

Abstract
The fusion of Abl with either Bcr or Tel in human leukaemia leads to the constitutive activation of Abl tyrosine kinase, which in turn induces growth-factor-independent proliferation and cell survival. However, the mechanism by which Bcr–Abl induces cellular transformation has not yet been well characterized. Here, we show that Bcr–Abl-expressing cells are resistant to growth inhibition and apoptosis mediated by transforming growth factor-β (TGF-β). Interestingly, we observed that the suppressive effects of Bcr–Abl on TGF-β responses were not mediated by an impairment of Smad signalling, which is believed to act as the principal mediator of TGF-β responses. In contrast, we found that Bcr–Abl can target the protein kinase AKT and the transcription factor FoxO3 to interfere with growth inhibition and apoptosis in response to TGF-β. Our results show a novel mechanism of cellular transformation by the oncogenic fusion protein Bcr–Abl through suppression of the cytostatic actions of TGF-β.
Keywords: AKT, apoptosis, Bcr–Abl, growth arrest, TGF-β
Introduction
Abl is a protein tyrosine kinase that is involved in cellular transformation induced by chromosomal translocations found in human leukaemia: Bcr–Abl and Tel–Abl (Crans & Sakamoto, 2001; Keung et al, 2002). It has been established that the coiled-coil domain of Bcr or the helix–loop–helix domain of Tel provokes dimerization of the fusion protein, with the subsequent constitutive activation of Abl. This leads to the activation of signalling pathways that are known to be responsible for suppression of apoptosis, as well as induction of growth-factor-independent proliferation. The best characterized of these is the phosphatidylinositol 3-OH-kinase (PI3K) signalling pathway. Once activated, PI3K activates the protein kinase AKT, which in turn phosphorylates several target molecules, including the transcription factor FoxO, the activities of which are related to cell proliferation, apoptosis and tumorigenesis (Accili & Arden, 2004). The consequence of AKT-induced phosphorylation of FoxO is to sequester these factors in the cytoplasm and thus negatively regulate their transcriptional activity. Loss of function of FoxO3 after its inactivation by AKT is linked directly to oncogenic transformation by Bcr–Abl (Kharas et al, 2004).
Transforming growth factor-β (TGF-β) is a potent negative growth-regulatory factor that inhibits cell proliferation by arresting progression through the cell cycle or by inducing apoptosis (Roberts, 1998). Signalling by TGF-β is initiated by the formation of a heteromeric complex consisting of two types of transmembrane serine/threonine kinase receptor. After ligand binding, the type II receptor kinase phosphorylates and activates the type I receptor kinase, which in turn phosphorylates Smad2 and Smad3. After phosphorylation, Smad2 and Smad3 form heteromeric complexes with Smad4 and translocate to the nucleus where they regulate gene expression (Massague & Wotton, 2000; Moustakas et al, 2001). The ability of Smad proteins to modulate transcription in response to ligand results from a functional association with distinct DNA-binding factors, including members of the forkhead superfamily such as FAST1, FAST2 and FoxO (Seoane et al, 2004).
In this study, we investigated whether cellular transformation by Bcr–Abl might interfere with TGF-β signalling in haematopoietic cells. We show that expression of Bcr–Abl inhibits TGF-β-mediated growth arrest and apoptosis. These findings shed light on the mechanism of cellular transformation by Bcr–Abl.
Results and Discussion
To investigate the effects of Bcr–Abl on TGF-β-mediated growth-inhibitory signals, we generated a pool of Ba/F3 cells that stably expressed Bcr–Abl (B/A; Fig 1A). As described previously (Maru et al, 1996), stable expression of Bcr–Abl rendered these cells independent of interleukin-3 (IL-3) for growth (Fig 2B). In Ba/F3 cells, TGF-β treatment induced growth arrest, as shown by an accumulation of cells in the G1 phase of the cell cycle at 24 h (Fig 1B), followed by a decrease in cell number at 48 h (Fig 1C). Interestingly, stable expression of Bcr–Abl in Ba/F3 cells blocked TGF-β-induced growth arrest (Fig 1B,C). In addition to inducing growth arrest, TGF-β can also induce death in Ba/F3 cells, which occurs at about 48 h. Analysis of DNA fragmentation (Fig 1D), cell shrinkage (Fig 1E) or cleavage of the caspase-3 substrate poly(ADP-ribose) polymerase (PARP; Fig 1F) indicated that the apoptotic effect of TGF-β was blocked in B/A cells. These results raised the possibility that the cellular transformation of Ba/F3 cells by Bcr–Abl might confer resistance to the negative growth-regulatory effects of TGF-β.
Figure 1.
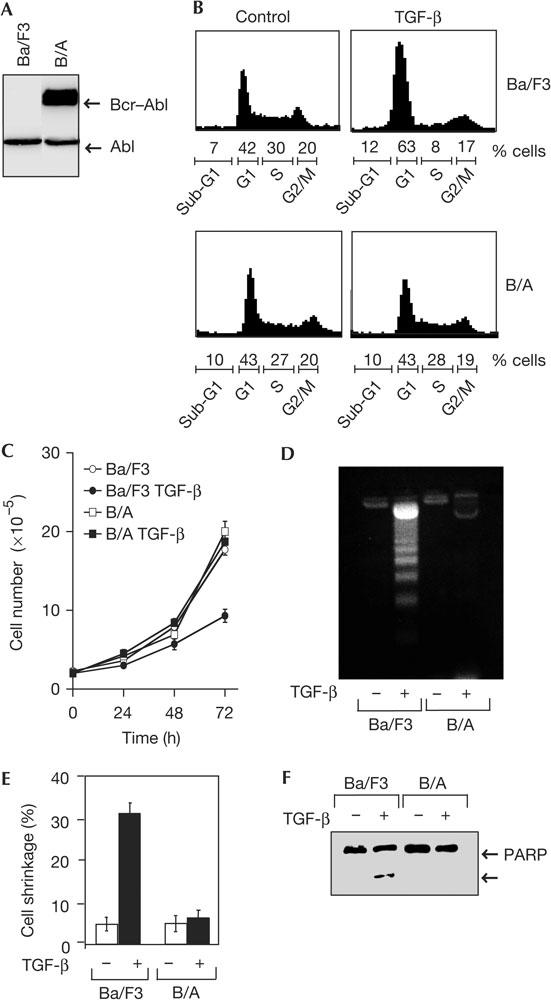
Expression of Bcr–Abl in Ba/F3 cells induces resistance to transforming growth factor-β-mediated growth inhibition and apoptosis. (A) Cell extracts from Ba/F3 cells that were stably transfected with empty vector (Ba/F3) or Bcr–Abl (B/A) were analysed by western blot with an anti-Abl antibody. (B) Ba/F3 or B/A cells cultured in the presence or absence of interleukin-3 (IL-3), respectively, were treated with or without transforming growth factor-β (TGF-β; 4 ng/ml) for 24 h and analysed by flow cytometry. (C) The effects of TGF-β on the proliferation of Ba/F3 or B/A cells were examined by determining cell numbers at different time periods. In all cases, the results shown are expressed as mean±s.d. of triplicates from a representative experiment performed at least three times. (D–F) Ba/F3 or B/A cells were incubated with or without TGF-β (4 ng/ml) for 48 h and apoptosis was determined by analysis of nucleosomal DNA fragmentation (D), cell shrinkage (E) or cleavage of the caspase-3 substrate poly(ADP-ribose) polymerase (PARP; F).
Figure 2.

The inhibitory effect of Bcr–Abl on transforming growth factor-β-induced growth-inhibitory signals is linked to the presence of Abl. (A) Cell extracts from Ba/F3 cells stably transfected with empty vector (Ba/F3), Tel–PDGFRβ (platelet-derived growth factor receptor-β; T/P), Tel–Abl (T/A) or Bcr–Abl (B/A) were analysed by immunoblotting with anti-Abl (left panel) or anti-PDGFRβ (right panel) antibodies. WB, western blot. (B) Ba/F3, T/P, T/A or B/A cells were cultured in the absence (Ba/F3, T/P, T/A and B/A) or presence (Ba/F3) of interleukin-3 (IL-3) and cell numbers were determined at different time periods. (C,D) Ba/F3, T/P, T/A or B/A cells were incubated with or without TGF-β, and cell number (C) or cell shrinkage and DNA fragmentation (D) were determined after 48 h of treatment. (E,F) Ba/F3 or B/A cells were cultured in the presence of IL-3 and the indicated combinations of transforming growth factor-β (TGF-β) and imatinib for 48 h, and cell numbers (E) or apoptosis (F) were examined. (G) K562 cells were cultured in Stem.AE medium in the presence of the indicated combinations of imatinib and TGF-β. Cells were analysed for relative DNA content by flow cytometry after 24 h of treatment.
Several oncogenic fusion proteins with tyrosine kinase activity have been identified in human leukaemia (Crans & Sakamoto, 2001). To determine whether the resistance to TGF-β-mediated growth arrest and apoptosis was specific to Bcr–Abl, we investigated the effects of TGF-β on Ba/F3 cells that stably expressed two other oncogenic fusion proteins, Tel–Abl and Tel–PDGFRβ (platelet-derived growth factor receptor-β). The expression of these fusion proteins is shown in Fig 2A, and their functionality is provided by the ability of the cell lines to proliferate in the absence of IL-3 (Fig 2B). As shown in Fig 2C,D, TGF-β-induced growth arrest and apoptosis were blocked by Bcr–Abl and Tel–Abl, but not by Tel–PDGFRβ, which suggests that the constitutive activation of the Abl kinase might have an important role in the suppression of TGF-β-mediated growth-inhibitory signals. To test this directly, we made use of an inhibitor of Abl kinase activity, imatinib. As expected, the growth of B/A cells was completely blocked by imatinib, but it could be rescued by the addition of IL-3 (data not shown). Interestingly, treatment of B/A cells with imatinib and IL-3 restored the sensitivity of cells to TGF-β-mediated growth inhibition (Fig 2E) and cell death (Fig 2F). Taken together, these results indicate that the presence of Abl in the fusion protein is responsible for the resistance to TGF-β-mediated growth arrest and apoptosis.
To obtain direct evidence that Bcr–Abl interferes with the negative growth-regulatory effects of TGF-β under physiological conditions, we took advantage of the availability of the leukaemia cell line K562, which was established from a patient with aberrant translocation, leading to generation of the oncogenic fusion protein Bcr–Abl. As shown in Fig 2G, the proliferation of K562 cells was not affected by TGF-β treatment. In contrast, the ability of TGF-β to induce growth arrest of K562 cells was restored when the cells were cultured with imatinib (Fig 2G). Together, these data indicate that Bcr–Abl might act under physiological conditions to suppress the negative growth-regulatory effects of TGF-β.
In an attempt to determine the mechanism underlying the inhibitory effects of Bcr–Abl, we found that expression of this oncogenic protein did not interfere with the ability of TGF-β to induce phosphorylation of Smad2 (Fig 3A) and its heterodimerization with Smad4 (Fig 3B). We also investigated whether the loss of the negative regulatory effects of TGF-β might be related to Bcr–Abl inhibition of the transcriptional functions of Smad, using the TGF-β-responsive reporter p3TP-Lux. As shown in Fig 3C, the TGF-β-dependent activation of p3TP-Lux was not lost in B/A cells. A similar conclusion could be drawn in gene reporter assays using the luciferase (CAGA)9-Lux reporter (Fig 3C), which contains concatamerized CAGA elements known to bind to the Smad3–Smad4 complex in response to TGF-β (Dennler et al, 1998). To provide further evidence that expression of Bcr–Abl failed to inhibit the transcriptional activity of Smad, we tested the effects of TGF-β on endogenous JunB expression, which contains the CAGA sequence in its promoter (Jonk et al, 1998). As shown in Fig 3D, Bcr–Abl had no effect on TGF-β-mediated endogenous JunB expression, which confirmed that Smad signalling was functional. We also investigated the ability of TGF-β to induce gene expression in the K562 cell line naturally bearing the Bcr–Abl translocation, and found that TGF-β-induced expression from the (CAGA)9-Lux reporter remained unchanged irrespective of whether K562 cells were cultured in the presence or absence of the Abl inhibitor imatinib (Fig 3E). Similarly, the ability of TGF-β to induce expression of endogenous JunB was not affected in K562 cells that were cultured in the absence or presence of imatinib (Fig 3F). Taken together, these data indicate that Bcr–Abl might suppress TGF-β-mediated cytostatic signals without interfering with the activation of Smad signalling.
Figure 3.

Bcr–Abl suppresses transforming growth factor-β-induced cytostatic responses without blocking the activation of the Smad pathway. (A) Ba/F3 or B/A cells were transfected with Myc–Smad2 and then treated with or without transforming growth factor-β (TGF-β) for 1 h. The phosphorylation of Smad2 was assessed by direct immunoblotting using a specific anti-phospho-Smad2 (p-Smad2) antibody. The blot was then reprobed with an anti-Myc antibody. (B) Ba/F3 or B/A cells were co-transfected with Myc–Smad2 and haemagglutinin (HA)–Smad4. Cells were then treated with or without TGF-β for 1 h and cell lysates were subjected to anti-Myc immunoprecipitation (IP) and then immunoblotted with the anti-HA antibody. Expression of transfected DNA was monitored by direct immunoblotting with anti-Myc or anti-HA antibodies. (C) Ba/F3 or B/A cells were transiently transfected with the indicated reporter constructs and treated with or without TGF-β for 18 h. Luciferase activity was normalized to β-galactosidase activity and was expressed as mean±s.d. of triplicates from a representative experiment performed at least three times. (D) Ba/F3 or B/A cells were treated with TGF-β for different time periods and expression of JunB was assessed by direct immunoblotting of cell lysates with an anti-JunB antibody. (E) K562 cells were transiently transfected with the (CAGA)9-Lux and cultured in Stem.AE medium in the presence of the indicated combinations of imatinib and TGF-β for 18 h before being analysed for luciferase activity. (F) K562 cells were cultured in the presence of the indicated combinations of imatinib and TGF-β for 3 h and expression of JunB was assessed by direct immunoblotting of cell lysates with an anti-JunB antibody. The blot was then reprobed with an anti-actin antibody.
The activation of the PI3K/AKT signalling pathway, which is essential for cellular transformation by Bcr–Abl, also leads to the suppression of TGF-β-mediated growth arrest and apoptosis (Komatsu et al, 2003; Seoane et al, 2004). We therefore investigated the effect of the chemical inhibitor of the PI3K signalling pathway, LY 294002, on the ability of Bcr–Abl to suppress TGF-β-dependent growth arrest. The results shown in Fig 4A,B indicate that LY 294002 was able to restore the sensitivity of B/A cells to TGF-β-induced growth arrest (Fig 4A) and apoptosis (Fig 4B). As the effects of LY 294002 and TGF-β are additive, this result indicates that TGF-β signalling might target other pathways that converge with the AKT pathway to regulate the cytostatic responses of TGF-β. Similar results were obtained when K562 cells were used instead of B/A cells (data not shown), which supports the idea that activation of the PI3K/AKT signalling pathway by Bcr–Abl under physiological conditions interferes with TGF-β-mediated cytostatic responses.
Figure 4.

Bcr–Abl activates the AKT pathway to suppress transforming growth factor-β-mediated growth-inhibitory signals. (A,B) Bcr–Abl (B/A) cells were treated with LY204009 (LY; 20 μM) in the presence or absence of transforming growth factor-β (TGF-β). Cell cycle was analysed 24 h later by flow cytometry (A). Apoptosis was determined by analysis of cell shrinkage or DNA fragmentation after 48 h of TGF-β treatment (B). (C) Ba/F3 or B/A cells were treated with TGF-β for the indicated periods of time and cell lysates were analysed by immunoblotting with an anti-phospho-AKT (p-AKT) antibody. The blot was then reprobed with an anti-AKT antibody. (D) Ba/F3, B/A or K562 cells were treated with imatinib or LY204009 for 1 h and then with TGF-β for 3 h. Cell lysates were analysed by immunoblotting with anti-phospho-AKT or anti-AKT antibodies.
Next, we explored how TGF-β regulates the AKT signalling pathway in Ba/F3 cells versus B/A cells. To this end, we examined the activation of AKT using an anti-phospho-AKT antibody that recognizes the phosphorylated Ser 473 and thus the activated AKT. We observed that TGF-β induced a strong decrease in the phosphorylation (activation) of AKT in Ba/F3 cells, which is consistent with its ability to induce cytostatic actions in these cells (Fig 4C). In marked contrast, treatment of B/A cells with TGF-β failed to induce any change in the phosphorylation of AKT (Fig 4C), which supports the idea that Bcr–Abl might function to restrain growth-inhibitory signals in response to TGF-β by interfering with the ability of TGF-β to limit the activation of the AKT pathway. Because Tel–PDGFRβ can also activate AKT (Dierov et al, 2002), it will be interesting to investigate in the future whether TGF-β induces growth-inhibitory signals in Tel–PDGFRβ-expressing cells by suppressing the Tel–PDGFRβ-dependent activation of AKT.
To provide further evidence that the activation of AKT by Bcr–Abl has a role in suppressing the cytostatic responses of TGF-β, we focused on the AKT substrate FoxO3, a transcriptional factor known to mediate growth inhibition as well as apoptosis after inhibition of the AKT pathway, and also in response to TGF-β signalling (Accili & Arden, 2004; Seoane et al, 2004). Evidence indicates that AKT-mediated phosphorylation of FoxO3 leads to inactivation of FoxO3 by facilitating its association with 14-3-3 proteins, which results in its retention in the cytoplasm (Accili & Arden, 2004). Furthermore, AKT-mediated phosphorylation of FoxO3 is activated by Bcr–Abl, and this process is essential in the cellular transformation by Bcr–Abl (Komatsu et al, 2003). In agreement with these observations, we found that treatment of B/A or K562 cells with the PI3K inhibitor LY 294002 or the Abl inhibitor imatinib suppressed the ability of Bcr–Abl to induce phosphorylation of AKT and FoxO3 (Figs 4D, 5B). Similarly to AKT, exposure of the Ba/F3 cell line to TGF-β resulted in a marked reduction in the phosphorylation of FoxO3, which was detectable after 1 h and persisted after 4 h (Fig 5A). However, in B/A cells, the phosphorylation of FoxO3 was unchanged at any time examined (Fig 5A). To confirm these results, we investigated the distribution of FoxO3 by immunofluorescence and found that exposure of Ba/F3 cells to TGF-β caused redistribution of FoxO3 from the cytoplasm to the nucleus. In contrast, the cytoplasmic localization of FoxO3 remained unchanged in B/A cells that were exposed to TGF-β (Fig 5C). We also used a luciferase reporter construct (FHRE-Luc) under the control of a fragment of the Fas ligand promoter containing the FoxO3-binding site (Brunet et al, 1999). We observed that TGF-β could stimulate transcription from the FHRE-Luc promoter in Ba/F3 cells, whereas it had no effect in B/A cells (Fig 5D). Thus, it is likely that expression of Bcr–Abl suppressed the ability of TGF-β to mediate inhibition of the PI3K/AKT pathway and thus the accumulation of the activated form of FoxO3. To investigate the implication of FoxO3 in Bcr–Ablsuppressed TGF-β effects, we made use of a constitutively activated mutant of FoxO3, FoxO3.TM, in which the sites of phosphorylation by AKT were mutated. Consistent with previous observations (Brunet et al, 1999), we found that expression of FoxO3.TM in B/A cells stimulated apoptosis. However, we consistently observed that TGF-β further enhanced the apoptotic effect of FoxO3.TM (Fig 5E), thereby providing evidence that Bcr–Abl inhibits TGF-β effects by inactivating FoxO3. On the basis of these observations and the finding that FoxO3 acts as an essential mediator in TGF-β-mediated growth-inhibitory signals (Accili & Arden, 2004; Seoane et al, 2004), we propose that Bcr–Abl might influence oncogenesis by disabling the cytostatic responses of TGF-β through the AKT/FoxO pathway. This mechanism of suppressing the ability of TGF-β to induce growth-inhibitory signals may not be restricted to the phosphorylation of FoxO3 by AKT because AKT can also sequester Smad3 in the cytoplasm, thereby preventing TGF-β from inducing apoptotic responses (Conery et al, 2004; Remy et al, 2004).
Figure 5.
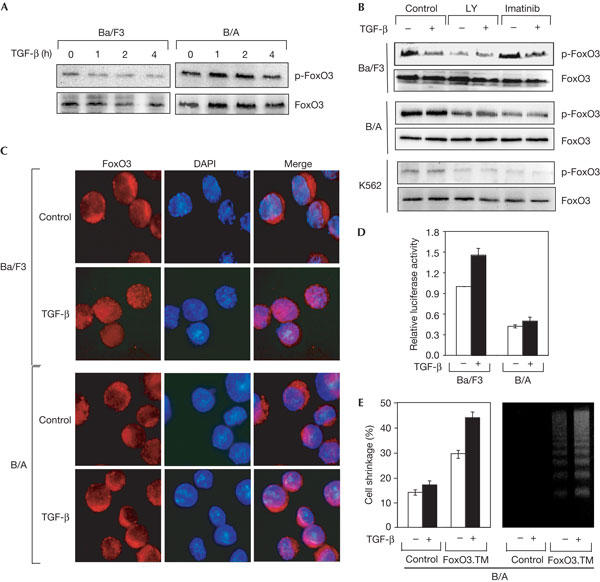
FoxO3 acts downstream of Bcr–Abl to suppress transforming growth factor-β-mediated cytostatic responses. (A) Ba/F3 or B/A cells were treated with transforming growth factor-β (TGF-β) for the indicated periods of time and cell lysates were analysed by immunoblotting with anti-phospho-FoxO3 (p-FoxO3) or anti-FoxO3 antibodies. (B) Ba/F3, B/A or K562 cells were treated with imatinib or LY204009 (LY) for 1 h and then with TGF-β for 3 h. Cell lysates were analysed by immunoblotting with anti-phospho-FoxO3 or anti-FoxO3 antibodies. (C) Ba/F3 or B/A cells were treated with or without TGF-β for 4 h and the distribution of FoxO3 was analysed by fluorescence microscopy after labelling with an anti-FoxO3 antibody and 4,6-diamidino-2-phenylindole (DAPI). (D) Cells were transfected with FoxO3 together with FHRE-Luc. After 24 h, they were treated with or without TGF-β for 18 h and then analysed for luciferase activity. (E) B/A cells were co-transfected with FoxO3.TM and green fluorescent protein (GFP). After sorting on the basis of GFP expression and treatment with or without TGF-β for 48 h, the extent of apoptosis was determined by cell shrinkage or DNA fragmentation.
Cancer cells can acquire resistance to the antiproliferative effects of TGF-β by different mechanisms, including defects in TGF-β receptors and mutational inactivation of downstream effectors of the TGF-β signalling pathway (Derynck et al, 2001). In the present study, we provide the first evidence that cells expressing Bcr–Abl and Tel–Abl lose control to the negative growth-regulatory effects of TGF-β. Furthermore, we show that Bcr–Abl functions to suppress TGF-β-mediated growth arrest and apoptosis without interfering with the activation of Smad proteins. In addition, we provide evidence that the AKT/FoxO3 signalling pathway acts downstream of Bcr–Abl to suppress the cytostatic actions of TGF-β. Therefore, these findings shed light on the mechanism of cellular transformation caused by aberrant chromosomal translocations found in chronic or acute myelogenous human leukaemia.
Methods
Expression vectors and cell culture. p3TP-Lux and (CAGA)9-Lux were kindly provided by Dr J. Massagué and Dr J.-M. Gauthier, respectively. Plasmids for FHRE-Luc, FoxO3 and FoxO3.TM were a gift from Dr A. Brunet. The expression vectors for Tel–PDGFRβ, Tel–Abl, Bcr–Abl, Myc–Smad2, haemagglutinin (HA)–Smad4 and enhanced green fluorescent protein (pEGFP) were described previously (Golub et al, 1996; Besancon et al, 1998; Prunier et al, 2001; Seo et al, 2004).
The murine IL-3-dependent Ba/F3 cells were maintained in RPMI containing 10% fetal calf serum and 1 ng/ml of murine recombinant IL-3 (PeproTech, London, UK). The human myelogenous leukaemia cell line K562 was maintained in RPMI containing 10% fetal calf serum. For experiments with imatinib, K562 cells were cultured in Stem.AE, a serum-free medium supplemented with different recombinant human cytokines (IL-3, IL-6, IL-9, stem-cell factor, erythropoietin; Stem Alpha France).
Cells were transfected with the different vectors by electroporation at 0.24 kV and 960 μF using a Bio-Rad apparatus. Selection of stable transfectants has been already described (Besancon et al, 1998).
Immunoprecipitation and western blot analyses. Cells were lysed and processed as described previously (Abecassis et al, 2004). Western blotting was performed with anti-c-Abl (Calbiochem, Darmstadt, Germany), anti-PDGFRβ and anti-phosphosmad2 (Upstate Biotechnology, Lake Placid, NY, USA), anti-Myc, anti-HA and anti-actin (Sigma, St Louis, MO, USA), anti-AKT, anti-phospho-AKT, anti-FoxO3 and anti-phospho-FoxO3 (Cell Signaling, Beverly, MA, USA) or anti-PARP and anti-JunB (Santa-Cruz Laboratories, Santa Cruz, CA, USA) antibodies. Blots were revealed with sheep anti-mouse or anti-rabbit IgG horseradish-peroxidase-linked antibodies and chemiluminescence (ECL Amersham, Orsay, France).
Cell-cycle analyses and apoptosis assays. Cells were treated with TGF-β for 24 h and the different phases of the cell cycle, including sub-G1 (proportion of apoptotic cells), were analysed with a FACScan cytometer. Shrunken cells were estimated after 48 h of TGF-β treatment and enumerated as a percentage of the total population. DNA fragmentation was analysed after 48 h of TGF-β treatment, as described previously (Besancon et al, 1998). Sorting of GFP-positive cells was carried out using the fluorescence-activated cell sorting (FACS) method.
Gene reporter assays. Cells were transfected by electroporation and incubated 24 h later with or without TGF-β (4 ng/ml) for 18 h. Luciferase activity was measured using a luciferase assay kit (Promega, Charbonnières, France) and was normalized for transfection efficiency using a β-galactosidase-expressing vector.
Immunofluorescence. Cells were stained with anti-FoxO3 and 4,6-diamidino-2-phenylindole and visualized under a fluorescence microscope, as described previously (Prunier et al, 2001).
Acknowledgments
This work was supported by Institut National de la Santé et de la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS) and l'Association de la Recherche sur le Cancer (ARC).
References
- Abecassis L, Rogier E, Vazquez A, Atfi A, Bourgeade MF (2004) Evidence for a role of MSK1 in transforming growth factor-β-mediated responses through p38α and Smad signaling pathways. J Biol Chem 279: 30474–30479 [DOI] [PubMed] [Google Scholar]
- Accili D, Arden KC (2004) FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 117: 421–426 [DOI] [PubMed] [Google Scholar]
- Besancon F, Atfi A, Gespach C, Cayre YE, Bourgeade MF (1998) Evidence for a role of NF-κB in the survival of hematopoietic cells mediated by interleukin 3 and the oncogenic TEL/platelet-derived growth factor receptor β fusion protein. Proc Natl Acad Sci USA 95: 8081–8086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868 [DOI] [PubMed] [Google Scholar]
- Conery AR, Cao Y, Thompson EA, Townsend CM Jr, Ko TC, Luo K (2004) Akt interacts directly with Smad3 to regulate the sensitivity to TGF-β induced apoptosis. Nat Cell Biol 6: 366–372 [DOI] [PubMed] [Google Scholar]
- Crans HN, Sakamoto KM (2001) Transcription factors and translocations in lymphoid and myeloid leukemia. Leukemia 15: 313–331 [DOI] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM (1998) Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 17: 3091–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A (2001) TGF-β signaling in tumor suppression and cancer progression. Nat Genet 29: 117–129 [DOI] [PubMed] [Google Scholar]
- Dierov J, Xu Q, Dierova R, Carroll M (2002) TEL/platelet-derived growth factor receptor β activates phosphatidylinositol 3 (PI3) kinase and requires PI3 kinase to regulate the cell cycle. Blood 99: 1758–1765 [DOI] [PubMed] [Google Scholar]
- Golub TR, Goga A, Barker GF, Afar DE, McLaughlin J, Bohlander SK, Rowley JD, Witte ON, Gilliland DG (1996) Oligomerization of the ABL tyrosine kinase by the Ets protein TEL in human leukemia. Mol Cell Biol 16: 4107–4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonk LJ, Itoh S, Heldin CH, ten Dijke P, Kruijer W (1998) Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-β, activin, and bone morphogenetic protein-inducible enhancer. J Biol Chem 273: 21145–21152 [DOI] [PubMed] [Google Scholar]
- Keung YK, Beaty M, Steward W, Jackle B, Pettnati M (2002) Chronic myelocytic leukemia with eosinophilia, t(9;12)(q34;p13), and ETV6-ABL gene rearrangement: case report and review of the literature. Cancer Genet Cytogenet 138: 139–142 [DOI] [PubMed] [Google Scholar]
- Kharas MG, Deane JA, Wong S, O'Bosky KR, Rosenberg N, Witte ON, Fruman DA (2004) Phosphoinositide 3-kinase signaling is essential for ABL oncogene-mediated transformation of B-lineage cells. Blood 103: 4268–4275 [DOI] [PubMed] [Google Scholar]
- Komatsu N et al. (2003) A member of Forkhead transcription factor FKHRL1 is a downstream effector of STI571-induced cell cycle arrest in BCR–ABL-expressing cells. J Biol Chem 278: 6411–6419 [DOI] [PubMed] [Google Scholar]
- Maru Y, Afar DE, Witte ON, Shibuya M (1996) The dimerization property of glutathione S-transferase partially reactivates Bcr–Abl lacking the oligomerization domain. J Biol Chem 271: 15353–15357 [DOI] [PubMed] [Google Scholar]
- Massague J, Wotton D (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J 19: 1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A, Souchelnytskyi S, Heldin CH (2001) Smad regulation in TGF-β signal transduction. J Cell Sci 114: 4359–4369 [DOI] [PubMed] [Google Scholar]
- Prunier C, Ferrand N, Frottier B, Pessah M, Atfi A (2001) Mechanism for mutational inactivation of the tumor suppressor Smad2. Mol Cell Biol 21: 3302–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy I, Montmarquette A, Michnick SW (2004) PKB/Akt modulates TGF-β signalling through a direct interaction with Smad3. Nat Cell Biol 6: 358–365 [DOI] [PubMed] [Google Scholar]
- Roberts AB (1998) Molecular and cell biology of TGF-β. Miner Electrolyte Metab 24: 111–119 [DOI] [PubMed] [Google Scholar]
- Seo SR, Lallemand F, Ferrand N, Pessah M, L'Hoste S, Camonis J, Atfi A (2004) The novel E3 ubiquitin ligase Tiul1 associates with TGIF to target Smad2 for degradation. EMBO J 23: 3780–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J, Le HV, Shen L, Anderson SA, Massague J (2004) Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117: 211–223 [DOI] [PubMed] [Google Scholar]