

Abstract
The imprint at the mat1 locus of Schizosaccharomyces pombe acts to initiate the replication-coupled recombination event that underlies mating-type switching. However, the nature of the imprint has been an area of dispute. Two alternative models have been proposed: one stated that the imprint is a nick in the DNA, whereas our data suggested that it consists of one or two ribonucleotides incorporated into the otherwise intact DNA duplex. Here, we verify key predictions of the RNA model by characterization of wild-type genomic DNA purified under conditions known to hydrolyse DNA–RNA–DNA hybrid strands. First, we observe one-nucleotide gap at the hydrolysed DNA, as expected from the presence of two ribonucleotides. Second, using a novel assay based on ligation-mediated PCR, a 3′-terminal ribonucleotide is detected at the hydrolysed imprint. Our observations allow the unification of available data sets characterizing the wild-type imprint.
Keywords: mat1, imprint, one-nucleotide gap, mating type, replication
Introduction
Mating-type switching in fission yeast occurs by a molecular mechanism that relies on the asymmetry of DNA replication. Schizosaccharomyces pombe switches mating type according to the ‘one-in-four' rule, where one of the two daughters of a newly switched cell is able to switch, and one of the four granddaughters has switched the mating type (Miyata & Miyata, 1981). Mating-type switching depends on the site- and strand-specific imprint at the mat1 locus; thus, the imprint marks the cells that are able to switch (Egel, 1984; Klar, 1990). The formation of the imprint requires that mat1 is replicated in the centromere-proximal direction, and it is introduced only in the DNA strand that is synthesized by the lagging-strand replication complex (Dalgaard & Klar, 1999). Imprinting depends on Swi7 (DNA polymerase-α) and the replication checkpoint proteins Swi1 and Swi3, and involves a lagging-strand-induced replication pause at mat1 (Egel et al, 1984; Singh & Klar, 1993; Dalgaard & Klar, 2000; Vengrova & Dalgaard, 2004). A recent study has shown that the imprint is formed during the S phase, and that its appearance coincides with pausing of the replication fork (Holmes et al, 2005). The imprint is maintained until the next S phase, when leading-strand replication complex is stalled at the imprint present in the template (Kaykov et al, 2004; Vengrova & Dalgaard, 2004). The resolution of this stalled replication fork is thought to induce recombination between mat1 and the donor cassettes mat2P or mat3 M (Fig 1A), which leads to the mating-type switching (Arcangioli & de Lahondes, 2000). The nature of the imprint, however, has been an area of dispute. A break is observed at mat1 when the S. pombe DNA is purified by many of the currently used methods (Arcangioli, 1998; Dalgaard & Klar, 1999). However, we have been able to purify the unbroken DNA, which could subsequently be broken/hydrolysed at the site of the imprint by alkali or RNase treatment. Our data suggested that the imprint consisted of one or two ribonucleotides incorporated into the otherwise intact DNA duplex (Fig 1A; Dalgaard & Klar, 1999; Vengrova & Dalgaard, 2004).
Figure 1.
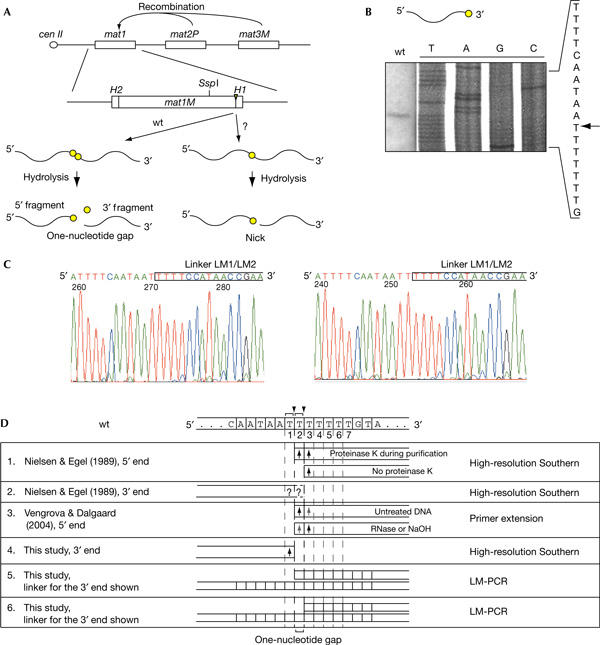
Defining the ends generated by hydrolysis of the mat1 imprint. (A) The imprint is located at the mat1 (P or M containing) locus, which defines the mating-type of the cell, whereas the donor loci mat2P and mat3 M contain genetic information necessary for switching. Hydrolysis of a two- or one-ribonucleotide imprint leads to the formation of a one-nucleotide gap or a nick, respectively. The imprint is shown as a triangle, and the ribonucleotides are shown as circles. The position of the centromere-proximal SspI site is indicated. (B) High-resolution mapping of the 3′ end at the imprint in the wild-type strain. The sequence of the region is given to the right, and the position of the band is indicated by an arrow. The gel is displayed with the low molecular bands at the top. As such the sequence is shown 5′–3′. (C) DNA sequencing of the PCR-amplified ligation products, generated by ligation of the linker LM1/LM2 (boxed) to the 3′ end at the hydrolysed imprint. (D) Summary of the published data, mapping the ends generated at the wild-type imprint. The DNA sequence of the imprinted strand is given above the table, where ribonucleotide positions are shown with brackets and arrowheads indicate putative RNA bonds broken by hydrolysis. Below, the ends mapped by the different studies and methods are aligned to the sequence. Black and grey arrows represent prominent and faint bands, respectively. LM-PCR, ligation-mediated PCR; wt, wild type.
In this study, which focuses on the mat1 M allele, we resolve the existing controversy concerning the nature of the imprint by verifying two key predictions of the RNA model for the fully hydrolysed wild-type mat1 imprint: (i) we show that for most of the molecules, complete hydrolysis of the imprint leads to the loss of one nucleotide, leaving a one-nucleotide gap at mat1; this is predicted by the presence of a two-ribonucleotide imprint and (ii) we establish that a 3′-terminal ribonucleotide is present at the end of the hydrolysed 5′ fragment.
Results
It is known that imprinted S. pombe mat1 DNA is fragile during most purification methods, as the imprint is converted into a break (Arcangioli, 1998; Dalgaard & Klar, 1999). Originally, the ends of this break were mapped by the high-resolution Southern blot method, in which the digested genomic DNA was resolved on a denaturing polyacrylamide gel next to the sequencing reactions (Nielsen & Egel, 1989). In this experiment, the 5′ end of the break at mat1 was detected as one band, corresponding to the third T in the imprinted sequence given in Fig 1D, line 1 (Nielsen & Egel, 1989). However, proteinase K treatment during purification led to the detection of a doublet, corresponding to the second and the third T in the imprinted sequence, which suggested that enzymatic hydrolysis of the imprinted DNA was taking place during purification. The position of these two bands corresponds precisely to those that we observed when the DNA, partially hydrolysed with RNase T2 or alkali, was analysed by primer extension (Fig 1D, line 3; Vengrova & Dalgaard, 2004). Thus, ample data are available defining the 5′ end of the hydrolysed mat1 imprint in wild-type strains. For replication intermediates, the 3′ end of the nascent leading strand, blocked at the imprint in the template strand, was mapped to the same position by high-resolution Southern blot (Vengrova & Dalgaard, 2004) and by ligation-mediated PCR (LM-PCR; Kaykov & Arcangioli, 2004). Contrary to this, the 3′ end of the hydrolysed imprint is not as well defined. In the original study by Nielsen & Egel (1989), the analysis of this end detected a single band, which could not be mapped precisely, but was ‘very close to its right-hand counterpart' (Fig 1D, line 2). We therefore set out to establish the precise position of the 3′ end at the hydrolysed mat1 imprint.
For this purpose, the DNA was digested with SspI and separated on a 6% denaturing polyacrylamide gel next to corresponding sequencing reactions (Fig 1B), allowing the 3′ end of the genomic DNA to be directly compared with the sequence. Using this method, the DNA end was mapped to the first T in the sequence (Fig 1D). To verify this result, the 3′ end at the imprint was further defined by the LM-PCR (Kaykov & Arcangioli, 2004). Two linkers were employed: one, LM1A/LM2, was used for the detection of the 3′ end corresponding to the first T (Fig 1D, line 5, and Fig 3C) and the other, LM1/LM2, was used for the detection of the 3′ end corresponding to the second T (Fig 1D, line 6, and Fig 3B; supplementary Fig D, line 1, online). The ligation was carried out using Escherichia coli DNA ligase, which less efficiently ligates over a one-nucleotide gap than T4 DNA ligase (data not shown; Goffin et al, 1987). The ligation product was amplified by PCR, and the resulting fragment was cloned and sequenced. Ten clones were sequenced for each linker. As expected, the ten sequences obtained using linker LM1A/LM2 showed amplification of the fragments ending at the first T. However, the sequences obtained using linker LM1/LM2 showed 50% of the genomic DNA fragments ending at the second T and the other 50% at the first T (Fig 1C,D). Thus, even when using E. coli ligase for the ligation reaction, for half of the amplified fragments, ligation over a one-nucleotide gap has occurred. This result confirms that after the standard purification procedure, most of the 5′ fragments at the imprint end at the first T, and thus one nucleotide is lost during hydrolysis.
Figure 3.
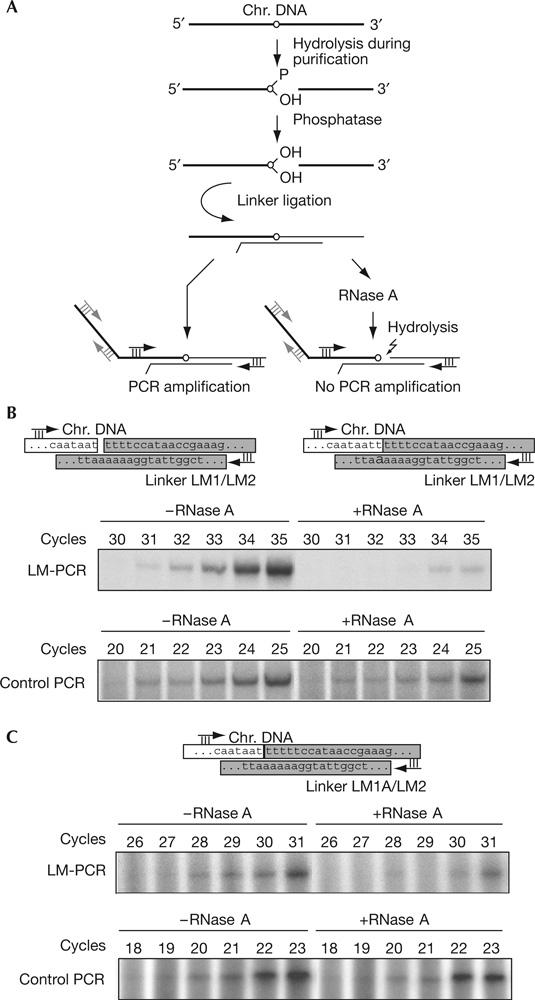
Detection of a 3′-terminal ribonucleotide in the 5′ fragment at the hydrolysed mat1 imprint. (A) Line drawing of the experimental strategy, using ligation-mediated PCR (LM-PCR) and RNase A digestion. Chromosomal and linker DNA are shown by thick and thin black lines, respectively. The ribonucleotide is indicated by a circle. The positions of the primers, used for LM-PCR and control PCR, are given as black and grey arrows, respectively. (B,C) LM-PCR of the 3′ end at the imprint using two different linkers (part of the given sequences). Linkers (grey boxes) were designed to allow ligation to nicked (B; primer LM1/LM2) or gapped (C; primer LM1A/LM2) molecules. The control amplification of a flanking sequence is shown. The number of cycles used in the PCR reaction is shown above each lane.
Another key prediction of the RNA model is that a single ribonucleotide is retained on the 3′ end of the 5′ fragment when the imprint is hydrolysed. We decided to use LM-PCR to test for the presence of this ribonucleotide. As ligation of such DNA–RNA hybrid to a DNA linker joins the 5′ phosphate of the linker to the 3′ hydroxyl of the ribonucleotide, the formed phosphodiester linkage is an RNA bond, which can by re-hydrolysed by RNases or alkali. RNase digestion would disrupt the ligation product and abolish amplification; therefore, the presence of a 3′ ribonucleotide can be detected by comparison of the amplification of the untreated and RNase-digested ligation products.
For the development of this method, we first tested the ability of different RNases to recognize a single ribonucleotide in an oligonucleotide duplex—the substrate, which would be generated by ligation of a double-stranded linker to the hydrolysed imprint. We found that RNase A and to some degree RNase T2, but not T1 or H, could hydrolyse such a substrate (supplementary Fig A online). Hydrolysis of a 3′ phosphodiester bond of a ribonucleotide by alkali or RNase A produces a 2′–3′ cyclic phosphate, which then forms equal amounts of 2′ and 3′ phosphate (Fig 2A). The presence of 3′ phosphate is known to prevent ligation of the deoxyribonucleotides; we tested whether a molecule resulting from the hydrolysis of a ribonucleotide bond in a DNA–RNA–DNA hybrid can be joined to a DNA linker by different ligases. An oligonucleotide with a single internal ribonucleotide was end labelled, hydrolysed using NaOH or RNase A (Fig 2B, lanes 2 and 6, respectively) and mixed with the double-stranded linker. It should be noted that only partial hydrolysis is achieved with NaOH under these conditions (supplementary Fig B online). Interestingly, when T4 DNA ligase was used, all labelled DNA, including the unhydrolysed oligonucleotide, was ligated to the linker (Fig 2B, band I in lane 2 and band II in lanes 2,6). Importantly, the substrate containing the 3′ ribonucleotide (band II) contains in equimolar proportion either a 2′ or a 3′ phosphate (Fig 2A). Thus, the efficient ligation observed using T4 ligase shows that this enzyme can efficiently join the 5′ phosphate of the linker to either a 2′- or a 3′-hydroxyl group in the presence of a 3′ or a 2′ phosphate, respectively. Such products will have an internal phosphate branching off at the newly formed junction (Fig 2B, line drawing, molecule III). We tested whether these ligation products are sensitive to NaOH and RNase A. As expected, the products were completely resistant to hydrolysis (Fig 2B, left panel, lanes 4,5,8,9); during RNA hydrolysis, a nucleophilic attack of the deprotonized 2′-hydroxyl group breaks the phosphate bond, and in the molecules analysed here, there is no free 2′- or 3′-hydroxyl group. When the same ligation experiment was carried out using E. coli DNA ligase, very little of the ligation product could be detected (Fig 2B, right panel). This shows that E. coli ligase is unable to efficiently use substrates with 2′- or 3′-terminal phosphates. We then removed the 2′- and 3′-terminal phosphates with Antarctic phosphatase and used the substrates thus obtained for ligation. Here, E. coli ligase generates products that are fully hydrolysable by RNase A (Fig 2C). Finally, we ensured that a DNA chain containing a single ribonucleotide could be used as a template by Taq DNA polymerase for subsequent LM-PCR. Using synthetic oligonucleotides, we found that Taq polymerase could efficiently elongate across up to three ribonucleotides (supplementary Fig C online).
Figure 2a.
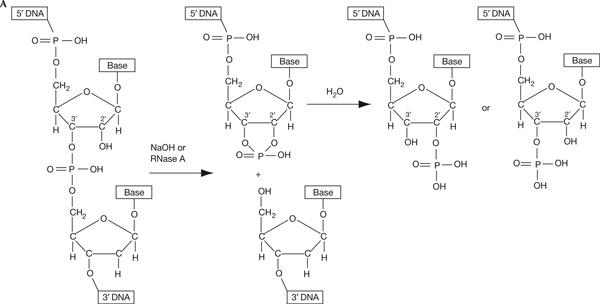
Escherichia coli DNA ligase, but not T4 DNA ligase, can be used to detect a 3′-terminal ribonucleotide on a DNA fragment. (A) NaOH or Rnase. A hydrolysis of a ribonucleotide phosphodiester bond results in the formation of equimolar amounts of 2′- and 3′-terminal phosphates and a 5′ hydroxyl. The 2′ and 3′ positions of the ribose are marked. The deoxyribonucleotide, following the ribonucleotide in the DNA–RNA–DNA chain, is depicted in grey. The intermediate 2′–3′ cyclic phosphate is shown.
Figure 2b.
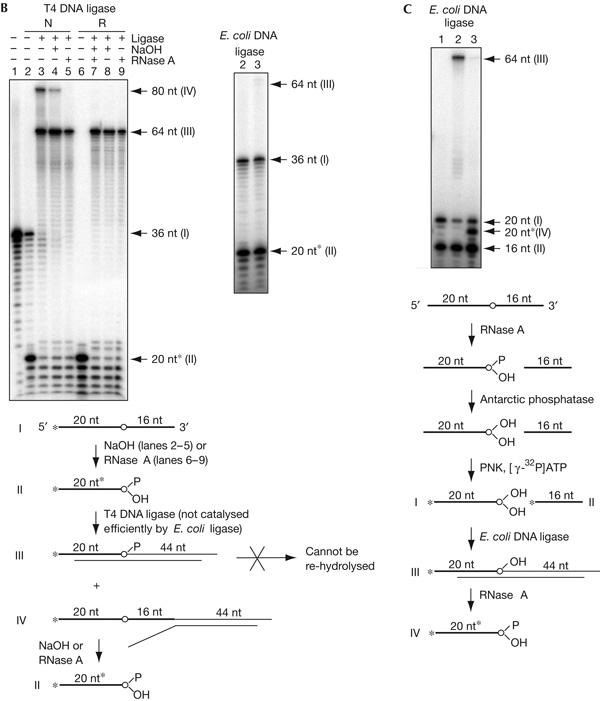
(B) A DNA linker can be efficiently ligated to a terminal ribonucleotide with 2′ or 3′ phosphate using T4, but not E. coli, DNA ligase, forming products resistant to re-hydrolysis. Ligation using T4 and E. coli ligases is shown in the left and right panels, respectively. An outline of the experiment is given in a line drawing below. Roman numbers and nucleotide length for the bands detected in the panel correspond to reaction substrates and products shown in the line drawing. The asterisk, marking the size of the oligonucleotide fragment generated by hydrolysis, indicates the increased mobility due to the extra phosphate (Tapper & Clayton, 1981). Radioactive 5′-end label is shown with an asterisk. The ribonucleotide is indicated by a circle. For both panels: lane 1, end-labelled oligonucleotide ART26; lanes 2–5, experiments using ART26 hydrolysed with NaOH as a substrate (N); lanes 6–9, experiments using ART26 hydrolysed with RNase A (R); lanes 2,6, hydrolysed ART26 substrate; lanes 3,7, linker ligation products; lanes 4,8, re-hydrolysis of the ligation product with NaOH; lanes 5,9, re-hydrolysis of the ligation product with RNase A. The fragment, marked IV (lane 3), formed from ligation of the linker to the unhydrolysed oligonucleotide I serves as an internal control; following NaOH or RNase A treatment (lanes 4,5), the band disappears, showing that the RNA bond with a hydroxyl at the 2′ position is efficiently hydrolysed under the conditions of the experiment. (C) A DNA oligonucleotide containing a 3′-terminal ribonucleotide devoid of 2′ or 3′ phosphate is a substrate for E. coli DNA ligase, and the ligation product can be re-hydrolysed using RNase A. Lane 1, substrate ART26 hydrolysed with NaOH, dephosphorylated with Antarctic phosphatase and 5′-end labelled; lane 2, ligation products; lane 3, ligation products re-hydrolysed with RNase A. An outline of the experiment is given below in a line drawing (description as in B).
Chromosomal DNA for the LM-PCR experiments was purified using the method that is known to yield a break at mat1, and was denatured with alkali to increase the amount of exposed 3′ ends at the imprint. The DNA was then treated with Antarctic phosphatase before ligation of the linker, to remove terminal phosphates from the 5′ fragment. Half the ligation product was digested with RNase A, and both treated and untreated samples were analysed by PCR (Fig 3A). Our previous data suggest that in the wild-type genomic mat1 DNA, there are ribonucleotides at positions 1 and 2 shown in Fig 1D (Vengrova & Dalgaard, 2004). We therefore used both linkers LM1/LM2 (Figs 1D, 3B) and LM1A/LM2 (Figs 1D, 3C) for the ligation. The RNase digest reduced amplification of the imprint-containing fragment obtained with both linkers; the difference of three cycles in the appearance of the amplification signal corresponds to more than 80% hydrolysis. No effect of the RNase treatment was observed for the amplification of a flanking sequence (Fig 3B,C, control PCR). Importantly, ligation products that are obtained with linker LM1/LM2 contain mat1 5′ fragments, in which ligation took place to either the first or the second T in the sequence (Fig 1C). The sensitivity of these products to hydrolysis by RNase A shows that the bonds formed to both nucleotides are RNA bonds. Finally, when a similar experiment was carried out on hydrolysed wild-type DNA, while excluding the Antarctic phosphatase treatment and using T4 DNA ligase, the RNase digest had little effect on the amplification (supplementary Fig D online). This result reflects the experiment shown in Fig 2B, in which the presence of 2′ or 3′ phosphate inhibits re-hydrolysis, suggesting that 2′ or 3′ phosphate is present at the 3′ ribonucleotide after hydrolysis of the imprint using this purification method.
Discussion
To verify the RNA nature of the S. pombe imprint, and for the unification of the data presented in the literature, we tested the key characteristics of the hydrolysis products of mat1: the presence of a 3′-terminal ribonucleotide and a one-nucleotide gap. Both these characteristics were observed for most of the molecules, establishing that the mat1 imprint is mainly constituted by two ribonucleotides (positions 1 and 2; Fig 1D). Although in this study we detected molecules containing a nick (one-ribonucleotide imprint) using PCR, such molecules cannot be detected by Southern blot and therefore they constitute only a minority. Importantly, this conclusion is consistent with previously published data. Arcangioli (1998) observed that on a native polyacrylamide gel, the SspI fragment of mat1, purified under conditions that allow RNA hydrolysis, ran as a doublet. Furthermore, when this fragment was analysed by native-to-denaturing polyacrylamide two-dimensional gel, the high-molecular-weight band and the low-molecular-weight band in the doublet were shown to represent non-imprinted and imprinted double-stranded molecules, respectively (Arcangioli, 1998). The increased mobility of the imprinted molecules is consistent with a one-nucleotide gap. In addition, the presence of two ribonucleotides at mat1 is also consistent with the observation that the 5′ end is represented by a doublet (Nielsen & Egel, 1989; Vengrova & Dalgaard, 2004). In both studies, two populations of 5′ ends, at the second and the third T, were detected in some preparations, suggesting that only a partial hydrolysis of the imprint had occurred. In the same studies, omission of proteinase K (Nielsen & Egel, 1989) or digestion of the DNA with RNase T2 (Vengrova & Dalgaard, 2004) led to full hydrolysis of the imprint, producing one band at the third T, establishing the 5′ end of the hydrolysed imprint. It should be noted that in a mutant strain in which the first T in the imprinted sequence is mutated, a nick is observed at mat1 (Kaykov & Arcangioli, 2004). The enzymatic characterization presented here (see also supplementary Figs E,F online; Fig 1D) shows that the results of Kaykov & Arcangioli can be explained by complete hydrolysis of this one-ribonucleotide imprint. Here, a nick would be generated, with a 5′ hydroxyl and a 2′ or 3′ phosphate at the 3′ ribonucleotide. Owing to the general difficulty in purifying the intact imprinted DNA, we expect that the imprint hydrolysis is mediated by cellular RNases.
In this study, we also develop a novel method for detecting 3′-terminal ribonucleotides left attached after hydrolysis of a DNA–RNA–DNA hybrid strand. In addition to designating the exact position at mat1 where switching is to be initiated, such 3′-terminal ribonucleotides could have other biological functions. At mat1, switching probably involves replication fork regression when the replication fork stalls at the imprint; however, the RNA bond of the imprint has to be hydrolysed at some point during the switching process to allow the removal of the outgoing cassette. Subsequent to this hydrolysis, a 3′-terminal nucleotide could act to recruit specific repair pathways to ensure the removal of this DNA strand.
An intriguing question remains of whether such ribonucleotide imprints are introduced elsewhere in the genome, and whether the novel LM-PCR-based method described here can be used for their detection. Importantly, this study demonstrates the presence of ribose at the site of the imprint; it is yet to be determined whether these ribonucleotides are constituted by uracil or by ribothymine, thus explaining the mechanism of their formation. Uracil would point at the polymerization event, perhaps carried out by a polymerase with broad substrate specificity (Joyce, 1997). The ribothymine nature of the imprint opens up the possibility of post-replicative oxidation, which has not been yet described.
Finally, our characterization of the ligase activities used in this study provides new insights into why ribonucleotides incorporated into the DNA can pose a danger to the cell. T4 ligase and, to a lesser degree, E. coli ligase can seal a nick at a hydrolysed ribonucleotide that possesses either a 2′ or a 3′ phosphate, probably by using the available 3′- or 2′-hydroxyl group. Thus, in such molecules, the DNA backbone will be distorted, causing problems during transcription or replication.
Methods
Purification of yeast chromosomal DNA. Strain JZ105 (mat1 M Δmat2,3∷LEU2, ade6-210, leu1-32, ura4-D18, his2) was used for all the experiments. The DNA was purified using DNAzol (Invitrogen, Paisley, UK). For a detailed description of the method, see the supplementary information online.
High-resolution Southern blot. The experiment was carried out as described previously (Vengrova & Dalgaard, 2004).
Hydrolysis and ligation of oligonucleotides. For the oligonucleotide ligation experiments, end-labelled ART26 (TTTATTTATTTTCAATAACrUGCAGTGTAATATAA AT) was hydrolysed with NaOH or RNase A. Hydrolysed ART26 (10 pmol) was mixed with 50 pmol of pre-annealed LM1/ART27 (LM1: P-TTTTCCATAACCGAAAGTAGTGACAAGTGTTGGCC ATGGAACAG; ART27: CACTACTTTCGGTTATGGAAAAAGTTATTGAAAAT AAATAAA) duplex. Ligation was carried out overnight at 16°C, using T4 DNA ligase or E. coli DNA ligase. The sample was split in three, and two aliquots were treated with NaOH or RNase A. For the experiment with dephosphorylated oligonucleotide, ART26 was digested with RNase A and treated with Antarctic phosphatase (New England Biolabs, Ipswich, MA, USA). The hydrolysed, dephosphorylated oligonucleotide was end labelled with [γ-32P]ATP, and the linker ligation was carried out. The ligation products were analysed on 10% denaturing polyacrylamide gel. A detailed description of the method is given in the supplementary information online.
Ligation-mediated PCR. The DNA was denatured with NaOH. LM-PCR was carried out using E. coli DNA ligase, as described previously (Kaykov & Arcangioli, 2004) with the following modifications. The following oligonucleotides were used for linkers: LM1 (as above), LM1A (P-TTTTTCCATAACCGAAAGTAGTGACAAGTGTTGGC CATGGAACAG) and LM2 (CACTACTTTCGGTTATGGAAAAAATTATTGAAAAT AAATAAAAACGAATATTC). After ligation, the sample was split in two and RNase A (Sigma-Aldrich, Dorset, UK) was added to one of the two fractions. The PCR on both fractions was carried out using Taq DNA polymerase (NEB) and oligonucleotides LM3D (Kaykov & Arcangioli, 2004) and LM4 (CTGTTCCATGGCCAACACTTG) under the following conditions: 95°C (5 min) and 15–34 cycles at 94°C (30 s)/65°C (30 s)/72°C (30 s). For the control PCR, oligonucleotides OK18 (Kaykov & Arcangioli, 2004) and mat1-NsiI-R (TTTGGGATGAGGTTTAATATATGTTTA) were used. PCR using these primers was performed as described above, except that primer annealing was carried out at 55°C. Further details of the method are given in the supplementary information online.
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/7400576-s1.pdf).
Supplementary Material
Supplementary Information
Acknowledgments
We thank R. Egel and S. Aligianni for helpful suggestions. This work was funded by Marie Curie Cancer Care and the Association for International Cancer Research (J.Z.D.).
References
- Arcangioli B (1998) A site- and strand-specific DNA break confers asymmetric switching potential in fission yeast. EMBO J 17: 4503–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangioli B, de Lahondes R (2000) Fission yeast switches mating type by a replication–recombination coupled process. EMBO J 19: 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgaard JZ, Klar AJ (1999) Orientation of DNA replication establishes mating-type switching pattern in S. pombe. Nature 400: 181–184 [DOI] [PubMed] [Google Scholar]
- Dalgaard JZ, Klar AJ (2000) swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell 102: 745–751 [DOI] [PubMed] [Google Scholar]
- Egel R (1984) The pedigree pattern of mating-type switching in Schizosaccharomyces pombe. Curr Genet 8: 205–210 [DOI] [PubMed] [Google Scholar]
- Egel R, Beach DH, Klar AJ (1984) Genes required for initiation and resolution steps of mating-type switching in fission yeast. Proc Natl Acad Sci USA 81: 3481–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin C, Bailly V, Verly WG (1987) Nicks 3′ or 5′ to AP sites or to mispaired bases, and one-nucleotide gaps can be sealed by T4 DNA ligase. Nucleic Acids Res 15: 8755–8771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes AM, Kaykov A, Arcangioli B (2005) Molecular and cellular dissection of mating-type switching steps in Schizosaccharomyces pombe. Mol Cell Biol 25: 303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce CM (1997) Choosing the right sugar: how polymerases select a nucleotide substrate. Proc Natl Acad Sci USA 94: 1619–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaykov A, Arcangioli B (2004) A programmed strand-specific and modified nick in S. pombe constitutes a novel type of chromosomal imprint. Curr Biol 14: 1924–1928 [DOI] [PubMed] [Google Scholar]
- Kaykov A, Holmes AM, Arcangioli B (2004) Formation, maintenance and consequences of the imprint at the mating-type locus in fission yeast. EMBO J 23: 930–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar AJ (1990) The developmental fate of fission yeast cells is determined by the pattern of inheritance of parental and grandparental DNA strands. EMBO J 9: 1407–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata H, Miyata M (1981) Mode of conjugation in homotallic cells of Schizosaccharomyces pombe. J Gen Appl Microbiol 27: 365–371 [Google Scholar]
- Nielsen O, Egel R (1989) Mapping the double-strand breaks at the mating-type locus in fission yeast by genomic sequencing. EMBO J 8: 269–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Klar AJ (1993) DNA polymerase-α is essential for mating-type switching in fission yeast. Nature 361: 271–273 [DOI] [PubMed] [Google Scholar]
- Tapper DP, Clayton DA (1981) Altered mobility of polydeoxyribonucleotides in high resolution polyacrylamide gels due to removal of terminal phosphates. Nucleic Acids Res 9: 6787–6794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengrova S, Dalgaard JZ (2004) RNase-sensitive DNA modification(s) initiates S. pombe mating-type switching. Genes Dev 18: 794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information