

Abstract
DNA repair by homologous recombination is involved in maintaining genome stability. Previous data report that wild-type p53 suppresses homologous recombination and physically interacts with Rad51. Here, we show the in vivo binding of wild-type p53 to a p53 response element in the promoter of Rad51 and the downregulation of Rad51 messenger RNA and protein by wild-type p53, favoured by DNA damage. Moreover, wild-type p53 inhibits Rad51 foci formation in response to double-strand breaks, whereas p53 contact mutant R280K fails to repress Rad51 mRNA and protein expression and Rad51 foci formation. We propose that transcriptional repression of Rad51 by p53 participates in regulating homologous recombination, and impaired Rad51 repression by p53 mutants may contribute to malignant transformation.
Keywords: p53, Rad51, transcriptional repression, homologous recombination, genome stability
Introduction
Double-strand breaks (DSBs) are lethal forms of DNA damage and homologous recombination (HR) is the main error-free DNA repair mechanism involved (van Gent et al, 2001). Both deficient and exacerbated HR activity determine genome instability (Tutt et al, 2001; Richardson et al, 2004), which is directly implicated in tumour progression (Liu et al, 2004). Rad51 protein has a central role in DSB repair by HR (Baumann & West, 1998), forming recombinatory structures known as Rad51 foci (Haaf et al, 1995; Raderschall et al, 1999). Moreover, Rad51 overexpression is found in tumours that show increased HR activity (Vispe et al, 1998). Thus, the requirement for a tight regulation of Rad51 and HR to maintain genome integrity is generally accepted (Bertrand et al, 2004).
p53 is the most frequently mutated gene in human cancer (Greenblatt et al, 1994). It exerts tumour-suppressor activity, regulating the cell cycle, programmed cell death and DNA repair. p53 functions are mediated by mechanisms that are transcriptional (Yu et al, 1999) and non-transcriptional (Mihara et al, 2003) dependent.
The p53 response to DNA damage involved in tumour suppression has been related to the activation of DNA repair mechanisms, such as base excision repair and nucleotide excision repair (Bernstein et al, 2002). However, p53 also inhibits inappropriate DNA repair by transactivating the base excision DNA repair inhibitory phosphatase PPM1D (Lu et al, 2004), and by negatively regulating key HR proteins such as RAD54 and RAD51 (Linke et al, 2003). The partial rescue of the lethal phenotype of Rad51 knockout (KO) mice on a p53-null background further suggests an interaction between Rad51 and p53 (Shu et al, 1999). Moreover, functional p53 is required to regulate HR activity, considering that cells harbouring p53 hotspot mutants show exacerbated HR (Saintigny & Lopez, 2002). Downregulation of HR activity by p53 has been ascribed to non-transcriptional-dependent mechanisms, because mutations in the transcriptional domain of p53 do not affect HR regulation (Bertrand et al, 2004). However, these p53 mutants were selected on the basis of impaired transactivation of p53 target genes, but transcriptional repression remains to be analysed.
In this work, we studied the downregulation of Rad51 by wild type (wt) p53. We show the transcriptional repression of the Rad51 gene by p53, which, together with the previously reported non-transcriptional regulation of RAD51 by p53 (Bertrand et al, 2004; Sengupta & Harris, 2005), may cooperate for efficient control of HR activity and the maintenance of genomic stability.
Results and Discussion
Characterization of wt p53 and p53R280K cells
We generated two Tet-Off-inducible cell lines in a p53-null cell background, expressing wt p53 (p53wt-B2I clone) or the p53 contact mutant p53R280K (p53R280K-A3 clone).
p53 protein expression was determined in total protein extracts recovered at different times after tetracycline withdrawal. As shown in Fig 1A, similar p53 expression was obtained in p53wt-B2I and p53R280K-A3 clones. p21 protein induction was analysed to evaluate the transcriptional activity of the p53 proteins; as expected, only wt p53 induces p21 (Fig 1A). Expression of wt p53 in this cell system induces G1 arrest, as described previously; however, DNA damage is needed to elicit a p53-dependent apoptotic response (Baptiste et al, 2002). Therefore, both cells were X-irradiated, and a sub-diploid population was induced in the p53wt-B2I cell line (Fig 1B).
Figure 1.
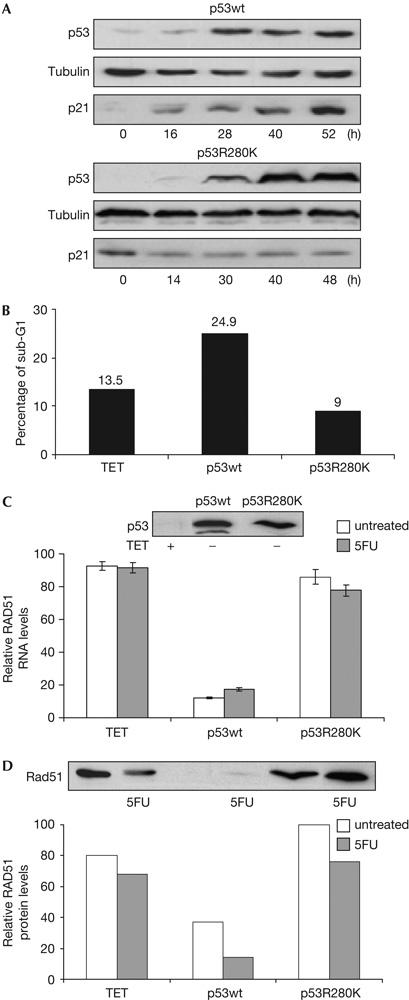
Characterization of Tet-Off p53-inducible cell lines. Modulation of Rad51 by wild-type p53. (A) p53, p21 and tubulin proteins detected with specific antibodies in the presence of tetracycline (0 h) and at indicated times after tetracycline removal. (B) Tet-Off-inducible cell lines were deprived of tetracycline for 28 h, irradiated (10 Gy) and recovered 12 h later. Tetracycline was maintained in control cells (TET). The percentage of sub-G1 population analysed by flow cytometry (propidium iodide staining) and representative of three independent experiments is shown. (C) Data of Rad51 messenger RNA levels by quantitative reverse transcription–PCR at 48 h after tetracycline removal and p53 protein expression by western blotting in p53wt and p53R280K Tet-Off-inducible clones. p53 protein expression was induced for 32 h, cells were then treated with 350 μM 5-fluorouracil (5FU) and recovered 16 h later. Tetracycline was maintained in control cells (TET). Error bars from three independent experiments are shown. (D) Rad51 protein from samples, treated as in (C), detected by western blotting. The histogram shows relative Rad51 protein levels normalized by tubulin expression. Relative levels in histograms (C,D) are calculated considering the maximum value as 100.
Wt p53 downregulates Rad51 messenger RNA and protein
We compared the gene expression patterns of cells expressing wt p53 and p53R280K to identify novel p53-dependent transcriptional activities. Complementary DNA microarray experiments were carried out with the Tet-Off-inducible cell lines at different times after tetracycline removal, and specifically activating p53 with 5-fluorouracil (5FU), an anti-metabolite chemotherapeutic drug (Baptiste et al, 2002).
We observed that Rad51 mRNA was downregulated 48 h after wt p53 protein induction (data not shown). To discriminate whether this effect was due to a gain of function of p53R280K, we analysed basal Rad51 mRNA levels in the p53-null background by quantitative reverse transcription–PCR (RT–PCR). As shown in Fig 1C, wt p53 protein downregulates Rad51 mRNA expression, whereas similar mRNA levels were obtained in the absence of p53 and with the p53R280K mutant. The reduction of Rad51 mRNA by 5FU requires wt p53 (Fig 1C), and correlates with a reduction of Rad51 protein in all conditions tested (Fig 1D).
We then analysed Rad51 regulation by p53wt and its role in HR repair by treatment with etoposide (VP16), to specifically induce DSBs (Lundin et al, 2003). As represented in Fig 2A, p53wt activation by VP16, as shown by induction of the p53 target gene p21, downregulates Rad51 mRNA and protein levels.
Figure 2.
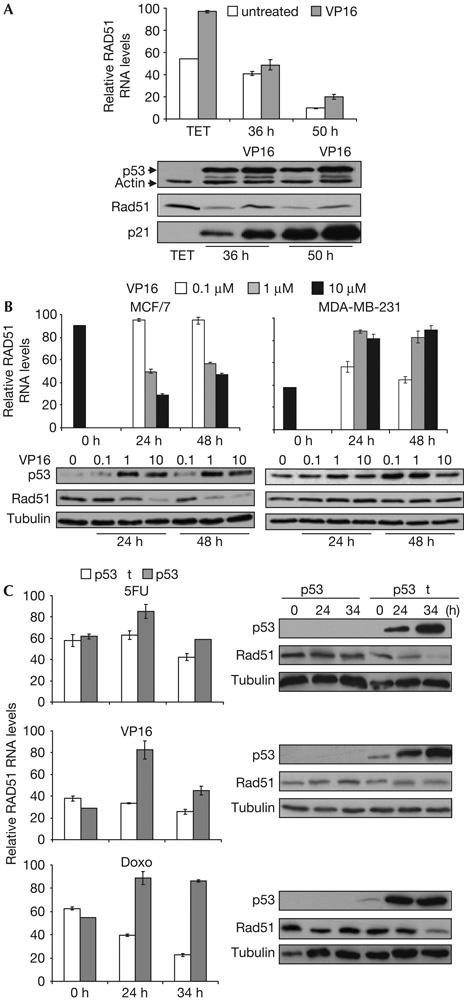
Wild-type p53 downregulates Rad51 messenger RNA and protein expression in response to DNA damage. (A) p53wt Tet-Off-inducible cells were grown for 28 h in the absence of tetracycline and subsequently treated with 15 μM VP16. RNA and protein extracts were recovered at 36 or 50 h after tetracycline removal. (B) MCF/7 and MDA-MB-231 cells were treated with VP16 (0, 0.1, 1 and 10 μM), and RNA and protein extracts were recovered at the indicated times. (C) Parental (p53wt) and somatic p53KO HCT116 cells were treated with 350 μM 5-fluorouracil (5FU), 15 μM VP16 or 0.5 μM doxorubicin (Doxo), and RNA and protein extracts were recovered at the indicated times. In all cases, Rad51 mRNA was evaluated by quantitative reverse transcription–PCR and relative RNA levels were obtained considering the maximum value as 100. Error bars from three experiments are shown. Rad51, p53, p21 and tubulin or actin proteins were detected by western blotting.
To show that endogenous p53 downregulates Rad51, we analysed Rad51 mRNA and protein expression in MCF/7 breast carcinoma cells expressing p53wt. We observed that VP16 treatment reduces Rad51 mRNA and protein expression in MCF/7 cells, in contrast to MDA-MB-231 breast carcinoma cells, which express p53R280K mutant protein (Fig 2B). Moreover, similar results were obtained with HCT116 parental cells (wt p53) versus the p53 somatic KO isogenic cells when treated with genotoxic agents. As shown in Fig 2C, induction of DSBs with doxorubicin significantly reduces both Rad51 mRNA and protein levels in the HCT116 wt p53 cells when compared with its p53KO counterpart.
Our results show the transcriptional downregulation of Rad51 by wt p53, which provides another mechanism for controlling Rad51 activity, and is in agreement with the dual role of p53. Thus, in response to DNA insult, p53 promotes DNA repair; however, if severe DNA damage is encountered, it inhibits inconvenient DNA repair, initiating apoptosis (Zhivotovsky & Kroemer, 2004).
Wt p53 binds to p53RE in the Rad51 promoter
To analyse whether the direct binding of wt p53 to the Rad51 promoter accounts for downregulation of Rad51 mRNA, we carried out chromatin immunoprecipitation (ChIP) assays, on the basis of a previous report describing potential p53-responsive elements in the Rad51 promoter (Wang et al, 2001). As shown in Fig 3B, wt p53 specifically binds to the Rad51-1 p53-responsive element, 160 bp upstream of the transcription starting point of the Rad51 promoter (Fig 3A). In the absence of DNA damage, binding was observed at 36 h after the removal of tetracycline, decreasing thereafter (Fig 3B), a result that is in agreement with previous data reporting the binding of non-activated p53 to chromatin-assembled promoters (Espinosa & Emerson, 2001). It is possible that the unusually high p53 protein levels attained in this cell system (Fig 1A) may account for downregulation of Rad51 in the absence of damage (Figs 1C,D, 2A). However, in p53wt-expressing MCF/7 and HCT116 tumour cells, Rad51 is only repressed in response to DNA damage (Fig 2B,C). Finally, binding of p53 to the Rad51 promoter correlates with delayed downregulation of Rad51 mRNA and protein expression (Fig 2A). In contrast, we observed that binding of wt p53 to the p21 promoter increases at 50 h after p53 protein expression, and parallels p21 protein induction (Figs 2A, 3B).
Figure 3.
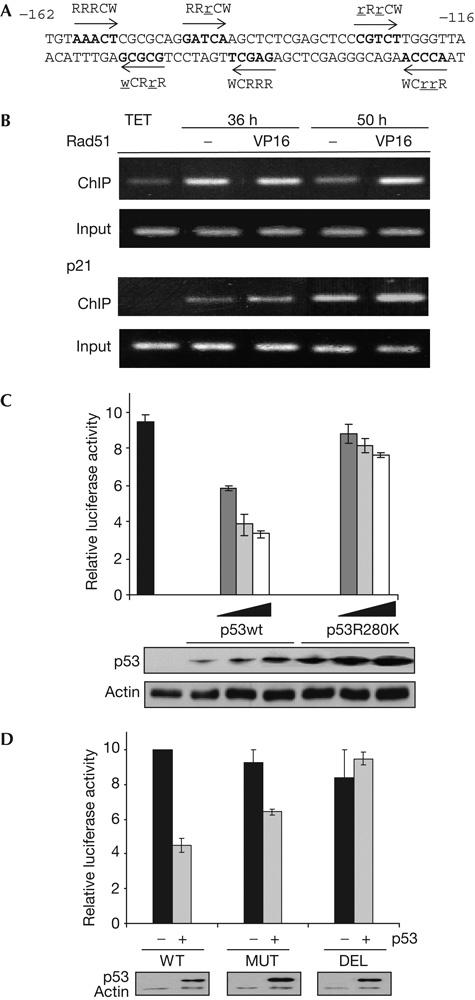
Wild-type p53 binds and represses Rad51 promoter. (A) The −162/−116 sequence of the Rad51 promoter is shown with quarter sites in the p53-binding consensus (Rad51-1) indicated by arrows and deviations from the consensus indicated by underlined lowercase letters. (B) Chromatin immunoprecipitation (ChIP) assays to amplify fragments of Rad51 and p21 promoter regions containing the p53REs. p53wt Tet-Off-inducible cells, grown for 28 h without tetracycline, were subsequently treated with15 μM VP16 and recovered at 36 or 50 h after tetracycline removal. Input represents specific amplification of sonicated chromatin before immunoprecipitation. p53-negative experimental control (TET) was obtained from cells grown with tetracycline. (C) H1299 cells were transfected with luciferase reporter plasmid Rad51-luc and p53wt or p53R280K coding plasmids (0, 0.4, 0.8 or 1.6 μg), and protein extracts were recovered 48 h later. The histogram shows relative luciferase activity considering the value of cells transfected with Rad51-luc plasmid alone as 10. Error bars from data of three experiments are shown. p53 and actin proteins were detected by western blotting. (D) H1299 cells were transfected with luciferase reporter Rad51-luc (WT), mutant Rad51-luc (MUT) or deletion mutant Rad51-luc (DEL) plasmids and p53wt coding plasmid (0 and 0.8 μg), and protein extracts were recovered 48 h later. The histogram shows relative luciferase activity considering the value in cells transfected with the reporter plasmids alone as 10. Error bars from data of three experiments are shown. p53 and actin proteins were detected by western blotting.
We then studied the effect of DSBs on the in vivo binding of wt p53 to the Rad51 promoter. VP16 treatment induced sustained binding of wt p53 to the Rad51 promoter, when compared with untreated cells (Fig 3B, 50 h), which suggests that p53 activation by DNA damage may promote the specific binding. Moreover, this result correlates with the downregulation of Rad51 mRNA and protein in response to VP16 treatment (Fig 2A). p53 binding to the p21 promoter and p21 protein induction in response to DNA damage were increased in all conditions tested (Figs 2A, 3B). Sustained binding of wt p53 to the Rad51 promoter and concomitant binding to the p21 promoter in response to DSBs suggest a cooperative cautionary effect between cell-cycle arrest and inhibition of DNA repair.
To show the regulation of Rad51 by specific binding of p53wt to the Rad51 promoter, we carried out luciferase reporter assays with the Rad51-Luc plasmid containing the p53-responsive element Rad51-1 from the Rad51 promoter. Coexpression of p53wt and Rad51-Luc resulted in p53-dependent repression of the reporter gene; however, expression of the contact mutant p53R280K had no significant effect (Fig 3C). These results were confirmed by transactivation experiments in which the reporter gene is mutated at the p53-binding sites of the Rad51 promoter. p53wt-mediated repression is abolished by deletion of the 42 bp promoter region containing the p53 response elements. Moreover, repression is partially reduced when introducing specific point mutations of the p53 consensus sequence but preserving the palindromic structure (Fig 3D).
Gene repression by p53, although poorly understood, is mediated by different mechanisms (Ho & Benchimol, 2003), which differ from those required for efficient transactivation, but are known to be relevant for p53 tumour-suppressor activity (Kho et al, 2004).
It has been previously reported that repression of HR by p53 is transactivation independent (Dudenhoffer et al, 1999; Willers et al, 2000; Linke et al, 2003). The transactivation-deficient p53 mutant (22Q, 23S; Lu & Levine, 1995) represses HR activity (Boehden et al, 2003); interestingly, the mouse homologue (25L, 26W) binds specifically to DNA (Jimenez et al, 2000). Moreover, transcriptional repression of cdc25c phosphatase by p53 requires specific DNA binding (St Clair et al, 2004). Thus, binding to p53REs in promoters or recruitment of transcriptional corepressors may account for preserved transcriptional repression in otherwise transactivation-impaired p53 mutants.
Inhibition of Rad51 foci formation by wt p53
Wt p53 inhibits Rad51-mediated HR (Linke et al, 2003; Bertrand et al, 2004; Yoon et al, 2004) and exacerbated HR is observed with p53 functional impairment (Saintigny & Lopez, 2002). To correlate the modulation of Rad51 transcription by p53 with HR regulation, we analysed Rad51 foci formation in cells expressing wt p53 or p53R280K mutant proteins in response to DSBs (Ivanov et al, 2003). As shown in Fig 4A, wt p53 inhibits Rad51 foci formation when compared with the p53R280K-expressing cells, in agreement with the downregulation of Rad51 mRNA and protein induced by wt p53 that we have previously reported (Fig 2A). Inhibition of Rad51 foci is wt p53 specific because no significant differences in the S/G2 population were detected (Fig 4B).
Figure 4.
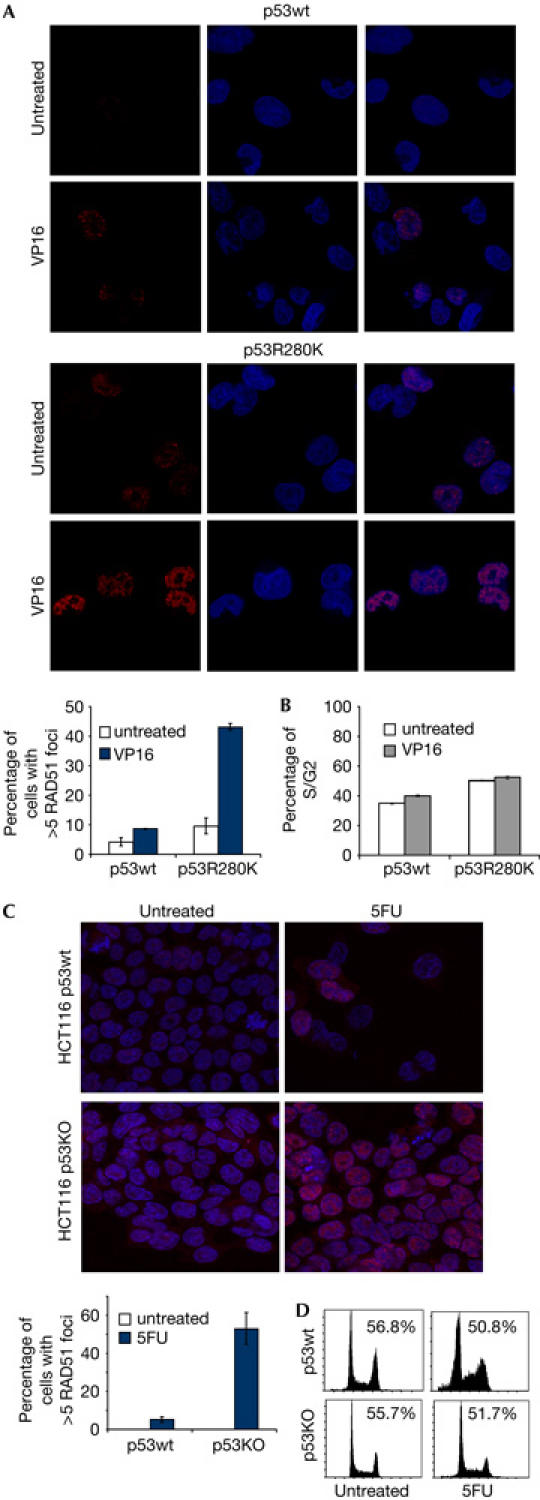
Wild-type p53 inhibits RAD51 foci formation in response to DNA damage. (A) Tet-Off p53wt- or p53R280K-inducible cell lines were grown for 24 h in the absence of tetracycline and treated for 8 h with 15 μM VP16. Subsequently, cells were fixed and labelled for DNA (blue) and Rad51 (red). A total of 100 cells with higher than five Rad51 foci per cell were analysed by confocal microscopy. Error bars from data of three separate experiments are shown. (B) Cell-cycle analysis by flow cytometry of propidium iodide (PI)-stained cells treated and recovered as in (A). The histogram shows the percentage of S/G2 population and error bars of data from three experiments. (C) HCT116 cells were treated with 350 μM 5-fluorouracil (5FU) for 32 h and Rad51 foci were analysed by confocal microscopy as in (A). Control cells (untreated) are grown without 5FU. (D) Cell-cycle analysis by flow cytometry of PI-stained cells treated and recovered as in (C). The percentage of G2/S population is shown.
We further confirmed the requirement of wt p53 to inhibit Rad51 foci in HCT116 p53KO versus the p53wt parental cells. We observed that less Rad51 foci were detected in the p53wt compared with the p53KO HCT116 cells (Fig 4D). Both cell lines had similar percentages of S/G2 population, as analysed by flow cytometry (Fig 4E). The inhibition of Rad51 foci observed in p53wt HCT116 cells correlates with our results reporting p53-dependent Rad51 downregulation in this cell line (Fig 2C).
The specific contribution of p53-dependent mechanisms in repressing HR remains to be established; however, cooperative efficient repression of HR by p53 may be required. For instance, p53 interacts with RPA, providing another mechanism of HR regulation by p53 (Romanova et al, 2004).
We propose a novel wt p53-dependent mechanism that contributes to controlling HR by downregulating Rad51 transcription, as demonstrated by the in vivo binding to a p53RE in the Rad51 promoter, the reduction of Rad51 mRNA and protein, and the inhibition of Rad51 foci formation. In this context, the transcriptional repression of Rad51 by p53 may cooperate with the previously reported inhibition of Rad51 assembly by p53 (Linke et al, 2003).
It has been suggested that induction of an apoptotic outcome in cells with abundant DNA damage requires repression of HR activity (Ivanov et al, 2003), consistent with a potential role of p53 in the tight control of HR. Moreover, Rad51 overexpression confers radio-resistance to the cell (Collis et al, 2001). These results thus indicate that the use of combined treatment with Rad51 inhibitors (Russell et al, 2003) may improve conventional current tumour therapies by restoring the repression of HR activity, which is impaired in cancer cells harbouring mutant p53 proteins.
Methods
Cell lines and plasmids. H24 cells and human wt p53 coding plasmid pUHD (10-3)-p53 were kindly provided by Dr X. Chen. R280K p53 mutation on pUHD(10-3)-p53 plasmid was generated with the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). Stable clones were obtained, as described (Niculescu et al, 1998). Growth conditions for HCT116 cell lines (a gift from Dr Vogelstein) and MDA-MB-231 and MCF/7 cells are described in the supplementary information online.
The luciferase reporter plasmid (Rad51-Luc) contains the −948/+1427 sequence from the human Rad51 promoter upstream of the coding region for firefly luciferase (pGL-3 Basic). The oligonucleotides, cloning strategy and reporter constructs are described in the supplementary information online.
Cell-cycle analysis. Fixed cells were treated with 0.25 mg/ml RNase A (Boehringer Mannheim, Basel, Switzerland) and 20 μg/ml propidium iodide (PI; Sigma, St Louis, MO, USA). Flow cytometry analysis was carried out with COULTER EPICS XL (EXPO 32 software), and 10,000 events were collected.
Western blotting. Total protein extract (50 μg) in RIPA lysis buffer was separated by SDS–polyacrylamide gel electrophoresis and analysed by western blotting with the following antibodies: anti-p53 monoclonal antibody (DO-1) and anti-p21 rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-Rad51 rabbit polyclonal antibody (BD-Biosciences, San Jose, CA, USA), anti-actin monoclonal antibody (clone AC-40) and anti-tubulin monoclonal antibody (DM-1 A; Sigma).
Quantitative reverse transcription–PCR. For details, see the supplementary information online.
Chromatin immunoprecipitation assay. ChIP experiments were performed according to the protocol of the Chromatin-Immunoprecipitation Assay Kit (Upstate Biotechnology, Charlottesville, VA, USA), with minor modifications. Detailed procedure and primers are provided in the supplementary information online.
Analysis of Rad51 foci formation. Fixed cells were stained with an anti-Rad51 antibody (1:100; BD Pharmingen, San Diego, CA, USA) and incubated with Alexa 555-conjugated goat anti-rabbit IgG (1:1,000). Nuclei were visualized with TOPRO-3 iodide (1:10,000; Molecular Probes, Eugene, OR, USA). Images were obtained with a Leica TCS-SP2 confocal microscope. See the supplementary information online.
Luciferase reporter assays. See the supplementary information online.
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/7400587-s1.pdf).
Supplementary Material
Supplementary Information
Acknowledgments
We are grateful to M.A. Gomez-Ferreria and V. Ruiz-Perez for valuable scientific advice. C.A.-L. was supported by predoctoral fellowships (Ministerio de Educación and Fundacion Ramón Areces) and I.L.-T. by a postdoctoral contract (Ministerio de Educación). CICYT grants (SAF2000-118-C0, SAF2003-00807) and the Breakthrough Breast Cancer Research Centre (Cancer Research UK) funded this work.
References
- Baptiste N, Friedlander P, Chen X, Prives C (2002) The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene 21: 9–21 [DOI] [PubMed] [Google Scholar]
- Baumann P, West SC (1998) Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem Sci 23: 247–251 [DOI] [PubMed] [Google Scholar]
- Bernstein C, Bernstein H, Payne CM, Garewal H (2002) DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat Res 511: 145–178 [DOI] [PubMed] [Google Scholar]
- Bertrand P, Saintigny Y, Lopez BS (2004) p53's double life: transactivation-independent repression of homologous recombination. Trends Genet 20: 235–243 [DOI] [PubMed] [Google Scholar]
- Boehden GS, Akyuz N, Roemer K, Wiesmuller L (2003) p53 mutated in the transactivation domain retains regulatory functions in homology-directed double-strand break repair. Oncogene 22: 4111–4117 [DOI] [PubMed] [Google Scholar]
- Collis SJ, Tighe A, Scott SD, Roberts SA, Hendry JH, Margison GP (2001) Ribozyme minigene-mediated RAD51 down-regulation increases radiosensitivity of human prostate cancer cells. Nucleic Acids Res 29: 1534–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudenhoffer C, Kurth M, Janus F, Deppert W, Wiesmuller L (1999) Dissociation of the recombination control and the sequence-specific transactivation function of P53. Oncogene 18: 5773–5784 [DOI] [PubMed] [Google Scholar]
- Espinosa JM, Emerson BM (2001) Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol Cell 8: 57–69 [DOI] [PubMed] [Google Scholar]
- Greenblatt MS, Bennett WP, Hollstein M, Harris CC (1994) Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 54: 4855–4878 [PubMed] [Google Scholar]
- Haaf T, Golub EI, Reddy G, Radding CM, Ward DC (1995) Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci USA 92: 2298–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J, Benchimol S (2003) Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ 10: 404–408 [DOI] [PubMed] [Google Scholar]
- Ivanov A, Cragg MS, Erenpreisa J, Emzinsh D, Lukman H, Illidge TM (2003) Endopolyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J Cell Sci 116: 4095–4106 [DOI] [PubMed] [Google Scholar]
- Jimenez GS, Nister M, Stommel JM, Beeche M, Barcarse EA, Zhang XQ, O'Gorman S, Wahl GM (2000) A transactivation-deficient mouse model provides insights into Trp53 regulation and function. Nat Genet 26: 37–43 [DOI] [PubMed] [Google Scholar]
- Kho PS, Wang Z, Zhuang L, Li Y, Chew JL, Ng HH, Liu ET, Yu Q (2004) p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J Biol Chem 279: 21183–21192 [DOI] [PubMed] [Google Scholar]
- Linke SP et al. (2003) p53 interacts with hRAD51 and hRAD54, and directly modulates homologous recombination. Cancer Res 63: 2596–2605 [PubMed] [Google Scholar]
- Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S, Lozano G (2004) Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 36: 63–68 [DOI] [PubMed] [Google Scholar]
- Lu H, Levine AJ (1995) Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc Natl Acad Sci USA 92: 5148–5154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Bocangel D, Nannenga B, Yamaguchi H, Appella E, Donehower LA (2004) The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol Cell 15: 621–634 [DOI] [PubMed] [Google Scholar]
- Lundin C, Schultz N, Arnaudeau C, Mohindra A, Hansen LT, Helleday T (2003) RAD51 is involved in repair of damage associated with DNA replication in mammalian cells. J Mol Biol 328: 521–535 [DOI] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577–590 [DOI] [PubMed] [Google Scholar]
- Niculescu AB, Chen X, Smeets M, Hengst L, Prives C, Reed SI (1998) Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol 18: 629–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raderschall E, Golub EI, Haaf T (1999) Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci USA 96: 1921–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C, Stark JM, Ommundsen M, Jasin M (2004) Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 23: 546–553 [DOI] [PubMed] [Google Scholar]
- Romanova LY, Willers H, Blagosklonny MV, Powell SN (2004) The interaction of p53 with replication protein A mediates suppression of homologous recombination. Oncogene 23: 9025–9033 [DOI] [PubMed] [Google Scholar]
- Russell JS, Brady K, Burgan WE, Cerra MA, Oswald KA, Camphausen K, Tofilon PJ (2003) Gleevec-mediated inhibition of Rad51 expression and enhancement of tumor cell radiosensitivity. Cancer Res 63: 7377–7383 [PubMed] [Google Scholar]
- Saintigny Y, Lopez BS (2002) Homologous recombination induced by replication inhibition, is stimulated by expression of mutant p53. Oncogene 21: 488–492 [DOI] [PubMed] [Google Scholar]
- Sengupta S, Harris CC (2005) p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol 6: 44–55 [DOI] [PubMed] [Google Scholar]
- Shu Z, Smith S, Wang L, Rice MC, Kmiec EB (1999) Disruption of muREC2/RAD51L1 in mice results in early embryonic lethality which can be partially rescued in a p53(−/−) background. Mol Cell Biol 19: 8686–8693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Clair S, Giono L, Varmeh-Ziaie S, Resnick-Silverman L, Liu WJ, Padi A, Dastidar J, DaCosta A, Mattia M, Manfredi JJ (2004) DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol Cell 16: 725–736 [DOI] [PubMed] [Google Scholar]
- Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, Griffin C, Thacker J, Ashworth A (2001) Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J 20: 4704–4716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gent DC, Hoeijmakers JH, Kanaar R (2001) Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2: 196–206 [DOI] [PubMed] [Google Scholar]
- Vispe S, Cazaux C, Lesca C, Defais M (1998) Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res 26: 2859–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wu Q, Qiu P, Mirza A, McGuirk M, Kirschmeier P, Greene JR, Wang Y, Pickett CB, Liu S (2001) Analyses of p53 target genes in the human genome by bioinformatic and microarray approaches. J Biol Chem 276: 43604–43610 [DOI] [PubMed] [Google Scholar]
- Willers H, McCarthy EE, Wu B, Wunsch H, Tang W, Taghian DG, Xia F, Powell SN (2000) Dissociation of p53-mediated suppression of homologous recombination from G1/S cell cycle checkpoint control. Oncogene 19: 632–639 [DOI] [PubMed] [Google Scholar]
- Yoon D, Wang Y, Stapleford K, Wiesmuller L, Chen J (2004) P53 inhibits strand exchange and replication fork regression promoted by human Rad51. J Mol Biol 336: 639–654 [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Rago C, Kinzler KW, Vogelstein B (1999) Identification and classification of p53-regulated genes. Proc Natl Acad Sci USA 96: 14517–14522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky B, Kroemer G (2004) Apoptosis and genomic instability. Nat Rev Mol Cell Biol 5: 752–762 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information