

Abstract
Ras-GTP imaging studies using the Ras-binding domain (RBD) of the Ras effector c-Raf as a reporter for overexpressed Ras have produced discrepant results about the possible activation of Ras at the Golgi apparatus. We report that RBD oligomerization provides probes for visualization of endogenous Ras-GTP, obviating Ras overexpression and the side effects derived thereof. RBD oligomerization results in tenacious binding to Ras-GTP and interruption of Ras signalling. Trimeric RBD probes fused to green fluorescent protein report agonist-induced endogenous Ras activation at the plasma membrane (PM) of COS-7, PC12 and Jurkat cells, but do not accumulate at the Golgi. PM illumination is exacerbated by Ras overexpression and its sensitivity to dominant-negative RasS17N and pharmacological manipulations matches Ras-GTP formation assessed biochemically. Our data illustrate that endogenous Golgi-located Ras is not under the control of growth factors and argue for the PM as the predominant site of agonist-induced Ras activation.
Keywords: Golgi, imaging, localization, plasma membrane, Ras
Introduction
En route to their final subcellular destination Ha-, Ki- and N-Ras (collectively referred to as Ras) experience post-translational modifications, which result in an increase in hydrophobicity of the holoprotein (Choy et al, 1999; Hancock, 2003). Ras GTPases contain a highly conserved, yet distinct, carboxy-terminal motif that targets them for prenylation and directs them to the endoplasmic reticulum for further post-translational modifications. At this point, routes bifurcate: Ha- and N-Ras become palmitoylated and transit through the Golgi to the plasma membrane (PM), whereas Ki-Ras does not experience palmitoylation and follows an as yet unknown path to the PM (Choy et al, 1999).
Recent imaging studies that used the Ras-binding domain (RBD) of the Ras effector c-Raf as a probe for Ras-GTP have raised much interest in this issue, as some reports have provided evidence that Ras may become activated and hence elicit differential signals at subcellular locations other than the PM. Notably, some investigators have documented activation of overexpressed Ha- and N-Ras at the Golgi apparatus by means of the guanine nucleotide exchange factor (GEF) RasGRP1 (Chiu et al, 2002; Bivona et al, 2003; Perez de Castro et al, 2004), whereas others failed to observe such an effect (Mochizuki et al, 2001; Bondeva et al, 2002; Jiang & Sorkin, 2002). All referred studies relied on the overexpression of Ras or Ras chimeric proteins, or the use of cell lines that stably overexpressed Ras. It has previously been appreciated that GTPase overexpression can potentially distort normal Ras behaviour (Hancock, 2003; Walker & Lockyer, 2004). As Ras is undoubtedly present in the Golgi at steady state (Choy et al, 1999), visualization of the live-cell distribution of endogenous Ras-GTP is required to unambiguously identify the subcellular platforms of Ras activation or, at least, to determine the relative degree of activation at the various sites.
Results and Discussion
Green fluorescent protein (GFP)–RBD has proven to be a useful live-cell biosensor for overexpressed Ras-GTP, but fails to report endogenous Ras activation (Chiu et al, 2002). We reasoned that probes with more inert binding to Ras-GTP should enable one to visualize endogenous Ras. We have created such probes by oligomerizing RBD. To characterize the RBD/Ras interaction, we carried out GAP protection assays (supplementary Fig 1A,B online). Monomeric (RBD-1), dimeric (RBD-2) and trimeric (RBD-3) RBD polypeptides all interfered with GAP-driven Ras- or Rap-bound GTP hydrolysis, consistent with the notion that RBD binding protects Ras/Rap from GAP action. Protection increased with the grade of RBD oligomerization in assays calibrated for equal numbers of binding valencies, indicating that oligomerization elevated RBD affinity for Ras/Rap-GTP (supplementary Fig 1B online). This finding was confirmed by isothermal titration calorimetry experiments (supplementary Fig 1C,D online). In conclusion, RBD oligomerization resulted in stronger binding to Ras-GTP.
To assess the usefulness of RBD-2/3 as imaging probes, we explored their subcellular distribution. GFP–RBD-1 distributed throughout the cytosol and nucleoplasm of serum-starved COS-7 cells (Fig 1A). In contrast, RBD-2 and RBD-3 expression resulted in cell death by apoptosis (Fig 1A; data not shown). As Ras reportedly protects cells from apoptosis induced by serum deprivation (Wolfman & Wolfman, 2000), we assumed that RBD-2/3 blocked endogenous Ras signalling. Concordantly, constitutively active Ha-RasG12V or Ki-RasG12V reverted RBD-3 cytotoxicity and recruited the probes to the PM (Fig 1B). Additionally, Ha-RasG12V caused probe accumulation at a perinuclear region that overlapped with Golgi markers (supplementary Fig 2 online; Chiu et al, 2002; Bivona et al, 2003). We conclude that oligomerized RBDs efficiently report overexpressed Ras-GTP, but the cytotoxicity precluded their use for visualization of endogenous Ras-GTP.
Figure 1.
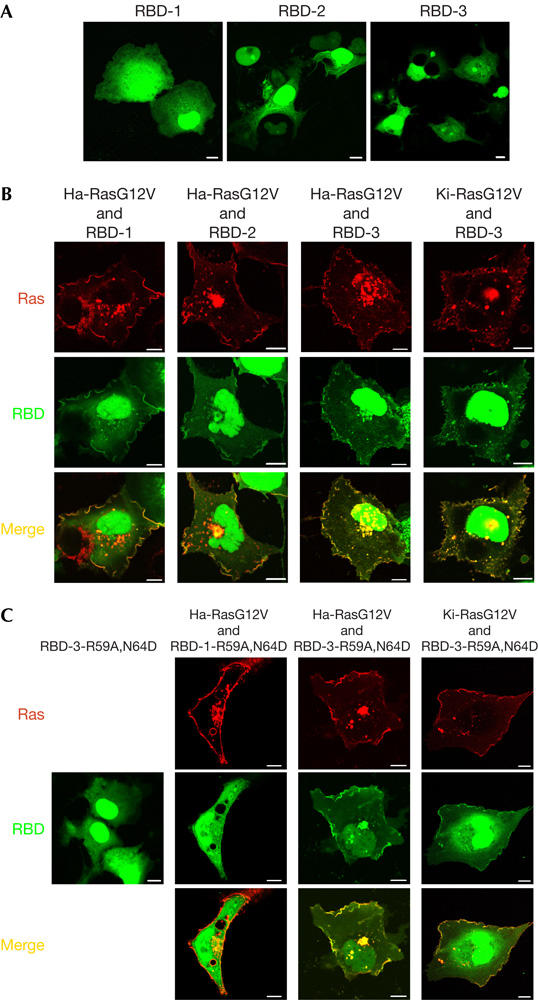
Cellular effects and distribution of oligomerized RBD polypeptides. (A) Distribution of GFP–RBD-1/2/3 in serum-starved COS-7 cells. More than 90% of transfected cells showed the described phenotype. GFP, green fluorescent protein; RBD, Ras-binding domain of the Ras effector c-Raf. (B) COS-7 cells were transfected with DsRed-tagged Ha-RasG12V or Ki-RasG12V in combination with GFP–RBD-1/2/3, as indicated. Cells were serum starved and imaged alive. Yellow pseudo-colour marks colocalization. (C) GFP–RBD-1-R59A,N64D or GFP–RBD-3-R59A,N64D was co-transfected with DsRed-Ha-RasG12V or DsRed-Ki-RasG12V, as indicated. Protein distribution was imaged confocally. Scale bars, 10 μm.
To generate non-toxic oligovalent probes, we introduced point mutations into RBD that attenuate binding to Ras-GTP (Nassar et al, 1996). All of the five mutant RBD-3 variants tested (R59A, R67A, T68A, N64D and K65M), including highly disruptive mutations such as R59A, colocalized with RasG12V and induced cell death if expressed on their own (data not shown). To further attenuate binding, we introduced further point mutations into RBD-3-R59A. Out of four double point mutants, RBD-3-R59A,N64D consistently colocalized with RasG12V and did not compromise cell viability (Fig 1C). The monomeric version, RBD-1-R59A,N64D, did not colocalize with RasG12V, underscoring the importance of oligomerization. Colocalization with RasG12V correlated with the ability of the probes to block RasG12V signalling (supplementary Fig 3 online). In conclusion, oligomerization converted the weakly interacting RBD-R59A,N64D module into a strong binding partner of Ras-GTP.
To assess the specificity of GFP–RBD-3-R59A,N64D, we explored its recruitment by other GTPase variants (Fig 2). Prenylation-deficient Ha-RasG12V,C186S did not alter the distribution of GFP–RBD-3-R59A,N64D. Palmitoylation-defective Ha-RasG12V,C181S,C184S recruited GFP–RBD-3-R59A,N64D to a reticular perinuclear structure, consistent with its reported accumulation at the Golgi/endoplasmic reticulum (Choy et al, 1999). GFP–RBD-3-R59A,N64D did not colocalize with the inactive Ha-RasS17N and Ha-RasG12V,D38A mutants. RBD-1-R59A,N64D shows 33-fold higher affinity for Ras-GTP than for Rap-GTP (Nassar et al, 1996). Concordantly, GFP–RBD-3-R59A,N64D did not colocalize with Rap1AG12V, whereas Rap1AG12V efficiently recruited trimeric RalGDS-RBD-3, a domain with high affinity for Rap-GTP (Nassar et al, 1996). Finally, GFP–RBD-3-R59A,N64D colocalized with GTP-loaded M-Ras and TC21, illustrating limitations in selectivity towards closely related Ras family GTPases. In sum, these data indicated that GFP–RBD-3-R59A,N64D was a non-toxic probe suited for visualization of active Ras, which, however, showed partial crossreactivity among Ras family members.
Figure 2.
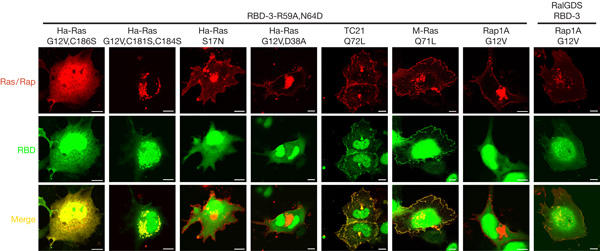
Specificity of GFP–RBD-3-R59A,N64D as a reporter for Ras-GTP. COS-7 cells were transfected with GFP–RBD-3-R59A,N64D or GFP–RalGDS-RBD-3 in combination with the indicated DsRed-tagged GTPases. Cells were imaged confocally. GFP, green fluorescent protein; RBD, Ras-binding domain of the Ras effector c-Raf. Scale bars, 10 μm.
The nature of the trimeric RBD-3/Ras-GTP interaction differs from that of RBD-1 in that it arises from the combined action of three binding valencies. As Ras-GTP-containing membranes essentially represent an oligovalent ligand for RBD probes, triple RBDs were likely to interact with higher affinity to cellular Ras-GTP. To test this notion, we challenged COS-7 cells with epidermal growth factor (EGF) and monitored GFP–RBD-3-R59A,N64D distribution (Fig 3A). EGF induced accumulation of the probe at the PM, prominently at membrane ruffles. We did not detect activation of TC21, M-Ras or R-Ras under these conditions (supplementary Fig 4 online). GFP–RBD-3-R59A,N64D ostensibly did not accumulate at endomembranes, although this was often hard to judge in COS-7 cells owing to overillumination of the perinuclear area. Ras overexpression exacerbated EGF-induced probe translocation to the PM (supplementary Fig 5 online). Stimulation with lysophosphatidic acid resulted in GFP–RBD-3-R59A,N64D translocation to the PM in the absence of membrane ruffling (supplementary Fig 6 online), consistent with the fact that lysophosphatidic acid does not induce Rac signalling in COS-7 cells (Van Leeuwen et al, 2003). To subject the specificity of GFP–RBD-3-R59A,N64D to further scrutiny, we investigated the effect of dominant-negative Ha-RasS17N. In COS-7 cells, Ha-RasS17N blocks Ras-dependent activation of Erk in response to EGF but not to 12-O-tetradecanoylphorbol-13-acetate (TPA; Howe et al, 1992; Ueda et al, 1996). Using COS-7 clones with inducible expression of Ha-RasS17N, we confirmed that Ha-RasS17N exerted its action at the level of Ras (Fig 3B). Consistently, Ha-RasS17N interrupted EGF- but not TPA-induced accumulation of GFP–RBD-3-R59A,N64D at the PM (Fig 3C). In conclusion, the pattern of GFP–RBD-3-R59A,N64D re-distribution to the PM in response to three Ras-activatory stimuli matched biochemically assessed Ras-GTP formation in all cases.
Figure 3.
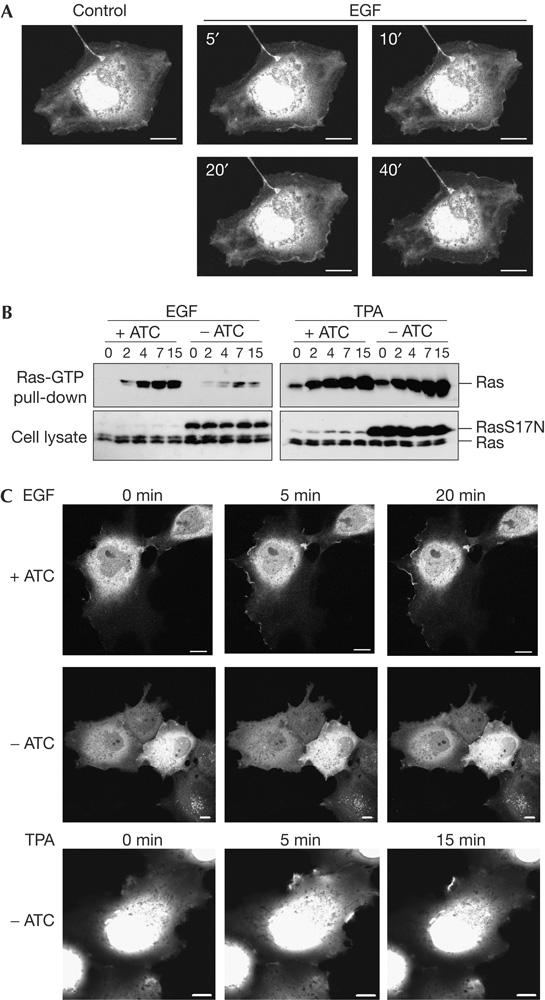
GFP–RBD-3-R59A,N64D reports activation of endogenous Ras. (A) COS-7 cells transfected with GFP–RBD-3-R59A,N64D were deprived of serum and challenged with 50 ng/ml epidermal growth factor (EGF). Confocal pictures were taken at the indicated time points. GFP, green fluorescent protein; RBD, Ras-binding domain of the Ras effector c-Raf. (B) COS-7 cells stably expressing Myc–Ha-RasS17N under the control of an anhydrotetracycline (ATC)-responsive promoter were cultured in the presence or absence of ATC (Myc–Ha-RasS17N expression off or on, respectively), serum starved and challenged with 50 ng/ml EGF or 100 nM 12-O-tetradecanoylphorbol-13-acetate (TPA). Cells were lysed and processed in a Ras-GTP pull-down assay. A representative result of four experiments carried out on two different COS-7/Ha-RasS17N clones is shown. (C) COS-7 cells with inducible expression of Myc–Ha-RasS17N were cultured as in B. Cells were transfected with GFP–RBD-3-R59A,N64D, deprived of serum and stimulated with 50 ng/ml EGF or 100 nM TPA. Confocal images were captured after the indicated time periods. More than 80% of transfected cells showed the same response. Scale bars, 10 μm.
As overillumination of the perinuclear area in COS-7 cells made it difficult to assess subcellular probe distribution, we used PC12 cells to explain whether endogenous Ras became activated at the Golgi. Ras-GTP visualization was accomplished with the triple wild-type GFP–RBD-3 reporter because PC12 cells tolerated its expression, in line with the fact that they do not require Ras signalling for proliferation or survival (Klesse et al, 1999). Cells were additionally stained with the vital Golgi marker BODIPY-TR-C5-ceramide. Nerve growth factor (NGF) induced a fast translocation of GFP–RBD-3 to the PM, which declined but remained detectable for up to 40 min (Fig 4A, quantified in Fig 4B). As expected, the attenuated GFP–RBD-3-R59A,N64D probe showed less pronounced agonist-induced re-distribution, which was arduous to quantify (data not shown). GFP–RBD-3 accumulation at the PM was accentuated by Ras overexpression (supplementary Fig 7 online). Agonist-dependent illumination of the Golgi was not detected. Probe re-distribution in response to EGF was largely indistinguishable from that caused by NGF, except that EGF-induced PM illumination declined to basal levels in 40 min (Fig 4A,B). This difference in the duration of GFP–RBD-3 accumulation at the PM matches biochemically determined Ras activation by EGF/NGF (data not shown; Qiu & Green, 1991).
Figure 4.
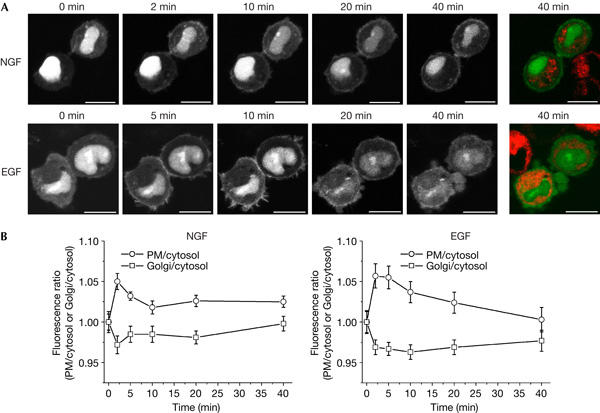
Ras activation in PC12 cells proceeds at the plasma membrane but not at the Golgi. (A) Serum-starved PC12 cells transfected with GFP–RBD-3 were stained with the Golgi marker BODIPY-TR-C5-ceramide, challenged with 50 ng/ml nerve growth factor (NGF) or epidermal growth factor (EGF) and imaged alive. White and green, GFP–RBD-3; red, BODIPY-TR-C5-ceramide. GFP, green fluorescent protein; RBD, Ras-binding domain of the Ras effector c-Raf. Scale bars, 10 μm. (B) Quantification of fluorescence associated with the plasma membrane (PM) and Golgi for the experiments shown in (A), plotted as the ratio of mean pixel fluorescence intensity at the PM or Golgi to cytosol versus time (mean±s.e.m.; NGF: n=6; EGF: n=9). The value for the unstimulated cell was set as 1. For a description of the quantification procedure, see supplementary Fig 8 online.
To extend these observations, we analysed Ras activation in Jurkat T-cells. Jurkat cells respond with strong and sustained Ras-GTP accumulation to the combined administration of TPA and the calcium ionophore ionomycin, whereas T-cell receptor activation of Ras is transient and less pronounced (Downward et al, 1990; Fig 5A). In these cells, the triple variant of RBD-R59A, which features affinity for Ras-GTP intermediate to that of RBD and RBD-R59A/N64D (Nassar et al, 1996), was best suited for visualization of Ras-GTP. TPA/ionomycin caused rapid and sustained accumulation of this probe at the PM (Fig 5B, quantified in Fig 5C). Probe re-distribution to the Golgi was not detected. TPA activation of Ras in T-cells is sensitive to PKC inhibitors (Downward et al, 1990; Roose et al, 2005). Application of the PKC inhibitor bisindolylmaleimide I during the course of stimulation rapidly reverted both biochemically assayed formation of Ras-GTP (Fig 5D) and GFP–RBD-3-R59A accumulation at the PM (Fig 5E), indicating that PKC activity is continuously required to maintain TPA-induced Ras-GTP levels. Taken together with the PC12 data, these findings illustrate that Golgi-located Ras-GTP levels did not rise in response to extracellular agonists.
Figure 5.
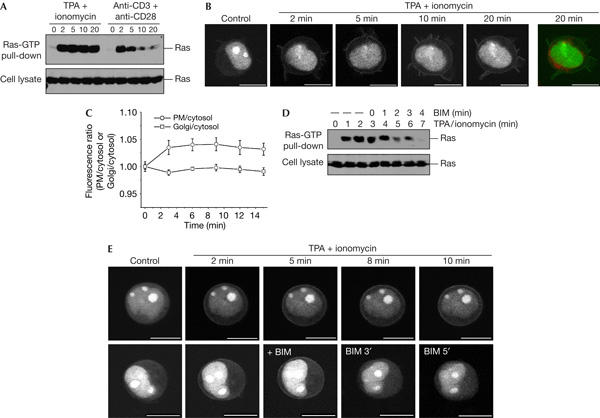
12-O-tetradecanoylphorbol-13-acetate/ionomycin activation of Ras in Jurkat cells does not occur at the Golgi. (A) Serum-starved Jurkat cells were challenged with 150 nM 12-O-tetradecanoylphorbol-13-acetate (TPA) plus 500 ng/ml ionomycin or anti-CD3 plus anti-CD28 (5 μg/ml each), to activate the T-cell receptor, lysed and processed in a Ras-GTP pull-down assay. (B) Serum-starved Jurkat cells expressing GFP–RBD-3-R59A were stained with BODIPY-TR-C5-ceramide, challenged with TPA/ionomycin and imaged alive. White and green, GFP–RBD-3-R59A; red, BODIPY-TR-C5-ceramide. GFP, green fluorescent protein; RBD, Ras-binding domain of the Ras effector c-Raf. (C) Quantification of fluorescence associated with the plasma membrane and Golgi for the experiment shown in (B) (n=8). Refer to legend to Fig 4B. (D) Serum-starved Jurkat cells were stimulated with TPA/ionomycin as before. At 3 min after stimulation, the cell suspension was made up to 5 μM bisindolylmaleimide I (BIM). After the indicated time points, samples were processed in a Ras-GTP pull-down assay. (E) Serum-starved Jurkat cells expressing GFP–RBD-3-R59A were challenged with TPA/ionomycin and imaged confocally. In the lower panel, the medium was made up to 5 μM BIM at the indicated time point. Scale bars, 10 μm.
An increasing body of genetic and biochemical data has made a strong case for RasGRP GEFs as mediators of TPA-induced Ras activation (Dower et al, 2000; Roose et al, 2005). Recently, RasGRP1 has been reported to mediate Ras activation at the Golgi following TPA administration or growth factor activation of phospholipase Cγ in COS, PC12 and Jurkat cells (Bivona et al, 2003; Caloca et al, 2003; Perez de Castro et al, 2004). In the current study, we have failed to observe probe recruitment to the Golgi in any of these cell types. Rather, the probe became targeted to the PM, indicating that RasGRP1 or other GEFs act primarily on PM-located endogenous Ras. With regard to Jurkat T-cells, this conclusion has as precedent two studies that reported exclusive PM recruitment of RasGRP1 in response to T-cell receptor crosslinking or TPA stimulation (Carrasco & Merida, 2004; Zugaza et al, 2004).
Along this line of reasoning and in agreement with previous findings (Jiang & Sorkin, 2002), we observed agonist-independent perinuclear staining by GFP–RBD-3-R59A,N64D in COS-7 cells overexpressing Ras under serum deprivation (supplementary Fig 5 online), which indicated that Golgi-located Ras escapes normal regulation if overexpressed in combination with an effector protein. We suggest that high levels of heterologous Ras and protection from GAPs exerted by RBD proteins coincidentally distort Ras-GDP/GTP levels, leading to aberrant accumulation of Ras-GTP.
In summary, the current study documents that Golgi-located endogenous Ras is not under the control of growth factor signalling, and argues for the PM as the predominant site of Ras activation.
Methods
Cell transfection and imaging. COS-7, PC12 (both seeded on collagen-coated glass-bottom dishes) and Jurkat cells were transfected with lipofectamine (Invitrogen, Carlsbad, CA, USA), PolyFect (Qiagen, Hilden, Germany) and DMRIE-C (Invitrogen) reagents, respectively. Cells were deprived of serum and treated with the Golgi marker BODIPY-TR-C5-ceramide (Molecular Probes, Eugene, OR, USA) before imaging. At this point, Jurkat cells were seeded on polylysine-coated glass-bottom dishes. Cells were challenged with agonists and confocal images were acquired with a Zeiss LSM 510 (Carl Zeiss GmbH, Oberkochen, Germany) inverted laser scanning microscope (LSM) using a C-Apochromat × 63 water immersion objective lens (Zeiss). GFP and DsRed/BODIPY-TR-C5-ceramide were excited with the Ar 488 nm and the HeNe 543 nm laser lines, respectively. GFP fluorescence was recorded with a 505–550 nm band-pass filter, and a 560 nm long-pass filter was used to record the BODIPY-TR-C5-ceramide fluorescence. The confocal aperture was adjusted to give optical sections of 0.8 μm (Jurkat) or 1 μm (COS-7 and PC12 cells). Images were captured at set time intervals for a period of at least 30 min after stimulation. LSM image files were processed using the Zeiss LSM image browser software (version 3.1).
For an extended and detailed description of methods, see the supplementary information online.
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/7400560-s1.pdf).
Supplementary Material
Supplementary Information
Acknowledgments
We acknowledge the generous provision of plasmids by J. de Gunzburg, C. Herrmann, J. Meng, G. Murphy and A. Wittinghofer. We thank O. Zschörnig, S. Heinemann, G. Nowak, P. Hemmerich, U. Horn and A. Berndt for technical help and for providing access to LSM instrumentation. This work was supported by the ‘Novartis-Stiftung für Therapeutische Forschung' and by ‘Deutsche Forschungsgemeinschaft' (DFG) grant RU860/1-1.
References
- Bivona TG, Perez De Castro I, Ahearn IM, Grana TM, Chiu V, Lockyer PJ, Cullen PJ, Pellicer A, Cox AD, Philips MR (2003) Phospholipase Cγ activates Ras on the Golgi apparatus by means of RasGRP1. Nature 424: 694–698 [DOI] [PubMed] [Google Scholar]
- Bondeva T, Balla A, Varnai P, Balla T (2002) Structural determinants of Ras–Raf interaction analyzed in live cells. Mol Biol Cell 13: 2323–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caloca MJ, Zugaza JL, Bustelo XR (2003) Exchange factors of the RasGRP family mediate Ras activation in the Golgi. J Biol Chem 278: 33465–33473 [DOI] [PubMed] [Google Scholar]
- Carrasco S, Merida I (2004) Diacylglycerol-dependent binding recruits PKCθ and RasGRP1 C1 domains to specific subcellular localizations in living T lymphocytes. Mol Biol Cell 15: 2932–2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson RL II, Cox AD, Philips MR (2002) Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol 4: 343–350 [DOI] [PubMed] [Google Scholar]
- Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR (1999) Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 98: 69–80 [DOI] [PubMed] [Google Scholar]
- Dower NA, Stang SL, Bottorff DA, Ebinu JO, Dickie P, Ostergaard HL, Stone JC (2000) RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat Immunol 1: 317–321 [DOI] [PubMed] [Google Scholar]
- Downward J, Graves JD, Warne PH, Rayter S, Cantrell DA (1990) Stimulation of p21ras upon T-cell activation. Nature 346: 719–723 [DOI] [PubMed] [Google Scholar]
- Hancock JF (2003) Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol 4: 373–384 [DOI] [PubMed] [Google Scholar]
- Howe LR, Leevers SJ, Gomez N, Nakielny S, Cohen P, Marshall CJ (1992) Activation of the MAP kinase pathway by the protein kinase raf. Cell 71: 335–342 [DOI] [PubMed] [Google Scholar]
- Jiang X, Sorkin A (2002) Coordinated traffic of Grb2 and Ras during epidermal growth factor receptor endocytosis visualized in living cells. Mol Biol Cell 13: 1522–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klesse LJ, Meyers KA, Marshall CJ, Parada LF (1999) Nerve growth factor induces survival and differentiation through two distinct signaling cascades in PC12 cells. Oncogene 18: 2055–2068 [DOI] [PubMed] [Google Scholar]
- Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A, Matsuda M (2001) Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature 411: 1065–1068 [DOI] [PubMed] [Google Scholar]
- Nassar N, Horn G, Herrmann C, Block C, Janknecht R, Wittinghofer A (1996) Ras/Rap effector specificity determined by charge reversal. Nat Struct Biol 3: 723–729 [DOI] [PubMed] [Google Scholar]
- Perez de Castro I, Bivona TG, Philips MR, Pellicer A (2004) Ras activation in Jurkat T cells following low-grade stimulation of the T-cell receptor is specific to N-Ras and occurs only on the Golgi apparatus. Mol Cell Biol 24: 3485–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu M-S, Green SH (1991) NGF and EGF rapidly activate p21ras in PC12 cells by distinct, convergent pathways involving tyrosine phosphorylation. Neuron 7: 937–946 [DOI] [PubMed] [Google Scholar]
- Roose JP, Mollenauer M, Gupta VA, Stone J, Weiss A (2005) A diacylglycerol–protein kinase C–RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T cells. Mol Cell Biol 25: 4426–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, Ohno S (1996) Protein kinase C activates the MEK–ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem 271: 23512–23519 [DOI] [PubMed] [Google Scholar]
- Van Leeuwen FN, Olivo C, Grivell S, Giepmans BN, Collard JG, Moolenaar W (2003) Rac activation by lysophosphatidic acid LPA1 receptors through the guanine nucleotide exchange factor Tiam1. J Biol Chem 278: 400–406 [DOI] [PubMed] [Google Scholar]
- Walker SA, Lockyer PJ (2004) Visualizing Ras signalling in real-time. J Cell Sci 117: 2879–2886 [DOI] [PubMed] [Google Scholar]
- Wolfman JC, Wolfman A (2000) Endogenous c-N-Ras provides a steady-state anti-apoptotic signal. J Biol Chem 275: 19315–19323 [DOI] [PubMed] [Google Scholar]
- Zugaza JL, Caloca MJ, Bustelo XR (2004) Inverted signaling hierarchy between Ras and Rac in T-lymphocytes. Oncogene 23: 5823–5833 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information