of APCs would depend on the health of cells in their neighbourhood.
What is the evidence in support of this idea? It has been extensively demonstrated that recognition of microbial molecular patterns by Toll-like receptors (TLRs) initiates immune responses that are essential for the control of infections (Kopp & Medzhitov, 2003). However, there is also evidence that TLR-mediated microbial recognition can lead to very different outcomes, depending on whether the stimulus originates from pathogenic or non-pathogenic organisms. Non-pathogenic commensal bacteria induce TLR-mediated signalling in the gut (Rakoff-Nahoum et al, 2004), but under normal conditions this does not trigger an inflammatory response comparable with that caused by pathogenic bacteria (Didierlaurent et al, 2002). The exacerbated response to pathogens might be explained, at least in part, by the ability of these organisms to penetrate into deeper tissue layers and produce effectors that directly stimulate inflammation (Galan, 2001). However, it has also been suggested that the absence of injury and/or the anti-inflammatory environment of the gut induces tolerance, because antigen presentation by dendritic cells occurs in the absence of costimulation (Sierro et al, 2001). Thus, in many instances the initiation of immune responses seems to require tissue damage, which is a speciality of pathogenic microorganisms. In addition to having mechanisms for invading and crossing cell barriers, pathogenic microbes produce a large number of membrane-damaging molecules, such as pore-forming toxins and specialized secretion systems, that puncture the membranes of eukaryotic cells. In addition to facilitating the contact of pathogens with deeper layers of mucosal surfaces, these mechanisms might promote the release of endogenous adjuvants from host cells.
There are several examples of situations in which injured cells stimulate immune responses in the absence of microorganisms. Immunity against tumours can be generated by experimentally inducing the death of cancer cells in situ (Barba et al, 1994; Caruso et al, 1993). CD4 T-cell responses are induced when mice are immunized with ovalbumin mixed with syngeneic cells that have been killed, but not when ovalbumin is injected alone (Gallucci et al, 1999). Although the exact mechanism by which injured cells influence immune responses is still not clear, they were shown to induce dendritic-cell maturation (Gallucci et al, 1999) and migration to draining lymph nodes in vivo (Shi & Rock, 2002). Thus, accumulating evidence suggests that both exogenous and endogenous adjuvants contribute to the initiation of immune responses by activating APCs (Rock et al, 2005).
Endogenous adjuvants
Although evidence indicates that injured cells release molecules with the capacity to modulate immune responses, little is known about the identity of these molecules. Heat-shock proteins (HSPs) were proposed to be endogenous adjuvant molecules due to their capacity to prime antigen-specific immunity in the absence of other adjuvants (Srivastava & Maki, 1991). However, subsequent studies raised the possibility that the observed immunostimulatory activity was a consequence of LPS contamination. LPS-free HSP60 and HSP70 were inactive, and very small amounts of LPS were able to restore adjuvant activity (Bausinger et al, 2002; Gao & Tsan, 2003). Additional studies in which HSPs are specifically inactivated or eliminated are required to clarify this issue. Such evidence has become available recently for the high mobility group box 1 (HMGB1) protein, which is both a nuclear factor and a secreted cytokine mediating responses to infection, injury and inflammation (Lotze & Tracey, 2005). Necrotic cells from HMGB1−/− mice were reported to have a reduced ability to promote inflammation, suggesting that release of this protein on cell death is a signal to alert the immune system (Scaffidi et al, 2002). Recently, two fractions with adjuvant activity were identified in the cytosol of ultraviolet-irradiated 3T3 mouse fibroblasts (Shi et al, 2003). Although the composition of the high molecular weight fraction is still unknown, the low molecular weight fraction was purified to homogeneity and identified as uric acid. The adjuvant activity of the purified fraction was abolished by treatment with the highly specific uric acid oxidase uricase, which eliminates the possibility of microbial contamination (Shi et al, 2003; for an excellent review, see Rock et al, 2005).
Maintenance of membrane integrity
The existence of endogenous adjuvants raises a puzzling question: what prevents the immune system from remaining in a state of constant activation, considering that cells are constantly dying and being replaced in vivo? One explanation is that pathological and non-pathological forms of cell death differ fundamentally in their capacity to trigger the release of endogenous adjuvants (Matzinger, 2001). In non-pathological situations cells die by apoptosis, a programmed death pathway in which cells, at least initially, maintain their membrane integrity. The large number of apoptotic cells that are generated in vivo are cleared rapidly by phagocytes in a non-inflammatory process (Henson et al, 2001). By contrast, necrotic death is characterized by plasma-membrane disruption and the release of intracellular contents, including the molecules that are responsible for sending 'danger' signals to the immune system. It is therefore not surprising that necrosis is usually associated with pathology and can lead to autoimmunity (Gallucci et al, 1999).
Both necrotic and apoptotic cells can be immunostimulatory (Rock et al, 2005). However, these observations are not inconsistent with the view that plasma-membrane disruption is required for the release of endogenous adjuvants. In most experimental settings, injury during cell manipulation cannot be ruled out, which possibly explains why apoptotic cell uptake by dendritic cells induces T-cell responses in some studies (Hoffmann et al, 2000), whereas in other studies only necrotic cells show immunostimulatory activity (Sauter et al, 2000). Apoptotic cells can also undergo secondary necrosis, with a concomitant release of their intracellular contents (Majno & Joris, 1995). This finding is consistent with the view that a delay in the clearance of apoptotic cells in vivo can lead to autoimmunity (Pisetsky, 2004). Apoptotic cells contain potent adjuvant activity (Shi et al, 2000), and so the maintenance of plasma-membrane integrity seems to be crucial for the prevention of undesired immune responses.
Plasma-membrane injury in vivo
Necrotic death is not the only mechanism by which endogenous adjuvants can be released in vivo. There is extensive evidence that the plasma membrane of cells in many tissues is injured and subsequently repaired without leading to cell death. In most of these studies, plasma-membrane wounding was monitored through the detection of membrane-impermeable molecules that entered the cytoplasm of intact cells. After imposition of a mechanical force, serum albumin, propidium iodide, lucifer yellow or fluorescent dextran were trapped inside viable cells from the skin (McNeil & Ito, 1990), lung (Doerr et al, 2005), skeletal muscle (McNeil & Khakee, 1992) and cardiac muscle (Clarke et al, 1995). Similar in vivo plasma-membrane wounds were detected in cells from experimentally undisturbed gastrointestinal tract (McNeil & Ito, 1989), endothelia (Yu & McNeil, 1992) and hair cells of the auditory papilla (Mulroy et al, 1998). The most pronounced detection of in vivo wounding was in the triceps muscle of mice subjected to downhill running, which reinforces the view that mechanical force is the main agent responsible for producing membrane disruptions in vivo (McNeil & Khakee, 1992). These studies show that injury to the plasma membrane of cells is physiologically a frequent event in vivo and they highlight the need for mechanisms to prevent endogenous adjuvant release and the development of autoimmunity.
Exocytosis-mediated plasma-membrane repair
Given the frequency by which cells are injured in vivo, it could be argued that this poses a problem for the containment of cytosolic molecules with strong immunostimulatory activity. However, it is important to realize that animal cells have a very efficient Ca2+-dependent mechanism for the rapid repair of wounds in their plasma membrane (Heilbrunn, 1956). In fibroblasts injured by a microneedle puncture, Ca2+ influx triggers plasma membrane resealing within 10–30 s (Steinhardt et al, 1994). Release of the cytosolic enzyme lactate dehydrogenase (LDH) is observed immediately after contraction wounding of fibroblasts, but further cytosol loss is rapidly stopped by membrane resealing (Chakrabarti et al, 2003; Reddy et al, 2001). Thus, the rapid repair of lesions in the plasma membrane could be a key mechanism for preventing the activation of immune responses under steadystate conditions (Fig 1).
Figure 1.
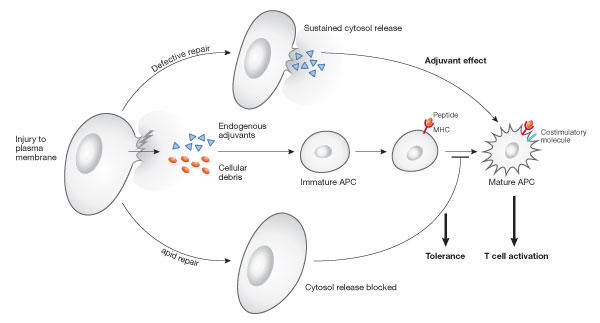
Resealing efficiency and release of endogenous adjuvants. Recent evidence suggests that the cytosol of mammalian cells contains molecules with adjuvant activity that are able to stimulate maturation of antigen-presenting cells (APCs) and activate naive T cells. Plasma-membrane repair might have a role in preventing immunostimulation, by limiting the release of cytosol from accidentally injured cells. (Modified from Rock et al, 2005).
It has long been known that the influx of Ca2+ occurring during injury to the plasma membrane triggers a resealing process that is mediated by exocytosis (McNeil & Steinhardt, 1997). This observation initially came as a surprise, as Ca2+-regulated exocytosis was largely regarded as a specialization of certain cell types (Stinchcombe & Griffiths, 1999). However, it soon became apparent that nonspecialized cells also contained Ca2+-regulated exocytic vesicles (Coorsen et al, 1996; Ninomiya et al, 1996). Subsequent studies identified lysosomes as the major vesicular population that fuses with the plasma membrane in response to Ca2+ influx (Jaiswal et al, 2002; Rodriguez et al, 1997).
A search for potential Ca2+ sensor molecules that regulate lysosomal exocytosis led to the detection of synaptotagmin VII (Syt VII) on lysosomes (Martinez et al, 2000). Dominant-negative inhibition or gene ablation showed that blocking Syt VII function inhibits both lysosomal exocytosis and membrane resealing (Chakrabarti et al, 2003; Martinez et al, 2000; Reddy et al, 2001). A recent report of the effects of the small molecule vacuolin 1 suggested that plasma-membrane repair could still occur in the absence of lysosomal exocytosis (Cerny et al, 2004). However, subsequent work showed that vacuolin 1, despite altering the morphology of lysosomes, does not inhibit the Ca2+-dependent fusion of lysosomes with the plasma membrane (Huynh & Andrews, 2005). Lysosomes therefore remain the most important candidates for the exocytic vesicular population that mediates membrane repair. Nevertheless, one aspect was apparently difficult to reconcile with such a role. Imaging experiments detected lysosomal exocytosis 30–150 s after stimulation with a Ca2+ ionophore (Jaiswal et al, 2002, 2004), which is slower than the kinetics of membrane resealing (Steinhardt et al, 1994). It is important to note, however, that ionophores cause a sharp decline in the ATP content of cells as a result of increased plasmalemmal Ca2+ ATPase activity and the dissipation of mitochondrial proton gradients (Gmitter et al, 1996). ATP depletion inhibits Ca2+-triggered lysosomal exocytosis (Rodriguez et al, 1997), which is expected given the requirement for the hexameric ATPase N-ethylmaleimidesensitive factor (NSF) in all intracellular membrane-fusion events (Littleton et al, 2001). Thus, the kinetics and properties of Ca2+ ionophore-triggered exocytosis should not be considered representative of what occurs under physiological conditions. This conclusion is reinforced by the fact that lysosomal exocytosis is reduced in Syt VII−/− cells stimulated by wounding (Chakrabarti et al, 2003) or by the receptor-mediated agonist bombesin (Jaiswal et al, 2004), but not after exposure to a Ca2+ ionophore (Jaiswal et al, 2004). A direct determination of the kinetics of lysosomal exocytosis triggered by membrane wounding should be possible as soon as the current technical obstacles are overcome.
Membrane repair: a defence against autoimmunity?
If plasma-membrane repair is important for limiting the release of endogenous adjuvants from cells, one prediction is that resealing defects might cause unregulated immunostimulation. There are two mouse models in which deletion of a single gene results in defective membrane repair: Syt VII−/− (Chakrabarti et al, 2003) and dysferlin−/− (Bansal et al, 2003) mice. Remarkably, Syt VII−/− mice develop an inflammatory process in the skin and skeletal muscle, with many similarities to the human autoimmune diseases polymyositis/dermatomyositis. Infiltration of inflammatory cells and foci of muscle-fibre destruction are observed in association with elevated serum creatine kinase activity, loss of muscle strength and the production of anti-nuclear antibodies—all of which are diagnostic features of autoimmune inflammatory myopathy. Interestingly, these animals also have a marked increase in skin collagen fibre content, consistent with what is observed in a number of human autoimmune syndromes (Chakrabarti et al, 2003). Other tissues show no detectable alterations, which suggests that the inflammatory process is restricted to tissues under mechanical stress. It is tempting to speculate that impaired plasma-membrane resealing in Syt VII−/− mice enhances the release of endogenous adjuvants in tissues in which injury is more frequent and that this results in local activation of the immune system. Testing this hypothesis requires the development of tools to inactivate endogenous adjuvants in vivo. There is already good evidence that dendritic cells activated by injured pancreatic-β cells can prime self-reactive T cells (Turley et al, 2003).
Defective membrane repair has also been shown in myofibres from dysferlin−/− mice (Bansal et al, 2003). Although present in other cell types, dysferlin is most abundantly expressed in skeletal and cardiac muscle. Interestingly, the cytosolic domain of dysferlin contains six C2 domains, some of which have predicted Ca2+ binding properties similar to the C2A and C2B domains of synaptotagmins (Bansal & Campbell, 2004; Davis et al, 2002). Dysferlin cycles between the sarcolemma and a population of intracellular vesicles the nature of which is still poorly understood. When muscle fibres are injured, Ca2+ binding to dysferlin C2 domains is thought to trigger the exocytosis of these vesicles, promoting membrane resealing (Bansal & Campbell, 2004; Bansal et al, 2003). Mutations in dysferlin are linked to two clinically distinct muscle-wasting diseases: limb-girdle muscular dystrophy type 2B and Miyoshi myopathy. Although autoimmunity is rare in human patients with these and other forms of muscular dystrophy (Confalonieri et al, 2003; Funauchi et al, 2002), SLJ/J mice that also carry mutations in dysferlin are considered a model for experimental autoimmune myopathy (Hart et al, 1987; Vafiadaki et al, 2001). It will be interesting to investigate how muscle fibres compare with other cell types, such as fibroblasts, regarding the amount of immunostimulatory molecules they contain and the rate by which these adjuvants are released on cell injury. Further studies using Syt VII- and dysferlin-deficient mice should provide important additional insights into the intriguing relationship between plasma membrane injury and repair, and how the immune system decides when to 'leap into action'.

Acknowledgments
I thank past and present members of my laboratory for their important contributions to the experiments and ideas discussed in this review, and Y. Shi (University of Massachusetts) for critical reading of the manuscript. Work in the Andrews laboratory is supported by National Institutes of Health (NIH) grants R37-AI34867 and RO1-GM064625.
References
- Bansal D, Campbell KP (2004) Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol 14: 206–213 [DOI] [PubMed] [Google Scholar]
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP (2003) Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423: 168–172 [DOI] [PubMed] [Google Scholar]
- Barba D, Hardin J, Sadelain M, Gage FH (1994) Development of anti-tumor immunity following thymidine kinase-mediated killing of experimental brain tumors. Proc Natl Acad Sci USA 91: 4348–4352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausinger H et al. (2002) Endotoxin-free heatshock protein 70 fails to induce APC activation. Eur J Immunol 32: 3708–3713 [DOI] [PubMed] [Google Scholar]
- Beutler B (2004) Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430: 257–263 [DOI] [PubMed] [Google Scholar]
- Caruso M, Panis Y, Gagandeep S, Houssin D, Salzmann JL, Klatzmann D (1993) Regression of established macroscopic liver metastases after in situ transduction of a suicide gene. Proc Natl Acad Sci USA 90: 7024–7028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerny J, Feng Y, Yu A, Miyake K, Borgonovo B, Klumperman J, Meldolesi J, McNeil PL, Kirchhausen T (2004) The small chemical vacuolin-1 inhibits Ca2+-dependent lysosomal exocytosis but not cell resealing. EMBO Rep 5: 883–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, Fowler KT, Gorelick FS, Andrews NW (2003) Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol 162: 543–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MS, Caldwell RW, Chiao H, Miyake K, McNeil PL (1995) Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circ Res 76: 927–934 [DOI] [PubMed] [Google Scholar]
- Confalonieri P, Oliva L, Andreetta F, Lorenzoni R, Dassi P, Mariani E, Morandi L, Mora M, Cornelio F, Mantegazza R (2003) Muscle inflammation and MHC class I up-regulation in muscular dystrophy with lack of dysferlin: an immunopathological study. J Neuroimmunol 142: 130–136 [DOI] [PubMed] [Google Scholar]
- Coorsen JR, Schmitt H, Almers W (1996) Ca2+ triggers massive exocytosis in Chinese hamster ovary cells. EMBO J 15: 3787–3791 [PMC free article] [PubMed] [Google Scholar]
- Davis DB, Doherty KR, Delmonte AJ, McNally EM (2002) Calciumsensitive phospholipid binding properties of normal and mutant ferlin C2 domains. J Biol Chem 277: 22883–22888 [DOI] [PubMed] [Google Scholar]
- Didierlaurent A, Sirard JC, Kraehenbuhl JP, Neutra MR (2002) How the gut senses its content. Cell Microbiol 4: 61–72 [DOI] [PubMed] [Google Scholar]
- Doerr CH, Gajic O, Berrios JC, Caples S, Abdel M, Lymp JF, Hubmayr RD (2005) Hypercapnic acidosis impairs plasma membrane wound resealing in ventilator injured lungs. Am J Respir Crit Care Med 171: 1371–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresser DW (1961) Effectiveness of lipid and lipidophilic substances as adjuvants. Nature 191: 1169–1171 [DOI] [PubMed] [Google Scholar]
- Dresser DW (1962) Specific inhibition of antibody production. II. Paralysis induced in adult mice by small quantities of protein antigen. Immunology 5: 378–388 [PMC free article] [PubMed] [Google Scholar]
- Dresser DW (1968) An assay for adjuvanticity. Clin Exp Immunol 3: 877–888 [PMC free article] [PubMed] [Google Scholar]
- Funauchi M, Nozaki Y, Yoo BS, Kinoshita K, Kanamaru A (2002) A case of limb-girdle muscular dystrophy with serum anti-nuclear antibody which led to a mistaken diagnosis of polymyositis. Clin Exp Rheumatol 20: 707–708 [PubMed] [Google Scholar]
- Galan JE (2001) Salmonella interactions with host cells: type III secretion at work. Annu Rev Cell Dev Biol 17: 53–86 [DOI] [PubMed] [Google Scholar]
- Gallucci S, Lolkema M, Matzinger P (1999) Natural adjuvants: endogenous activators of dendritic cells. Nat Med 5: 1249–1255 [DOI] [PubMed] [Google Scholar]
- Gao B, Tsan MF (2003) Recombinant human heat shock protein 60 does not induce the release of tumor necrosis factor alpha from murine macrophages. J Biol Chem 278: 22523–22529 [DOI] [PubMed] [Google Scholar]
- Gmitter D, Brostrom CO, Brostrom MA (1996) Translational suppression by Ca2+ ionophores: reversibility and roles of Ca2+ mobilization, Ca2+ influx, and nucleotide depletion. Cell Biol Toxicol 12: 101–113 [DOI] [PubMed] [Google Scholar]
- Hart MN, Linthicum DS, Waldschmidt MM, Tassell SK, Schelper RL, Robinson RA (1987) Experimental autoimmune inflammatory myopathy. J Neuropathol Exp Neurol 46: 511–521 [DOI] [PubMed] [Google Scholar]
- Heilbrunn L (1956) The Dynamics of Living Protoplasm. New York, NY, USA: Academic [Google Scholar]
- Henson PM, Bratton DL, Fadok VA (2001) Apoptotic cell removal. Curr Biol 11: R795–R805 [DOI] [PubMed] [Google Scholar]
- Hoffmann TK, Meidenbauer N, Dworacki G, Kanaya H, Whiteside TL (2000) Generation of tumorspecific T-lymphocytes by cross-priming with human dendritic cells ingesting apoptotic tumor cells. Cancer Res 60: 3542–3549 [PubMed] [Google Scholar]
- Huynh C, Andrews NW (2005) The small chemical vacuolin-1 alters the morphology of lysosomes without inhibiting Ca2+-regulated exocytosis. EMBO Rep 6: 843–847 (in this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal JK, Andrews NW, Simon SM (2002) Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol 159: 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal JK, Chakrabarti S, Andrews NW, Simon SM (2004) Synaptotagmin VII restricts fusion pore expansion during lysosomal exocytosis. PLoS Biol 2: 1224–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA Jr (1989) Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54: 1–13 [DOI] [PubMed] [Google Scholar]
- Kopp E, Medzhitov R (2003) Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol 15: 396–401 [DOI] [PubMed] [Google Scholar]
- Littleton JT, Barnard RJ, Titus SA, Slind J, Chapman ER, Ganetzky B (2001) SNARE-complex disassembly by NSF follows synaptic-vesicle fusion. Proc Natl Acad Sci USA 98: 12233–12238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5: 331–342 [DOI] [PubMed] [Google Scholar]
- Majno G, Joris I (1995) Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 146: 3–15 [PMC free article] [PubMed] [Google Scholar]
- Martinez I, Chakrabarti S, Hellevik T, Morehead J, Fowler K, Andrews NW (2000) Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol 148: 1141–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P (1994) Tolerance, danger, and the extended family. Annu Rev Immunol 12: 991–1045 [DOI] [PubMed] [Google Scholar]
- Matzinger P (2001) Essay 1: the Danger model in its historical context. Scand J Immunol 54: 4–9 [DOI] [PubMed] [Google Scholar]
- McNeil PL, Ito S (1989) Gastrointestinal cell plasma membrane wounding and resealing in vivo. Gastroenterology 96: 1238–1248 [DOI] [PubMed] [Google Scholar]
- McNeil PL, Ito S (1990) Molecular traffic through plasma membrane disruptions of cells in vivo. J Cell Sci 96: 549–556 [DOI] [PubMed] [Google Scholar]
- McNeil PL, Khakee R (1992) Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am J Pathol 140: 1097–1109 [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Steinhardt RA (1997) Loss, restoration and maintenance of plasma membrane integrity. J Cell Biol 137: 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulroy MJ, Henry WR, McNeil PL (1998) Noise-induced transient microlesions in the cell membranes of auditory hair cells. Hear Res 115: 93–100 [DOI] [PubMed] [Google Scholar]
- Ninomiya Y, Kishimoto T, Miyashita Y, Kasai H (1996) Ca2+-dependent exocytic pathways in Chinese hamster ovary fibroblasts revealed by a caged-Ca2+ compound. J Biol Chem 271: 17751–17754 [DOI] [PubMed] [Google Scholar]
- Pisetsky DS (2004) The immune response to cell death in SLE. Autoimmun Rev 3: 500–504 [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118: 229–241 [DOI] [PubMed] [Google Scholar]
- Reddy A, Caler E, Andrews N (2001) Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 106: 157–169 [DOI] [PubMed] [Google Scholar]
- Rock KL, Hearn A, Chen CJ, Shi Y (2005) Natural endogenous adjuvants. Springer Semin Immunopathol 26: 231–246 [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Webster P, Ortego J, Andrews NW (1997) Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol 137: 93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N (2000) Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med 191: 423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418: 191–195 [DOI] [PubMed] [Google Scholar]
- Shi Y, Evans JE, Rock KL (2003) Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425: 516–521 [DOI] [PubMed] [Google Scholar]
- Shi Y, Rock KL (2002) Cell death releases endogenous adjuvants that selectively enhance immune surveillance of particulate antigens. Eur J Immunol 32: 155–162 [DOI] [PubMed] [Google Scholar]
- Shi Y, Zheng W, Rock KL (2000) Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc Natl Acad Sci USA 97: 14590–14595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierro F, Dubois B, Coste A, Kaiserlian D, Kraehenbuhl JP, Sirard JC (2001) Flagellin stimulation of intestinal epithelial cells triggers CCL20-mediated migration of dendritic cells. Proc Natl Acad Sci USA 98: 13722–13727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava PK, Maki RG (1991) Stress-induced proteins in immune response to cancer. Curr Top Microbiol Immunol 167: 109–123 [DOI] [PubMed] [Google Scholar]
- Steinhardt RA, Guoqiang B, Alderton JM (1994) Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 263: 390–393 [DOI] [PubMed] [Google Scholar]
- Stinchcombe JC, Griffiths GM (1999) Regulated secretion from hemopoietic cells. J Cell Biol 147: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Rev Immunol 21: 335–376 [DOI] [PubMed] [Google Scholar]
- Turley S, Poirot L, Hattori M, Benoist C, Mathis D (2003) Physiological β cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med 198: 1527–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafiadaki E et al. (2001) Cloning of the mouse dysferlin gene and genomic characterization of the SJL-Dysf mutation. Neuroreport 12: 625–629 [DOI] [PubMed] [Google Scholar]
- Yu QC, McNeil PL (1992) Transient disruptions of aortic endothelial cell plasma membranes. Am J Pathol 141: 1349–1360 [PMC free article] [PubMed] [Google Scholar]