

Abstract
Insulin promotes glucose uptake into muscle and adipose tissues through glucose transporter 4 (GLUT4). In unstimulated cells, rapid endocytosis, slow exocytosis and dynamic or static retention cause GLUT4 to concentrate in early recycling endosomes, the trans-Golgi network and vesicle-associated protein 2-containing vesicles. The coordinated action of phosphatidylinositol 3-kinase effectors, protein kinase Akt, atypical protein kinase C (aPKC) and Akt substrate of 160-kDa (AS160), regulates the GLUT4 cycle by affecting its translocation, fusion with the plasma membrane, internalization and sorting. We review the evidence that supports such cycling, evaluate current models proposing static or dynamic retention, and highlight how distinct steps of GLUT4 transport are regulated by insulin signals. In particular, fusion seems to be regulated by aPKC (via munc18) and Akt (via syntaxin4-interacting protein (synip)). AS160 participates in GLUT4 intracellular retention, and possibly fusion, through candidate ras-related GTP-binding protein (Rab)2, Rab8, Rab10 and/or Rab14. The localization of the insulin-sensitive GLUT4 compartment and the precise target of insulin-derived signals remain open for future investigation.
Keywords: glucose uptake, GLUT4 compartments, GLUT4 traffic, insulin signalling, type 2 diabetes mellitus
Introduction
The global prevalence of type 2 diabetes will reach 300 million cases by 2025 (Zimmet et al, 2001). This metabolic disease results from defective tissue sensitivity to insulin and subsequent impairment in insulin secretion. Insulin maintains glucose homeostasis largely by enhancing glucose uptake into muscle and adipose tissues, which is a process mediated by glucose transporter 4 (GLUT4). Since the cloning of GLUT4 in 1989, numerous studies have attempted to dissect the molecular basis of its regulation by insulin and stimuli such as muscle contraction. Most of our understanding of these phenomena stems from studies in cultured cells, as reviewed here. In unstimulated adipose and muscle cells, GLUT4 constitutively cycles to and from the plasma membrane (PM), but the extent of such cycling is currently debated. In both cell types, the steady-state distribution of GLUT4 favours intracellular compartments, and there is a general agreement that insulin largely promotes the exocytic arm of GLUT4 cycling and, to a lesser extent, reduces the endocytic arm (Bryant et al, 2002; Rudich & Klip, 2003; Watson et al, 2004a). Unlike secretory granule exocytosis, which is unidirectional, regulated GLUT4 cycling is more akin to the regulated secretory exocytosis shown by synaptic vesicles, which, following neurotransmitter release, reform by membrane internalization and protein sorting (Südhof, 2004). Insulin resistance—particularly in skeletal muscle—is associated with insufficient recruitment of GLUT4 to the PM despite normal GLUT4 expression (Björnholm & Zierath, 2005). This finding emphasizes the importance of understanding GLUT4 dynamics in designing strategies to bypass or resolve insulin resistance. The regulation of glucose influx might also involve steps at the level of GLUT4 and hexokinase activities (Antonescu et al, 2005; Fueger et al, 2005), which are beyond the scope of this review. Here, we focus on the current understanding and ongoing debate regarding GLUT4 cycling.
Intracellular localization of GLUT4
GLUT4 constitutively cycles to and from the PM through slow exocytosis and fast endocytosis (Satoh et al, 1993; Li et al, 2001). Diverse microscopy approaches have localized GLUT4 to tubulo-vesicular structures in the perinuclear region and in distinct foci throughout the cytosol (Slot et al, 1991; Malide et al, 2000). Perinuclear GLUT4 partially colocalizes with markers of the endosomal recycling compartment (ERC), the Golgi complex and the trans-Golgi network (TGN; Bryant et al, 2002; Ploug & Ralston, 2002). Given the dynamic behaviour of GLUT4, it is not possible to discern by static microscopy analysis whether perinuclear or cytosolic depots encompass the insulin-sensitive GLUT4-donor compartment. These and subsequent studies involving the chemical ablation of transferrin receptor (TfR)-containing compartments (Martin et al, 1996; Zeigerer et al, 2002) using peroxidase-loaded TfR consistently identified two populations of GLUT4 pools: one overlapping with TfR, which is an ERC marker, and one segregating away from it (non-ERC pool). There is consensus that, directly or indirectly, insulin mobilizes GLUT4 from the non-ERC pool, which has therefore been dubbed the GLUT4 ‘specialized compartment (SC)' or ‘GLUT4 storage vesicle' (GSV).
Despite extensive studies, it has been challenging to define the biochemical nature of the SC/GSV. The insulin-responsive aminopeptidase (IRAP) co-segregates with GLUT4 and similarly redistributes to the PM in response to insulin. Although IRAP might regulate the retention/sequestration of GLUT4 vesicles (Hosaka et al, 2005), it does not identify a specialized pool of GLUT4 (Keller, 2003), but rather parallels GLUT4 distribution across compartments. A more selective marker of SC/GSV might be vesicle-associated protein 2 (VAMP2), which is a vesicular soluble N-ethylmaleimide-sensitive factor attachment protein receptor (v-SNARE) that is exclusively required for GLUT4 fusion with the membrane in the insulin-stimulated, but not the basal, state. Interestingly, VAMP2 is found only in a subset of GLUT4 vesicles that largely segregate away from TfR (Bryant et al, 2002; Watson et al, 2004a). Moreover, stimuli such as hypertonic shock and platelet-derived growth factor do not rely on VAMP2 to increase surface GLUT4 levels, and instead depend on the v-SNARE VAMP7 (Randhawa et al, 2004; Török et al, 2004). Future work should confirm whether the SC/GSV constitutes a pre-formed compartment of VAMP2 and GLUT4 or whether GLUT4 vesicles acquire VAMP2 during insulin-induced translocation to the PM.
How is the SC/GSV formed? The Golgi-localized γ-ear-containing Arf-binding protein GGA is required for nascent GLUT4 sorting from the TGN to the SC/GSV (Watson et al, 2004b). GGA interacts with sortilin, which is a TGN and endosomal membrane protein proposed to be necessary and sufficient for the formation of small GLUT4-enriched vesicles and for GLUT4 protein stability (Shi & Kandror, 2005). Accordingly, co-expression of sortilin and GLUT4 tagged with myc7 epitopes in 3T3-L1 fibroblasts generated such vesicles, and, conversely, reducing endogenous sortilin through RNA interference (RNAi) prevented their constitutive formation in 3T3-L1 adipocytes. Whether such vesicles conform to other characteristics of the SC/GSV—such as enrichment in VAMP2—and respond to insulin-derived signals requires further investigation.
GLUT4 cycling: a Ptolemy versus Copernicus analogy
Are there static GLUT4 compartments or do they continuously revolve to and from the PM? Two models have been proposed to explain insulin-responsive GLUT4 storage and cycling in 3T3-L1 adipocytes (Fig 1). Model 1, which is endorsed by McGraw and colleagues, proposes that the entire complement of GLUT4 eventually recycles to the PM in the basal state, that GLUT4 intracellular storage is dynamic and that the SC/GSV is distinct from the furin-positive TGN (Karylowski et al, 2004). The last of these conclusions derives from the inability to ablate GLUT4 with peroxidase-loaded furin beyond that caused by peroxidase-loaded TfR. According to this model, insulin promotes two routes for GLUT4 mobilization towards the PM, a direct one from the SC/GSV and an indirect one from the SC/GSV via the ERC. Exit from the ERC is supported by a reduction in insulin-induced GLUT4 translocation to the PM on ablation of TfR-containing compartments, as well as by the participation of the GTPase Rab11 (which controls ERC protein sorting) in the insulin response (Zeigerer et al, 2002). The behaviour of GLUT4 in muscle cells is consistent with tenets of this model (Rudich & Klip, 2003). Indeed, in unstimulated muscle cells, all GLUT4 molecules cycle to the PM (Foster et al, 2001), yet only 50% of GLUT4 is sensitive to TfR-mediated ablation (V.K. Randhawa, T.E. McGraw & A. Klip, unpublished data). The concept that GLUT4 can follow two distinct exit routes towards the PM is buttressed by the participation of VAMP2 in insulin-dependent, but not constitutive, GLUT4 recycling (Cheatham et al, 1996; Olson et al, 1997; Martin et al, 1998; Randhawa et al, 2004).
Figure 1.
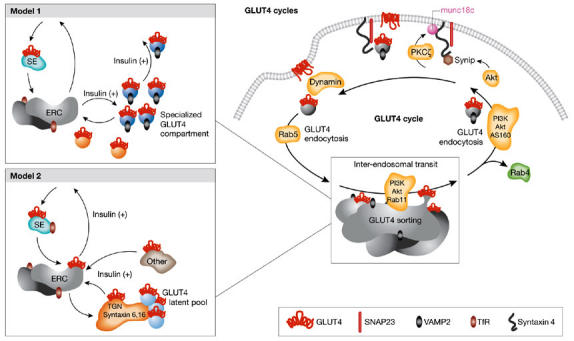
Models of insulin-responsive glucose transporter 4 (GLUT4) compartments and cycling. Illustrated is a hypothetical version of GLUT4 cycling summarizing the current literature. GLUT4 cycles between intracellular compartments and the plasma membrane (PM) by the coordinated regulation of exocytic mobilization, fusion with the PM, endocytosis, and inter-endosomal transit and sorting. Each stage of GLUT4 transport is regulated by insulin-derived signals. The contents of the expanded box illustrate aspects of the two debated models of GLUT4 compartments and exit modalities. The models differ in negating or supporting the presence of a static/latent pool of GLUT4, in the contribution of the trans-Golgi network (TGN) as a storage site for GLUT4, and in the number of GLUT4 exit routes towards the PM. The plus symbol (+) indicates stimulation. AS160, Akt-substrate of 160 kDa; ERC, endosomal recycling compartment; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; SE, sorting syndrome; TfR, transferrin receptor.
Model 2, championed by James and colleagues, proposes that only a fraction of GLUT4 recycles to the PM in the basal state, that insulin increases the quantity of GLUT4 available for translocation, and that part of the TfR-negative GLUT4 compartment interfaces with the TGN (Coster et al, 2004). Therefore, increasing insulin doses progressively engages a larger fraction of the non-cycling pool, which is then mobilized towards the PM through a single exit route—that is, from the ERC. Intriguingly, a component of the non-cycling pool contains ‘latent GLUT4 molecules' that are not mobilized, even in response to insulin. It is speculated that the latent pool contains transporters that are synthesized early in cellular life and are preferentially excluded from the insulin response (Govers et al, 2004). The TGN participation in GLUT4 storage is inferred from the partial colocalization of GLUT4 and IRAP with the mannose 6-phosphate receptor, the adaptor-related protein complex-1 (Bryant et al, 2002), syntaxin6 and syntaxin16, and with the acquisition of sialic acid during the recycling of IRAP treated with neuraminidase while at the cell surface (Shewan et al, 2003). Notably, however, there was no colocalization of GLUT4 with TGN38, which is an established TGN marker. The TGN has heterogeneous functional domains and, hence, whether it interfaces with the SC/GSV remains an open question.
Models 1 and 2 differ in the extent to which cellular GLUT4 recycles in the basal state, in the presence of a static/latent pool of GLUT4, in the contribution of the TGN as a storage site for GLUT4 and in the number of insulin-dependent GLUT4 exit routes towards the PM. Model 1 rests on studies performed in 3T3-L1 adipocytes transiently expressing haemagglutinin (HA)– GLUT4–enhanced green fluorescent protein (eGFP) or L6 muscle cells stably expressing GLUT4myc. Conversely, Model 2 arises from studies in 3T3-L1 adipocytes stably expressing HA–GLUT4. The observed discrepancies are, therefore, not likely to be caused by differences between stable or transient expression of the transporter, and the possible contribution of cell clonal background or GLUT4-expression levels must instead be considered.
Insulin signals directing GLUT4 cycling
The intricacies of insulin signalling have been reviewed recently (Gual et al, 2005; Thong et al, 2005). Insulin binding to, and activation of, its receptor is rapidly followed by docking of insulin-receptor substrates and activation of Class IA phosphatidylinositol 3-kinase (PI3K). The latter catalyses the formation of phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3), which, in turn, leads to the activation of atypical protein kinase C (aPKC)λ/ζ and Akt/PKB, enzymes required for insulin-induced GLUT4 translocation (Farese et al, 2005; Welsh et al, 2005). Akt/PKB acts on more than 35 substrates that are involved in various metabolic and mitogenic processes. Of these, the Rab–GTPase-activating protein (GAP) Akt substrate of 160-kDa (AS160) participates in GLUT4 translocation to the PM (Sano et al, 2003), presumably through its recently identified targets Rab2, Rab8, Rab10 and/or Rab14 (Miinea et al, 2005). Supporting these observations, Rab10, Rab11 and Rab14 were found on GLUT4 vesicles, and silencing of the AS160 gene by small-hairpin RNAs (shRNAs) elevated the insulin-dependent increase in surface levels of GLUT4 (Larance et al, 2005). Bolstering these studies, it was recently shown that knockdown of AS160 in unstimulated adipocytes increases surface levels of GLUT4 and glucose uptake, suggesting that AS160 contributes to GLUT4 retention in the basal state (Eguez et al, 2005). Additionally, a PI3K-independent pathway involving c-Cbl associated protein (CAP), Cbl and the GTPase TC10 could regulate GLUT4 translocation (Saltiel & Pessin, 2002), but apparently this is specific to adipocytes (JeBailey et al, 2004). We next focus on the input of insulin-derived signals in several discrete steps of the GLUT4 cycle: exocytic mobilization, fusion with the PM, and internalization and intracellular sorting (Fig 1).
GLUT4 exocytic mobilization. Insulin increases the rate of GLUT4 exocytosis in a PI3K-dependent manner (Yang et al, 1996). Accordingly, delivery of PI(3,4,5)P3 increases surface GLUT4 levels, whereas PI3P (the product of Class II and Class III PI3K) facilitates the arrival of GLUT4 vesicles at the PM without allowing fusion (Maffucci et al, 2003; Ishiki et al, 2005; Kanda et al, 2005). Therefore, distinct phosphoinositides regulate the arrival and fusion of GLUT4 vesicles with the PM. Insulin-activated PI3K also results in the activation of the Rho-family protein Rac, which, in turn, leads to actin remodelling beneath the membrane of muscle cells (JeBailey et al, 2004). A PI(3,4,5)P3-independent input similarly activates the GTPase TC10 to reorganize actin in adipocytes (Saltiel & Pessin, 2002). Remodelled actin sites could contribute to GLUT4 mobilization by promoting GLUT4 and signal sorting, guiding myosin motors on GLUT4 vesicles or positioning GLUT4 near fusion sites on the PM (Patel et al, 2005). Insulin-induced actin remodelling does not require Akt, but this enzyme (predominantly Akt2) is required for the net GLUT4 translocation process, as illustrated by the use of dominant-negative mutants, gene silencing via RNAi and Akt2-gene knockout (Welsh et al, 2005). Akt directly targets GLUT4-containing endomembranes, as constitutively active or inactive mutants of the enzyme targeted to GLUT4-containing compartments respectively emulate or reduce the insulin response of GLUT4/IRAP. Accordingly, AS160 has been implicated in the GLUT4-mobilization step on the basis of a reduction in GLUT4–eGFP signal beneath the PM induced by overexpression of a non-phosphorylatable mutant of AS160, as detected by total internal reflection fluorescence microscopy (Zeigerer et al, 2004). The aPKCs also affect GLUT4 mobilization towards the PM by promoting association between the GTPase Rab4, the motor protein KIF3 and the microtubules required for such mobilization (Imamura et al, 2003). In summary, these studies imply that the PI3K→Akt→AS160 and PI3K→aPKC axes regulate GLUT4 mobilization towards the PM. How individual, complementing or redundant these two axes are remains to be defined.
GLUT4-vesicle fusion with the PM. In addition to regulating GLUT4 vesicle mobilization, insulin-derived signals also impinge on their fusion with the PM. At 19 °C, insulin elicits GLUT4 mobilization towards the PM without causing Akt activation or GLUT4 fusion, which instead rapidly ensue on re-warming to 37 °C (van Dam et al, 2005). Akt can target components of the vesicle–PM fusion machinery, which comprises the vesicular VAMP2, the target membrane SNAP23 and syntaxin4 (SNAREs), as well as synip, tomosyn and munc18c that bind syntaxin4 to modulate the insulin-dependent gain in surface GLUT4 (Widberg et al, 2003; Hodgkinson et al, 2005). Akt-dependent synip phosphorylation liberates syntaxin4, presumably to promote SNARE-complex formation (Yamada et al, 2005); however, the need for synip phosphorylation in insulin-dependent GLUT4 translocation has been contested by mutagenesis studies (Sano et al, 2005). As in the case of Akt, aPKCs have an input in GLUT4 fusion. PKCζ seems to phosphorylate VAMP2 (Braiman et al, 2001), and insulin promotes the formation of a complex that includes PKCζ and munc18c, thereby dissociating munc18c from syntaxin4 (Hodgkinson et al, 2005). Collectively, these molecular events could underpin the recent observation that insulin slows down the constitutive rapid movement of GLUT4 beneath the PM, and promotes vesicle tethering and fusion to increase surface GLUT4 levels (Lizunov et al, 2005). The regulation of fusion has been recently emphasized by in vitro studies whereby only PM isolated from insulin–stimulated cells were able to bind purified membrane vesicles containing GLUT4 (Koumanov et al, 2005).
GLUT4 endocytosis and inter-endosomal transit. GLUT4 internalization from the cell surface is reduced on microinjection into adipocytes of dynamin or amphiphysin peptides (Volchuk et al, 1998), and by expression of dynamin (Al-Hasani et al, 1998; Kao et al, 1998) or caveolin-3 mutants (Cohen et al, 2003). These results indicate that GLUT4 internalizes through both clathrin-coated pits and caveolae. Insulin modestly reduces GLUT4 endocytosis, in part by inhibiting Rab5 activity (Huang et al, 2001). In muscle cells, internalized GLUT4 reaches the early endosome in 2 minutes and the ERC in 20 minutes. Insulin accelerates GLUT4 arrival to, and departure from, the ERC, which is a process that requires Akt (Foster et al, 2001) and PIKfyve (Berwick et al, 2004). Such acceleration might contribute to the increased exocytosis of GLUT4 observed in response to insulin. More molecular detail of intracellular GLUT4 sorting in insulin-stimulated cells is required to improve our understanding of the unique action of this hormone.
Concluding remarks and open questions
GLUT4 cycling is regulated at the levels of its exocytosis, fusion, endocytosis and inter-endosomal transit. Most studies have assessed the impact of interfering with specific signalling molecules on particular segments of GLUT4 traffic. However, for the most part, this approach does not establish the precise target of the insulin-derived signals. It is pressing to identify the steps that are the primary recipients of insulin-derived signals and to differentiate them from secondary responses. For example, an insulin-derived signal might act on GLUT4-vesicle fusion at the PM, secondarily backing up the exocytic arm of GLUT4 cycling. Another potentially related question concerns the location of the SC/GSV in muscle and fat cells: is it perinuclear or found in the cytosolic pool of small vesicles? In addition, mapping the entire signalling cascades from receptor to GLUT4 and the precise site-of-action of each insulin-derived signal will be paramount to designing therapeutic strategies to relieve insulin resistance. Finally, it is important to identify molecules that bind to GLUT4, thereby determining its location, retention, and release from endomembranes and the PM. Faithful tracking of GLUT4 molecules in space and time might still reveal unexpected nuances of GLUT4 cycling in live muscle and adipose cells.


Acknowledgments
We thank P. Bilan for carefully reading the manuscript. We apologize to colleagues whose work was not cited owing to space limitations. Work reviewed from our laboratory was supported by grants from the Canadian Institutes of Health Research and the Canadian Diabetes Association. A Natural Sciences and Engineering Research Council of Canada studentship supported C.B.D.
References
- Al-Hasani H, Hinck CS, Cushman SW (1998) Endocytosis of the glucose transporter GLUT4 is mediated by the GTPase dynamin. J Biol Chem 273: 17504–17510 [DOI] [PubMed] [Google Scholar]
- Antonescu CN, Thong FSL, Niu W, Karneli E, Klip A (2005) To be or not to be: regulation of the intrinsic activity of GLUT4. Curr Med Chem Immunol Endo Metab Agents 5: 175–187 [Google Scholar]
- Berwick DC, Dell GC, Welsh GI, Heesom KJ, Hers I, Fletcher LM, Cooke FT, Tavare JM (2004) Protein kinase B phosphorylation of PIKfyve regulates the trafficking of GLUT4 vesicles. J Cell Sci 117: 5985–5993 [DOI] [PubMed] [Google Scholar]
- Björnholm M, Zierath JR (2005) Insulin signal transduction in human skeletal muscle: identifying the defects in Type II diabetes. Biochem Soc Trans 33: 354–357 [DOI] [PubMed] [Google Scholar]
- Braiman L, Alt A, Kuroki T, Ohba M, Bak A, Tennenbaum T, Sampson SR (2001) Activation of protein kinase Cz induces serine phosphorylation of VAMP2 in the GLUT4 compartment and increases glucose transport in skeletal muscle. Mol Cell Biol 21: 7852–7861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant NJ, Govers R, James DE (2002) Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 3: 267–277 [DOI] [PubMed] [Google Scholar]
- Cheatham B, Volchuk A, Kahn CR, Wang L, Rhodes CJ, Klip A (1996) Insulin-stimulated translocation of GLUT4 glucose transporters requires SNARE-complex proteins. Proc Natl Acad Sci USA 93: 15169–15173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AW, Combs TP, Scherer PE, Lisanti MP (2003) Role of caveolin and caveolae in insulin signaling and diabetes. Am J Physiol Endocrinol Metab 285: E1151–E1160 [DOI] [PubMed] [Google Scholar]
- Coster AC, Govers R, James DE (2004) Insulin stimulates the entry of GLUT4 into the endosomal recycling pathway by a quantal mechanism. Traffic 5: 763–771 [DOI] [PubMed] [Google Scholar]
- Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE (2005) Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab 2: 263–272 [DOI] [PubMed] [Google Scholar]
- Farese RV, Sajan MP, Standaert ML (2005) Atypical protein kinase C in insulin action and insulin resistance. Biochem Soc Trans 33: 350–353 [DOI] [PubMed] [Google Scholar]
- Foster LJ, Li D, Randhawa VK, Klip A (2001) Insulin accelerates inter-endosomal GLUT4 traffic via phosphatidylinositol 3-kinase and protein kinase B. J Biol Chem 276: 44212–44221 [DOI] [PubMed] [Google Scholar]
- Fueger PT, Shearer J, Bracy DP, Posey KA, Pencek RR, McGuinness OP, Wasserman DH (2005) Control of muscle glucose uptake: test of the rate-limiting step paradigm in conscious, unrestrained mice. J Physiol 562: 925–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govers R, Coster AC, James DE (2004) Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol Cell Biol 24: 6456–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gual P, Le Marchand-Brustel Y, Tanti JF (2005) Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 87: 99–109 [DOI] [PubMed] [Google Scholar]
- Hodgkinson CP, Mander A, Sale GJ (2005) Identification of 80K-H as a protein involved in GLUT4 vesicle trafficking. Biochem J 388: 785–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka T, Brooks CC, Presman E, Kim SK, Zhang Z, Breen M, Gross DN, Sztul E, Pilch PF (2005) p115 interacts with the GLUT4 vesicle protein, IRAP, and plays a critical role in insulin-stimulated GLUT4 translocation. Mol Biol Cell 16: 2882–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Imamura T, Olefsky JM (2001) Insulin can regulate GLUT4 internalization by signaling to Rab5 and the motor protein dynein. Proc Natl Acad Sci USA 98: 13084–13089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura T, Huang J, Usui I, Satoh H, Bever J, Olefsky JM (2003) Insulin-induced GLUT4 translocation involves protein kinase C-λ-mediated functional coupling between Rab4 and the motor protein kinesin. Mol Cell Biol 23: 4892–4900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiki M, Randhawa VK, Poon V, Jebailey L, Klip A (2005) Insulin regulates the membrane arrival, fusion, and C-terminal unmasking of glucose transporter-4 via distinct phosphoinositides. J Biol Chem 280: 28792–28802 [DOI] [PubMed] [Google Scholar]
- JeBailey L, Rudich A, Huang X, Di Ciano-Oliveira C, Kapus A, Klip A (2004) Skeletal muscle cells and adipocytes differ in their reliance on TC10 and Rac for insulin-induced actin remodeling. Mol Endocrinol 18: 359–372 [DOI] [PubMed] [Google Scholar]
- Kanda H, Tamori Y, Shinoda H, Yoshikawa M, Sakaue M, Udagawa J, Otani H, Tashiro F, Miyazaki J, Kasuga M (2005) Adipocytes from Munc18c-null mice show increased sensitivity to insulin-stimulated GLUT4 externalization. J Clin Invest 115: 291–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao AW, Ceresa BP, Santeler SR, Pessin JE (1998) Expression of a dominant interfering dynamin mutant in 3T3L1 adipocytes inhibits GLUT4 endocytosis without affecting insulin signaling. J Biol Chem 273: 25450–25457 [DOI] [PubMed] [Google Scholar]
- Karylowski O, Zeigerer A, Cohen A, McGraw TE (2004) GLUT4 is retained by an intracellular cycle of vesicle formation and fusion with endosomes. Mol Biol Cell 15: 870–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SR (2003) The insulin-regulated aminopeptidase: a companion and regulator of GLUT4. Front Biosci 8: s410–s420 [DOI] [PubMed] [Google Scholar]
- Koumanov F, Jin B, Yang J, Holman GD (2005) Insulin signaling meets vesicle traffic of GLUT4 at a plasma-membrane-activated fusion step. Cell Metab 2: 179–189 [DOI] [PubMed] [Google Scholar]
- Larance M et al. (2005) Characterisation of the role of the RabGAP AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem [epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Li D, Randhawa VK, Patel N, Hayashi M, Klip A (2001) Hyperosmolarity reduces GLUT4 endocytosis and increases its exocytosis from a VAMP2-independent pool in l6 muscle cells. J Biol Chem 276: 22883–22891 [DOI] [PubMed] [Google Scholar]
- Lizunov VA, Matsumoto H, Zimmerberg J, Cushman SW, Frolov VA (2005) Insulin stimulates the halting, tethering, and fusion of mobile GLUT4 vesicles in rat adipose cells. J Cell Biol 169: 481–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffucci T, Brancaccio A, Piccolo E, Stein RC, Falasca M (2003) Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J 22: 4178–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malide D, Ramm G, Cushman SW, Slot JW (2000) Immunoelectron microscopic evidence that GLUT4 translocation explains the stimulation of glucose transport in isolated rat white adipose cells. J Cell Sci 113: 4203–4210 [DOI] [PubMed] [Google Scholar]
- Martin LB, Shewan A, Millar CA, Gould GW, James DE (1998) Vesicle-associated membrane protein 2 plays a specific role in the insulin-dependent trafficking of the facilitative glucose transporter GLUT4 in 3T3-L1 adipocytes. J Biol Chem 273: 1444–1452 [DOI] [PubMed] [Google Scholar]
- Martin S, Tellam J, Livingstone C, Slot JW, Gould GW, James DE (1996) The glucose transporter (GLUT-4) and vesicle-associated membrane protein-2 (VAMP-2) are segregated from recycling endosomes in insulin-sensitive cells. J Cell Biol 134: 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS, Lienhard GE (2005) AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase activating protein domain. Biochem J 391: 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson AL, Knight JB, Pessin JE (1997) Syntaxin 4, VAMP2, and/or VAMP3/cellubrevin are functional target membrane and vesicle SNAP receptors for insulin-stimulated GLUT4 translocation in adipocytes. Mol Cell Biol 17: 2425–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N, Huang C, Klip A (2005) Cellular location of insulin-triggered signals and implications for glucose uptake. Pflugers Arch (in the press) [DOI] [PubMed] [Google Scholar]
- Ploug T, Ralston E (2002) Exploring the whereabouts of GLUT4 in skeletal muscle (review). Mol Membr Biol 19: 39–49 [DOI] [PubMed] [Google Scholar]
- Randhawa VK, Thong FS, Lim DY, Li D, Garg RR, Rudge R, Galli T, Rudich A, Klip A (2004) Insulin and hypertonicity recruit GLUT4 to the plasma membrane of muscle cells by using N-ethylmaleimide-sensitive factor-dependent SNARE mechanisms but different v-SNAREs: role of TI-VAMP. Mol Biol Cell 15: 5565–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudich A, Klip A (2003) Push/pull mechanisms of GLUT4 traffic in muscle cells. Acta Physiol Scand 178: 297–308 [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Pessin JE (2002) Insulin signaling pathways in time and space. Trends Cell Biol 12: 65–71 [DOI] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Lienhard GE (2005) Synip phosphorylation does not regulate insulin-stimulated GLUT4 translocation. Biochem Biophys Res Commun 332: 880–884 [DOI] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE (2003) Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278: 14599–14602 [DOI] [PubMed] [Google Scholar]
- Satoh S, Nishimura H, Clark AE, Kozka IJ, Vannucci SJ, Simpson IA, Quon MJ, Cushman SW, Holman GD (1993) Use of bismannose photolabel to elucidate insulin-regulated GLUT4 subcellular trafficking kinetics in rat adipose cells. Evidence that exocytosis is a critical site of hormone action. J Biol Chem 268: 17820–17829 [PubMed] [Google Scholar]
- Shewan AM, van Dam EM, Martin S, Luen TB, Hong W, Bryant NJ, James DE (2003) GLUT4 recycles via a trans-Golgi network (TGN) subdomain enriched in syntaxins 6 and 16 but not TGN38: involvement of an acidic targeting motif. Mol Biol Cell 14: 973–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Kandror KV (2005) Sortilin is essential and sufficient for the formation of Glut4 storage vesicles in 3T3-L1 adipocytes. Dev Cell 9: 99–108 [DOI] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE (1991) Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol 113: 123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2004) The synaptic vesicle cycle. Annu Rev Neurosci 27: 509–547 [DOI] [PubMed] [Google Scholar]
- Thong FS, Dugani CB, Klip A (2005) Turning signals on and off: GLUT4 traffic in the insulin-signaling highway. Physiology (Bethesda) 20: 271–284 [DOI] [PubMed] [Google Scholar]
- Török D, Patel N, Jebailey L, Thong FS, Randhawa VK, Klip A, Rudich A (2004) Insulin but not PDGF relies on actin remodeling and on VAMP2 for GLUT4 translocation in myoblasts. J Cell Sci 117: 5447–5455 [DOI] [PubMed] [Google Scholar]
- van Dam EM, Govers R, James DE (2005) Akt activation is required at a late stage of insulin-induced GLUT4 translocation to the plasma membrane. Mol Endocrinol 19: 1067–1077 [DOI] [PubMed] [Google Scholar]
- Volchuk A, Narine S, Foster LJ, Grabs D, De Camilli P, Klip A (1998) Perturbation of dynamin II with an amphiphysin SH3 domain increases GLUT4 glucose transporters at the plasma membrane in 3T3-L1 adipocytes. Dynamin II participates in GLUT4 endocytosis. J Biol Chem 273: 8169–8176 [DOI] [PubMed] [Google Scholar]
- Watson RT, Kanzaki M, Pessin JE (2004) Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr Rev 25: 177–204 [DOI] [PubMed] [Google Scholar]
- Watson RT, Khan AH, Furukawa M, Hou JC, Li L, Kanzaki M, Okada S, Kandror KV, Pessin JE (2004) Entry of newly synthesized GLUT4 into the insulin-responsive storage compartment is GGA dependent. EMBO J 23: 2059–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh GI, Hers I, Berwick DC, Dell G, Wherlock M, Birkin R, Leney S, Tavare JM (2005) Role of protein kinase B in insulin-regulated glucose uptake. Biochem Soc Trans 33: 346–349 [DOI] [PubMed] [Google Scholar]
- Widberg CH, Bryant NJ, Girotti M, Rea S, James DE (2003) Tomosyn interacts with the t-SNAREs syntaxin4 and SNAP23 and plays a role in insulin-stimulated GLUT4 translocation. J Biol Chem 278: 35093–35101 [DOI] [PubMed] [Google Scholar]
- Yamada E, Okada S, Saito T, Ohshima K, Sato M, Tsuchiya T, Uehara Y, Shimizu H, Mori M (2005) Akt2 phosphorylates Synip to regulate docking and fusion of GLUT4-containing vesicles. J Cell Biol 168: 921–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Clarke JF, Ester CJ, Young PW, Kasuga M, Holman GD (1996) Phosphatidylinositol 3-kinase acts at an intracellular membrane site to enhance GLUT4 exocytosis in 3T3-L1 cells. Biochem J 313: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigerer A, Lampson MA, Karylowski O, Sabatini DD, Adesnik M, Ren M, McGraw TE (2002) GLUT4 retention in adipocytes requires two intracellular insulin-regulated transport steps. Mol Biol Cell 13: 2421–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigerer A, McBrayer MK, McGraw TE (2004) Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell 15: 4406–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmet P, Alberti KG, Shaw J (2001) Global and societal implications of the diabetes epidemic. Nature 414: 782–787 [DOI] [PubMed] [Google Scholar]