

Abstract
Given that ligand binding is essential for the rapid internalization of epidermal growth factor receptor (EGFR), the events induced by ligand binding probably contribute to the regulation of EGFR internalization. These events include receptor dimerization, activation of intrinsic tyrosine kinase activity and autophosphorylation. Whereas the initial results are controversial regarding the role of EGFR kinase activity in EGFR internalization, more recent data suggest that EGFR kinase activation is essential for EGFR internalization. However, we have shown here that inhibition of EGFR kinase activation by mutation or by chemical inhibitors did not block EGF-induced EGFR internalization. Instead, proper EGFR dimerization is necessary and sufficient to stimulate EGFR internalization. We conclude that EGFR internalization is controlled by EGFR dimerization, rather than EGFR kinase activation. Our results also define a new role for EGFR dimerization: by itself it can drive EGFR internalization, independent of its role in the activation of EGFR kinase.
Keywords: EGF receptor, endocytosis, receptor dimerization, receptor tyrosine kinase activation
Introduction
The internalization of constitutively internalized receptors is largely mediated by sorting signals such as YXXΦ and NPXY (Bonifacino & Dell'Angelica, 1999). However, for the receptors that are internalized in response to ligand binding, there is probably some means of switching their sorting signals on and off (Trowbridge et al, 1993). Given that ligand binding is essential for the rapid internalization of epidermal growth factor receptor (EGFR), the events induced by ligand binding probably contribute to the regulation of ligand-induced EGFR internalization. These events include receptor dimerization, activation of intrinsic tyrosine kinase activity and autophosphorylation. Whereas the initial results are controversial regarding the role of EGFR kinase activity in EGFR internalization (Chen et al, 1987; Honegger et al, 1987; Glenney et al, 1988; Felder et al, 1990, 1992), recent studies suggest that EGFR kinase activity is required for EGF-induced EGFR internalization (Lamaze & Schmid, 1995; Wilde et al, 1999; Sorkina et al, 2002; Schmidt et al, 2003). However, we recently showed that inhibition of EGFR kinase activation by the specific inhibitor AG1478 did not block EGFR internalization, which indicates that EGFR kinase activation is not required for EGFR internalization (Wang et al, 2002). As the only well-defined event that occurs in response to ligand binding and upstream of kinase activation is receptor dimerization, we propose that EGF-induced EGFR dimerization is the event that triggers EGFR internalization and that EGFR kinase activation and the consequent downstream signalling events are not required. In this communication, we tested this hypothesis and showed that elimination of EGFR kinase activation by mutation or chemical blockers does not inhibit EGFR internalization, whereas deletion of the extracellular dimerization loop of EGFR blocks dimerization and internalization. We conclude that dimerization of the receptor, not its kinase activity, is the trigger for ligand-induced endocytosis of EGFR.
Results
EGFR kinase activation and EGFR internalization
To determine whether EGFR kinase activation is required for EGF-induced EGFR internalization, we first revisited the previous controversy regarding the internalization of kinase-dead EGFR with a single mutation of lysine 721 to alanine (K721A). We have constructed a yellow fluorescent protein (YFP)-tagged K721A (K721A–YFP). Wild-type EGFR tagged to YFP (EGFR–YFP) was used as a control. When expressed in 293T cells, K721A–YFP was not phosphorylated by EGF stimulation (Fig 1A). In 293T cells transfected with EGFR–YFP, EGF strongly stimulated the phosphorylation of EGFR–YFP, and the phosphorylation was completely inhibited by the addition of EGFR kinase inhibitor AG1478.
Figure 1.
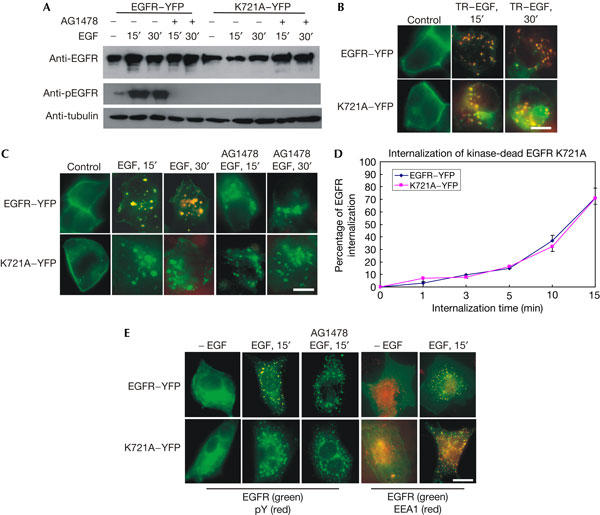
Internalization of kinase-dead epidermal growth factor receptor K721A. 293T cells (A–D) or Chinese hamster ovary (CHO) cells (E) were transiently transfected with epidermal growth factor receptor (EGFR)–YFP (yellow fluorescent protein) or K721A–YFP. (A) Immunoblotting. After EGF (100 ng/ml) stimulation, 293T cells were lysed and immunoblotted with indicated antibodies. (B) Intrinsic fluorescence. The cells were stimulated with Texas red (TR)-conjugated EGF (TR–EGF; 100 ng/ml), and the internalization of both EGFR and EGF was viewed by intrinsic fluorescence. (C) Indirect immunofluorescence. After EGF stimulation, the localization of EGFR (green) and phospho-tyrosine (pTyr; red) was shown by indirect immunofluorescence. (D) Quantitative analysis of EGFR–YFP and K721A–YFP internalization by flow cytometry. Data are the mean of at least three experiments performed in triplicate. (E) Indirect immunofluorescence analysis of the internalization of EGFR–YFP and K721A–YFP in CHO cells. CHO cells were transiently transfected with EGFR–YFP or K721A–YFP. After EGF (100 ng/ml) stimulation, the localization of EGFR (green) and pTyr (red) or EEA1 (red) was shown by indirect immunofluorescence. Scale bars, 20 μm.
The internalization of K721A–YFP was first examined by fluorescence microscopy. In 293T cells expressing K721A–YFP (control), YFP was detected at the plasma membrane. After incubation with Texas red (TR)-conjugated EGF (TR–EGF) at 37°C for 15 and 30 min, both TR and YFP were detected in endosomes (Fig 1B). Indirect immunofluorescence showed that EGF did not stimulate phosphorylation of K721A–YFP but stimulated its internalization (Fig 2C). EGFR–YFP was phosphorylated and internalized after EGF stimulation. The addition of AG1478 inhibited EGF-induced phosphorylation of EGFR–YFP, but it did not block its internalization (Fig 2C). The internalization of K721A–YFP and EGFR–YFP was further quantified by flow cytometry. As shown in Fig 1D, K721A–YFP was internalized after EGF stimulation in a similar pattern to EGFR–YFP. To determine further whether K721A is internalized in endosomes and that our observation is not limited only to 293T cells, we transfected Chinese hamster ovary (CHO) cells with K721A–YFP or EGFR–YFP. The localization of EGFR was shown by the intrinsic fluorescence of YFP, and the endosome marker early endosome antigen 1 (EEA1) was localized by indirect immunofluorescence with an anti-EEA1 antibody (Fig 1E). We showed that both K721A–YFP and EGFR–YFP were internalized in EEA1-positive endosomes in response to EGF.
Figure 2.
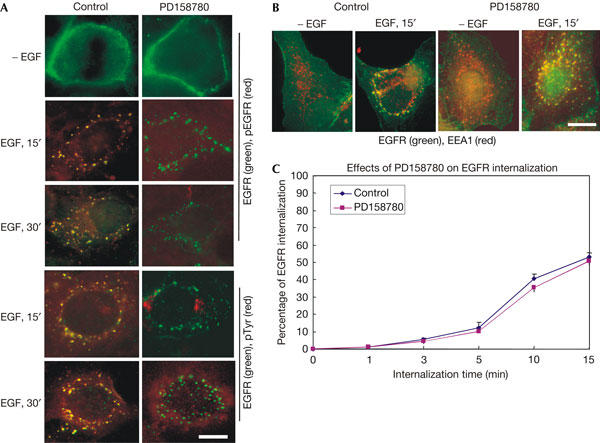
Internalization of epidermal growth factor receptor after treatment with PD158780. (A) BT20 cells were treated with PD158780 (100 nM) for 15 min and then stimulated with epidermal growth factor (EGF) (100 ng/ml) at 37°C for the indicated time. EGF receptor (EGFR; green) and phospho-tyrosine (pTyr; red) or phospho-EGFR (pEGFR; red) localization was determined by double indirect immunofluorescence. (B) BT20 cells were treated with PD158780 (100 nM) for 15 min and then stimulated with EGF (100 ng/ml) at 37°C for the indicated time. EGFR (green) and EEA1 (red) localization was determined by double indirect immunofluorescence. (C) Quantitative analysis of the effects of PD158780 on EGFR internalization in BT20 cells by flow cytometry. Data are the means of at least three experiments performed in triplicate. Scale bars, 20 μm.
We also tested the effect of another EGFR inhibitor, PD158780, on EGFR internalization. We treated BT20 cells with PD158780 and then stimulated cells with EGF. The phosphorylation and internalization of EGFR were examined by fluorescence microscopy and flow cytometry. We showed that PD158780 inhibited EGFR phosphorylation, but did not block EGFR internalization in EEA1-positive endosomes (Fig 2). Together, our results indicate that EGF-induced EGFR kinase activation is not required for EGFR internalization.
EGF-induced EGFR dimerization and internalization
As the only well-defined event between ligand binding and kinase activation is receptor dimerization, our results suggest that receptor dimerization is the crucial post-ligand-binding event in regulating EGFR internalization. To test this possibility, we first examined whether EGF-induced dimerization of EGFR is sufficient to stimulate EGFR internalization. We show in Fig 1 that inhibition of EGFR kinase activation did not block EGF-induced EGFR internalization. Then, we further determined whether EGFR is dimerized under these conditions. EGFR dimerization was examined by crosslinking with bis-(sulphosuccinimidyl suberate) (BS3) followed by immunoblotting. We showed that kinase-dead EGFR K721A is dimerized as wild-type EGFR after EGF stimulation for 15 min (Fig 3A). We also showed that inhibition of EGFR kinase activity in BT20 cells by AG1478 and PD158780 did not block EGF-induced EGFR dimerization (Fig 3B). Thus, EGF-induced EGFR dimerization without subsequent kinase activation is sufficient to stimulate EGFR internalization. It is interesting to note that both AG1478 and PD158780 induce the dimerization of EGFR in the absence of EGF stimulation, which is consistent with a previous report (Arteaga et al, 1997).
Figure 3.

Epidermal growth factor-induced receptor dimerization in the inhibition of EGFR kinase. (A) EGF-induced dimerization of K721A–YFP (yellow fluorescent protein). 293T cells were transiently transfected with EGFR–YFP or K721A–YFP. After EGF stimulation for 15 min, 293T cells were crosslinked with bis-(sulphosuccinimidyl suberate) (BS3). The cell lysates were immunoblotted with an anti-EGFR antibody. (B) EGF-induced EGFR dimerization in BT20 cells treated with either PD158780 or AG1478. BT20 cells were treated with either PD158780 or AG1478, and then stimulated with EGF for 15 min. The cells were crosslinked with bis-(sulphosuccinimidyl suberate) (BS3) and the cell lysates were immunoblotted with an anti-EGFR antibody.
Dimerization is sufficient for EGFR internalization
We next examined whether EGFR dimerization by itself, without ligand binding, is sufficient to stimulate EGFR internalization. EGFR was artificially dimerized using the receptor dimerization kit ARGENT™ Regulated Homodimerization kit from ARIAD Pharmaceuticals Inc. (Cambridge, MA, USA). The reagents in the kit are based on the human protein FKBP12 (FKBP for FK506-binding protein) and its small molecule ligands (Spencer et al, 1993). We first constructed the fusion protein by fusing the full-length EGFR with FKBP (EGFR–FKBP) according to the instructions provided by the company (Fig 4A). The fusion proteins were expressed in 293T cells. Crosslinking experiments showed that this fusion protein is dimerized and phosphorylated after the addition of AP20187 or EGF (Fig 4B). We also showed that AP20187-induced phosphorylation of EGFR–FKBP is inhibited by pretreatment with AG1478 (Fig 4C). This indicated that EGFR–FKBP was phosphorylated by its intrinsic kinase activity. The internalization of EGFR–FKBP was examined by indirect immunofluorescence. We showed that with or without inhibition of its kinase activation, EGFR–FKBP is internalized efficiently after its dimerization by AP20187 (Fig 4D). These results indicate that, in the absence of ligand binding and kinase activation, a proper dimerization itself is sufficient to stimulate EGFR internalization.
Figure 4.
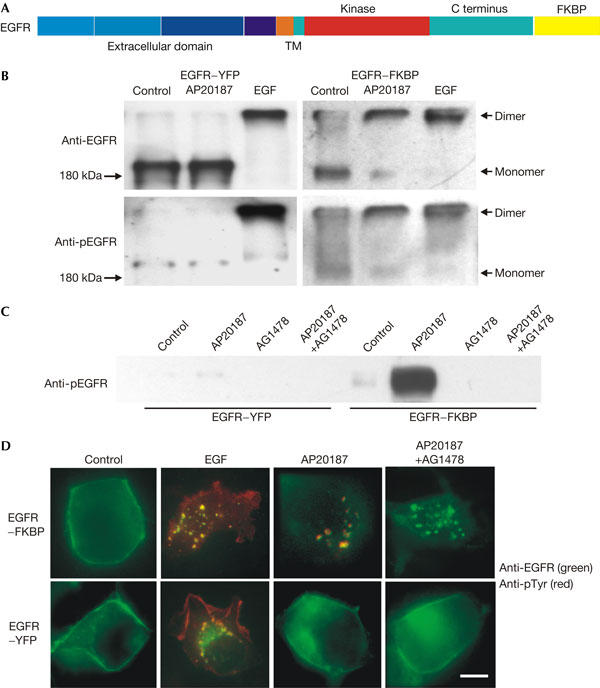
Epidermal growth factor receptor internalization after non-ligand-induced dimerization. (A) Schematic representation of full-length epidermal growth factor receptor (EGFR) fused with FKBP (EGFR–FKBP). C terminus, carboxyl terminus; TM, transmembrane. (B) EGFR–FKBP was expressed in 293T cells by transient transfection. After treatment with either AP20187 (100 nM) or EGF (100 ng/ml), the proteins were crosslinked with disuccinimidyl suberate (DSS). The cells were then lysed and the protein samples were immunoblotted with anti-EGFR and anti-pEGFR antibodies. (C) EGFR–FKBP was expressed in 293T cells by transient transfection. After treatment with AP20187 (100 nM) and/or AG1478 (0.5 μM), the cells were lysed and the protein samples were immunoblotted with anti-phospho-EGFR (pEGFR) antibodies. (D) Double indirect immunofluorescence. EGFR–FKBP was expressed in 293T cells by transient transfection. After treatment with EGF (100 ng/ml), AP20187 (100 nM) and AG1478 (0.5 μM), the localization of EGFR (green) and phospho-tyrosine (pTyr; red) was shown by double indirect immunofluorescence. Scale bar, 20 μm.
Inhibition of EGFR dimerization and internalization
We then examined whether EGF-induced EGFR dimerization is essential for EGF-induced internalization of EGFR. It was recently shown that an EGFR mutant with the deletion of its dimerization loop of domain II still bound to EGF, but was unable to dimerize and autophosphorylate after EGF binding (Garrett et al, 2002). However, it is not known whether this deletion mutant is able to internalize in response to EGF. We constructed the same mutant with the deletion of its dimerization loop and tagged this mutant with YFP (ΔCR1–YFP). We transiently expressed this mutant in Cos7 cells. Immunoblotting with an anti-phospho-EGFR (pEGFR) antibody showed that ΔCR1–YFP was not phosphorylated in response to EGF (Fig 5A). As Cos7 cells also express endogenous EGFR, we detected two bands in the anti-EGFR blot: the higher band is ΔCR1–YFP and the lower band is endogenous EGFR. Only one band, ΔCR1–YFP, was detected in immunoblot with antibody to GFP. Immunoblotting with anti-pEGFR showed that, in Cos7 cells transfected with EGFR–YFP, both endogenous and transfected EGFR were phosphorylated; however, in cells transfected with ΔCR1–YFP, only the endogenous EGFR (lower band) was phosphorylated (Fig 5A).
Figure 5.
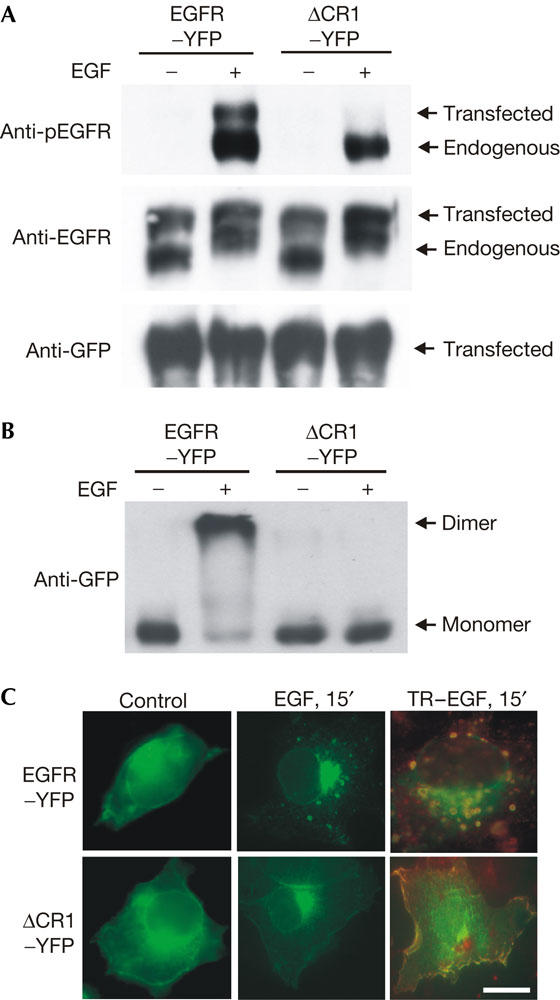
Inhibition of epidermal growth factor receptor internalization by blocking EGFR dimerization. The EGFR dimerization loop (amino acids 244–259) was deleted and the deletion mutant was tagged with enhanced yellow fluorescent protein (EYFP; ΔCR1–YFP). ΔCR1–YFP or EGFR–YFP was expressed in Cos7 cells by transient transfection and the cells were treated with EGF (100 ng/ml) or Texas red (TR)-conjugated EGF (TR–EGF; 100 ng/ml). (A) The cells were lysed and the protein samples were immunoblotted with indicated antibodies. (B) The cells were crosslinked with bis-(sulphosuccinimidyl suberate) (BS3) and lysed. The protein samples were immunoblotted with an anti-green fluorescent protein (GFP) antibody. (C) The localization of ΔCR1–YFP (green), EGFR–YFP (green) and TR–EGF (red) was shown by intrinsic fluorescence. Scale bar, 20 μm.
EGF-induced dimerization of ΔCR1–YFP was examined by crosslinking, followed by immunoblotting with antibodies to GFP. We showed that ΔCR1–YFP was not dimerized after EGF stimulation (Fig 5B). The internalization of ΔCR1–YFP was examined by fluorescence microscopy (Fig 5C). Cos7 cells were transfected with either EGFR–YFP or ΔCR1–YFP. Without EGF stimulation, both EGFR–YFP and ΔCR1–YFP were localized at the plasma membrane. After EGF stimulation for 15 min, EGFR–YFP was internalized into endosomes as expected. However, ΔCR1–YFP remained at the plasma membrane after the addition of EGF (Fig 5C). Moreover, after treatment with TR–EGF, both EGFR–YFP and the bound TR–EGF were internalized into endosomes, but ΔCR1–YFP and most of the bound TR–EGF were not internalized (Fig 5C).
We have also examined the dimerization, phosphorylation and internalization of ΔCR1 in BaF/3 cells stably expressing ΔCR1. We showed by immunoblotting that this mutant is not phosphorylated in response to EGF (Fig 6A). Crosslinking experiments further showed that ΔCR1 is not dimerized after EGF stimulation (Fig 6B). Moreover, flow cytometry showed that ΔCR1 was not internalized in response to EGF (Fig 6C). Together, these results indicate that EGF-induced EGFR dimerization is necessary for EGF-induced EGFR internalization.
Figure 6.
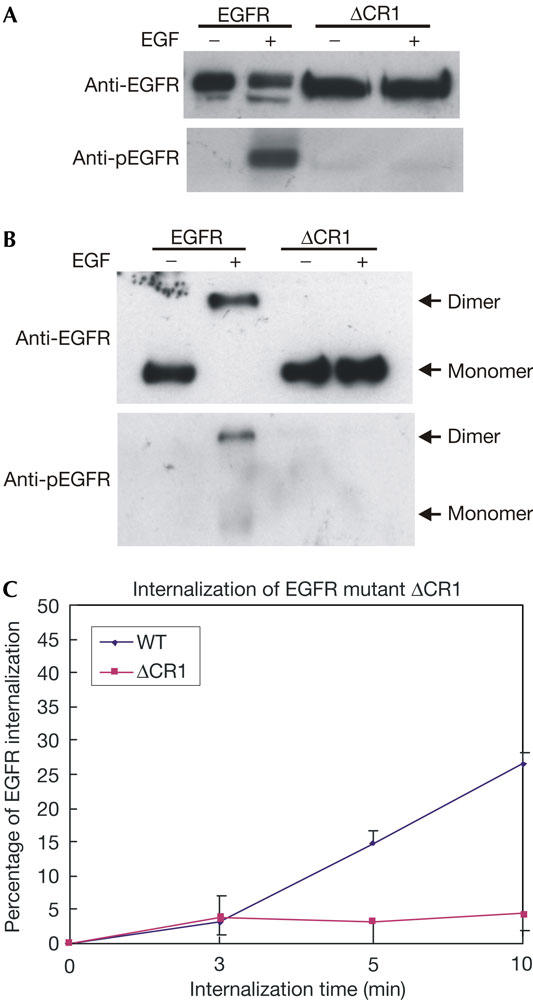
Inhibition of epidermal growth factor-induced internalization of ΔCR1 in BaF/3 cells stably expressing ΔCR1. BaF/3 cells stably expressing epidermal growth factor receptor (EGFR) or ΔCR1 were stimulated with EGF (100 ng/ml) for 15 min. (A) The cells were lysed and the protein samples were immunoblotted with anti-EGFR and anti-phospho-EGFR (pEGFR) antibodies. (B) The cells were crosslinked with bis-(sulphosuccinimidyl suberate) (BS3) and lysed. The protein samples were immunoblotted with anti-EGFR and anti-pEGFR antibodies. (C) Quantitative analysis of EGFR internalization in BaF/3 cells by flow cytometry. BaF/3 cells stably expressing EGFR or ΔCR1 were stimulated with EGF (100 ng/ml) for the indicated time and the internalization of ΔCR1 was analysed by flow cytometry. WT, wild type.
Discussion
In this paper, we further studied the role of EGFR kinase activity in EGFR internalization. We showed that inhibition of EGFR kinase activation by mutation or chemical inhibitors AG1478 and PD158780 did not block EGF-induced EGFR internalization (Figs 1, 2). Our results indicate that EGFR kinase activation is dispensable in EGF-induced EGFR internalization. Although our results contradict some recently published data (Lamaze & Schmid, 1995; Sorkina et al, 2002; Schmidt et al, 2003), they are consistent with some of the early data published by Schlessinger's group (Honegger et al, 1987; Felder et al, 1990, 1992). We are not sure why some other groups' results are different to ours. However, several factors may be involved. First, as the inhibition of EGFR kinase results in the recycling of the receptor instead of the degradation of the receptor in lysosomes, it is possible that some studies missed the recycling process and thus concluded that EGFR is not internalized at all. Second, the cell lines and assay methods used in different labs vary greatly. It has been suggested that the requirement for EGFR kinase in EGFR internalization may vary significantly in different cell lines (Jiang et al, 2003). Third, EGFR kinase activation, although dispensable, may have subtle roles in EGFR internalization.
The most important and novel finding of this study is that EGFR dimerization is necessary and sufficient for EGF-induced EGFR internalization (Figs 3, 4, 5 and 6). As EGFR kinase activation is not essential for EGFR internalization, the driving force of EGFR internalization might be (i) the ligand binding itself, (ii) the dimerization of EGFR, or (iii) ligand binding and the dimerization of EGFR.
We showed here that inhibition of EGFR dimerization by deleting the dimerization loop inhibited EGF-induced EGFR internalization (Figs 5, 6). This indicates that EGF binding by itself, in the absence of EGFR dimerization, is not able to stimulate EGFR endocytosis. A previous study has shown that ligand binding in the absence of receptor dimerization is not sufficient to activate EGFR and downstream signalling (Garrett et al, 2002). These results suggest that dimerization is essential for EGF to initiate EGFR internalization and EGFR-mediated cell signalling. Moreover, we also showed that non-ligand-induced dimerization of EGFR in the absence of EGF is sufficient to stimulate EGFR internalization. This suggests that proper dimerization itself is the driving force for EGFR internalization.
Our data define a novel function for EGFR dimerization: EGF-induced EGFR dimerization by itself triggers the internalization of EGFR. Previously, the crucial role of receptor dimerization in EGF- stimulated EGFR signalling has been exclusively linked to its role in the activation of EGFR tyrosine kinase (Heldin, 1995; Schlessinger, 2002). Our results indicate that EGFR dimerization can mediate the EGF-induced cellular process independently of its role in activating EGFR tyrosine kinases.
This finding is also important in guiding future research efforts in explaining the molecular mechanisms that regulate EGFR internalization. So far, studies have focused on the role of EGFR tyrosine kinase activity and the phosphorylation of the EGFR carboxyl terminus and other downstream proteins in regulating EGFR internalization. Our results indicate that signals other than tyrosine phosphorylation are essential in regulating EGFR internalization. Thus, future research should be directed at finding out how EGFR dimerization triggers EGFR internalization. There are two possibilities. It is possible that EGF-induced EGFR dimerization causes necessary conformational changes of the receptor, to expose the cryptic internalization codes. Alternatively, the internalization-regulating proteins essential for EGFR internalization may have a dimeric nature and can only bind to dimerized EGFR.
Many receptors including platelet-derived growth factor receptor, nerve growth factor receptor and hepatocyte growth factor receptor are dimerized and internalized after ligand binding (Oved & Yarden, 2002; Schlessinger, 2002). Thus, it is possible that our finding for EGFR may also be applicable to these receptor tyrosine kinases.
Methods
Cell culture, treatment and transfection. Cos7, 293T, CHO and BT20 cells were grown in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. BaF/3 cells stably expressing wild-type EGFR or mutant EGFR ΔCR1 were cultured as described previously (Garrett et al, 2002). For the inhibition experiments with PD158780 or AG1478, BT-20 cells were serum starved for 24 h, and PD158780 (100 nM) or AG1478 (0.5 μM) was applied. EGF was then added to a final concentration of 100 ng/ml for the indicated time. For the transient transfection, 293T cells were transfected using the calcium phosphate method and Cos7 cells were transfected with lipofectin 2000. To dimerize EGFR–FKBP expressed in 293T cells, the cells were treated with AP20187 (100 nM) for 30 min.
Antibodies and chemicals. Rabbit anti-EGFR (1005), goat anti-phospho-EGFR (pEGFR), mouse anti-phospho-tyrosine (pTyr) and mouse anti-tubulin antibodies were from Santa Cruz Biotech (Santa Cruz, CA, USA). Mouse anti-pEGFR antibody was from Upstate Biotechnology Inc. (Lake Placid, NY, USA). Rabbit anti-GFP antibody and pEYFP-N1 (enhanced YFP) vector were from Clonetech (Palo Alto, CA, USA). Alexa Fluor 647-labelled EGF and TR-labelled EGF were from Molecular Probes Inc. (Eugene, OR, USA). Unless otherwise specified, all the chemicals were from Sigma (St Louis, MO, USA).
Plasmid construction. EGFR–FKBP was constructed using the receptor dimerization kit ARGENT™ Regulated Homodimerization kit from ARIAD Pharmaceuticals Inc. The EYFP-tagged dimerization loop deletion mutant EGFR (ΔCR1–YFP) was generated according to Garrett et al (2002). The only modification was that we used our previously constructed EGFR–YFP, instead of untagged EGFR, as a template.
Flow cytometry. EGFR internalization was assayed by flow cytometry modified from that previously described (Duan et al, 2003). Briefly, cells with 80% confluence were serum starved for 6 h. The cells were then incubated with Alexa Fluor 647-labelled EGF at 4°C for 30 min. After washing, cells were incubated at 37°C for indicated durations to allow internalization. After acidic wash (0.2 M acetic acid and 0.5 M NaCl (pH 2.8)) to remove non-internalized EGF, the fluorescence emission of internalized EGF was detected by flow cytometry.
Crosslinking immunoblotting and fluorescence microscopy. 293T, Cos7, BaF/3 and BT20 cells were cultured in 60 mm dishes to subconfluency. The cells with or without transfection were serum starved overnight. After treatment with EGF for 30 min at 4°C and then 5 min at 37°C, the cells were collected in 0.2–0.5 ml PBS. Disuccinimidyl suberate (DSS; for cells transfected with the EGFR–FKBP construct) or BS3 (for cells transfected with other constructs) was added to a final concentration of 2.5–5 mM and incubated on ice for 2 h. The quench solution (1 M Tris, pH 7.5, 1:100 dilution) was then added to a final concentration of 10 mM and incubated for 15 min on ice. The cells were then lysed with 1% NP-40 and EGFR dimerization was analysed by immunoblotting. Immunoblotting and fluorescence microscopy were carried out as described previously (Wang et al, 2002).
Acknowledgments
We thank R. Campenot for reading the manuscript, C.W. Ward for providing BaF/3 cells stably expressing ΔCR1 and ARIAD Pharmaceuticals Inc. for providing the ARGENT™ Regulated Homodimerization kit. This work was supported in part by grants from the Canadian Institutes of Health Research (CIHR) and the Alberta Heritage Foundation for Medical Research (AHFMR). Z.W. is an AHFMR Senior Scholar.
References
- Arteaga CL, Ramsey TT, Shawver LK, Guyer CA (1997) Unliganded epidermal growth factor receptor dimerization induced by direct interaction of quinazolines with the ATP binding site. J Biol Chem 272: 23247–23254 [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Dell'Angelica EC (1999) Molecular bases for the recognition of tyrosine-based sorting signals. J Cell Biol 145: 923–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WS, Lazar CS, Poenie M, Tsien RY, Gill GN, Rosenfeld MG (1987) Requirement for intrinsic protein tyrosine kinase in the immediate and late actions of the EGF receptor. Nature 328: 820–823 [DOI] [PubMed] [Google Scholar]
- Duan L et al. (2003) Cbl-mediated ubiquitinylation is required for lysosomal sorting of epidermal growth factor receptor but is dispensable for endocytosis. J Biol Chem 278: 28950–28960 [DOI] [PubMed] [Google Scholar]
- Felder S, Miller K, Moehren G, Ullrich A, Schlessinger J, Hopkins CR (1990) Kinase activity controls the sorting of the epidermal growth factor receptor within the multivesicular body. Cell 61: 623–634 [DOI] [PubMed] [Google Scholar]
- Felder S, LaVin J, Ullrich A, Schlessinger J (1992) Kinetics of binding, endocytosis, and recycling of EGF receptor mutants. J Cell Biol 117: 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett TP et al. (2002) Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor α. Cell 110: 763–773 [DOI] [PubMed] [Google Scholar]
- Glenney JR Jr, Chen WS, Lazar CS, Walton GM, Zokas LM, Rosenfeld MG, Gill GN (1988) Ligand-induced endocytosis of the EGF receptor is blocked by mutational inactivation and by microinjection of anti-phosphotyrosine antibodies. Cell 52: 675–684 [DOI] [PubMed] [Google Scholar]
- Heldin CH (1995) Dimerization of cell surface receptors in signal transduction. Cell 80: 213–223 [DOI] [PubMed] [Google Scholar]
- Honegger AM, Szapary D, Schmidt A, Lyall R, Van Obberghen E, Dull TJ, Ullrich A, Schlessinger J (1987) A mutant epidermal growth factor receptor with defective protein tyrosine kinase is unable to stimulate proto-oncogene expression and DNA synthesis. Mol Cell Biol 7: 4568–4571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Huang F, Marusyk A, Sorkin A (2003) Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol Biol Cell 14: 858–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamaze C, Schmid SL (1995) Recruitment of epidermal growth factor receptors into coated pits requires their activated tyrosine kinase. J Cell Biol 129: 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oved S, Yarden Y (2002) Signal transduction: molecular ticket to enter cells. Nature 416: 133–136 [DOI] [PubMed] [Google Scholar]
- Schlessinger J (2002) Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 110: 669–672 [DOI] [PubMed] [Google Scholar]
- Schmidt MH, Furnari FB, Cavenee WK, Bogler O (2003) Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc Natl Acad Sci USA 100: 6505–6510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina T, Huang F, Beguinot L, Sorkin A (2002) Effect of tyrosine kinase inhibitors on clathrin-coated pit recruitment and internalization of epidermal growth factor receptor. J Biol Chem 277: 27433–27441 [DOI] [PubMed] [Google Scholar]
- Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR (1993) Controlling signal transduction with synthetic ligands. Science 262: 1019–1024 [DOI] [PubMed] [Google Scholar]
- Trowbridge IS, Collawn JF, Hopkins CR (1993) Signal-dependent membrane protein trafficking in the endocytic pathway. Annu Rev Cell Biol 9: 129–161 [DOI] [PubMed] [Google Scholar]
- Wang Y, Pennock S, Chen X, Wang Z (2002) Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol Cell Biol 22: 7279–7290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde A, Beattie EC, Lem L, Riethof DA, Liu SH, Mobley WC, Soriano P, Brodsky FM (1999) EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell 96: 677–687 [DOI] [PubMed] [Google Scholar]