

Abstract
The pRb (retinoblastoma protein) tumour suppressor protein has a crucial role in regulating the G1- to S-phase transition, and its phosphorylation by cyclin-dependent kinases is an established and important mechanism in controlling pRb activity. In addition, the targeted acetylation of lysine (K) residues 873/874 in the carboxy-terminal region of pRb located within a cyclin-dependent kinase-docking site hinders pRb phosphorylation and thereby retains pRb in an active state of growth suppression. Here, we report that the acetylation of pRb K873/874 occurs in response to DNA damage and that acetylation regulates the interaction between the C-terminal E2F-1-specific domain of pRb and E2F-1. These results define a new role for pRb acetylation in the DNA damage signalling pathway, and suggest that the interaction between pRb and E2F-1 is controlled by DNA-damage-dependent acetylation of pRb.
Keywords: Rb, E2F, acetylation, DNA damage
Introduction
The pRb (retinoblastoma protein) tumour suppressor protein has a crucial role in control of the G1- to S-phase transition in mammalian cells (Weinberg, 1995; Dyson, 1998). This is mediated in part by its ability to target key transcription factors, such as E2F, which modulate the expression of genes required for cell-cycle progression. In turn, the activity of pRb is regulated by post-translational mechanisms, primarily phosphorylation, directed by cyclin-dependent kinases (CDKs), including cyclin D/Cdk4 and cyclin E/Cdk2 (Sherr, 2000). In targeting E2F, pRb uses a multiplicity of mechanisms to control transcription. While the pRb-binding domain in E2F overlaps its transcriptional activation domain, enabling pRb to hinder the interaction of E2F with the transcription apparatus (Stevens & La Thangue, 2003), a dominantly repressing complex of chromatin modulators assembles with pRb, including histone deacetylase and methyltransferases, allowing pRb to enforce transcriptional inactivity by regulating chromatin (Laj et al, 1999; Robertson et al, 2000; Vandel et al, 2001).
The inactivation of pRb has a crucial role during tumorigenesis (Weinberg, 1995). Indeed, pRb activity is believed to be under aberrant control in most tumour cells, causing the release of E2F either through mutation in Rb or through increased activity of cyclin/CDK complexes, which thereafter contributes to the enhanced growth rate by inactivating pRb (Stevens & La Thangue, 2003). Moreover, pRb is a target for viral oncoproteins, such as adenovirus E1A, where its sequestration facilitates de-regulated growth control (Dyson, 1998).
The DNA damage signalling pathway is an evolutionarily conserved response that acts in checkpoint control to maintain genome integrity in response to genotoxic stress. Classically, the response is divided into two arms, with each arm involving one of two groups of protein kinases, the ataxia-telangiectasia-mutated (ATM)/ATM- and Rad3-related protein (ATR) family of phosphoinositide 3-kinases and checkpoint kinases (Chks; Bartek & Lukas, 2003). Within the pathway, ATM/ATR kinases are believed to act as sensor kinases, which, following activation, signal to a variety of downstream targets including Chk1 and Chk2. Furthermore, while there is considerable crosstalk between each pathway, ATM and ATR are believed to signal to Chk2 and Chk1, respectively. This involves a linear pathway, which, for ATM, is activated by ionizing radiation and other agents that cause DNA double-strand breaks, including radiomimetic drugs, and, for ATR, is activated by ultraviolet light and replication stress (Bartek & Lukas, 2003). Chks, which are generally regarded as effectors of the DNA damage response, phosphorylate diverse substrates, including p53, E2F-1, BRCA1, Mdm2, cdc25C and promyelocytic leukaemia protein (PML), which thereafter leads to cell-cycle arrest or apoptosis (Peng et al, 1997; Shieh et al, 2000; Maya et al, 2001; Yang et al, 2002; Stevens et al, 2003; Zhang et al, 2004).
While the phosphorylation of pRb by CDKs is an established and important mechanism in the control of pRb tumour suppressor activity, a further level of regulation is mediated through the targeted acetylation of lysine (K) residues in the carboxy-terminal region of pRb. In previous studies, we proposed that the acetylation of lysine residues within a docking site that mediates the interaction between pRb and cyclin E/Cdk2, namely K873/874, retains pRb in a hypo-phosphorylated state by interfering with the subsequent phosphorylation of pRb by cyclin E/Cdk2 (Chan et al, 2001). In adenovirus-transformed cells, the E1A oncoprotein, which binds to p300/CBP proteins through an amino-terminal transformation-sensitive domain, stimulates pRb acetylation by recruiting p300 and pRb into a multimeric protein complex (Chan et al, 2001). Furthermore, pRb acetylation is regulated during cellular differentiation (Chan et al, 2001; Nguyen et al, 2004), which is consistent with a role for pRb acetylation in mediating growth arrest.
On the basis of these observations, we reasoned that pRb acetylation may be regulated under physiological situations in which cell-cycle progression is affected. To this end, we explored the possibility that pRb acetylation is regulated in response to DNA damage. Our results show that pRb is acetylated at K873/874 in response to DNA damage and that acetylation governs the interaction of the C-terminal E2F-1-specific domain of pRb with E2F-1. Specifically, DNA-damage-dependent acetylation of pRb releases E2F-1 from the C-terminal binding domain. These results show that pRb acetylation is under DNA damage response control and identify E2F-1 as a key target in the response to pRb acetylation.
Results
An antibody against acetylated lysines 873/874
To assess whether the acetylation of pRb K873/874 is under physiological control, we sought to generate an antibody against acetylated K873/874 (supplementary Fig 1A online). To this end, we prepared the anti-peptide antibody SK37, which selectively recognizes acetylated K873/874 in the context of a pRb peptide (supplementary Fig 1B online). By immunoblotting, antibody SK37 detected glutathione S-transferase (GST)–Rb acetylated by p300 but not its non-acetylated counterpart, whereas both acetylated GST–Rb and its non-acetylated counterpart were readily detectable using the general anti-Rb antibody C15 (supplementary Fig 1C online).
To further examine the specificity of antibody SK37, the pRb peptide acetylated at K873/874 and the corresponding non-acetylated peptide were each used in a competition immunoblot assay with antibody SK37 and acetylated pRb (supplementary Fig 1D online). Antibody SK37 bound to acetylated pRb, and the binding activity was specifically competed by the acetylated peptide, whereas the non-acetylated peptide failed to compete (supplementary Fig 1D online). Furthermore, a GST–pRb fusion protein, in which K873/874 residues were altered to arginine (R) residues (GST–Rb 873/874RR) or glutamine (GST–Rb 873–874QQ), that had been similarly acetylated by p300 failed to bind to SK37 (supplementary Fig 1E online), confirming that SK37 selectively binds to acetylated K873/874 residues.
DNA damage causes increased acetylation of K873/874
Having shown that antibody SK37 selectively detects acetylated residues K873/874, we addressed the physiological control of pRb acetylation. As post-translational control of effector proteins is a key mechanism of control in the DNA damage response, and because pRb activity is implicated in the DNA damage response through the p53-dependent induction of p21 (Harrington et al, 1998), we surmised that pRb may be subject to acetylation control under DNA damage conditions. In this regard, we examined the effect of DNA damage on pRb acetylation at K873/874 in NIH3T3 fibroblasts treated with etoposide, which primarily activates the ATM/Chk2 arm of the DNA damage response by causing double-strand breaks in DNA (Darbon et al, 2000; Shiloh, 2003). Treating NIH3T3 cells with increasing concentrations of etoposide caused a corresponding increase in the level of acetylated pRb (Fig 1A), in which the accumulation of acetylated pRb occurred up to 8 h after treatment (Fig 1B). In contrast, in cells treated with ultraviolet light, the effect on acetylation at K873/874 was significantly reduced (Fig 1C). Confirmation that this was not merely due to ineffective ultraviolet treatment was demonstrated through the stabilization of p53 under the same conditions (Fig 1C). Acetylated pRb was also detected in a variety of other cell types, including WI38, U937, AT and F9EC cells (data not shown). We conclude that acetylation of pRb K873/874 is under DNA-damage-responsive control and that the acetylation process is activated in response to particular DNA-damaging agents.
Figure 1.
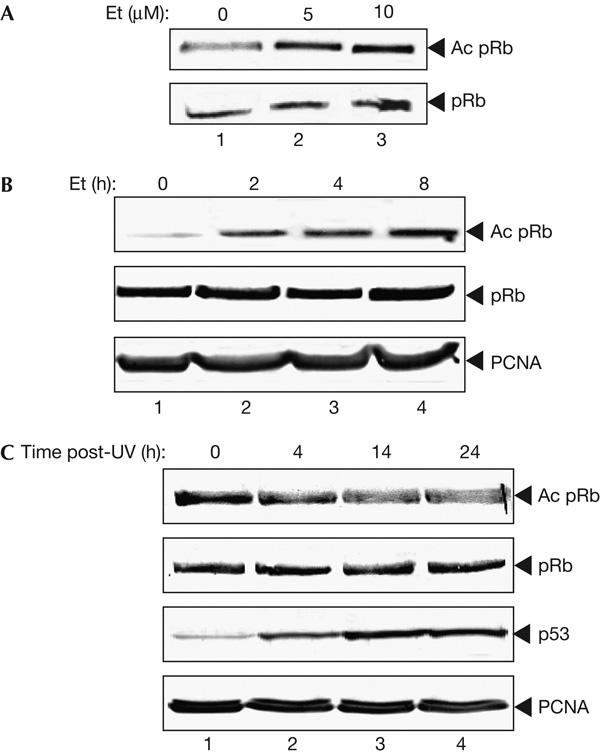
Damage-inducible acetylation of the Rb protein. (A) Immunoblot depicting the damage-responsive accumulation of acetylated (Ac) pRb in NIH3T3 cells. Cells were treated with 5 μM (track 2) or 10 μM (track 3) etoposide (Et) for 8 h and immunoblotted with anti-acetylated pRb (top) or anti-pRb (lower). (B) Immunoblot depicting the accumulation of acetylated pRb over time in NIH3T3 cells after treatment with 10 μM etoposide. Extracts (5 μg) were immunoblotted with anti-acetylated pRb (top panel), anti-pRb (middle panel) or anti-proliferating-cell nuclear antigen (PCNA; bottom panel). (C) Immunoblot depicting the effect of exposure to 50 J/cm2 ultraviolet in NIH3T3 cells, collected at the indicated times after ultraviolet exposure (post-UV). Extracts (5 μg) were blotted with anti-acetylated pRb (top panel), anti-pRb (middle panel), anti-p53 (third panel) or anti-PCNA (bottom panel).
Altered nuclear distribution of acetylated pRb
We investigated the intracellular distribution of acetylated pRb and found that within the nucleus the distribution was altered following etoposide treatment. Thus, in untreated NIH3T3 cells, there was low but detectable nuclear staining with a uniform distribution (Fig 2A), consistent with the detection of acetylated pRb by immunoblotting (Fig 1). However, following etoposide treatment, there was a substantial increase in staining intensity coupled with a speckled distribution, which was excluded from nucleoli (Fig 2A). Similar to the earlier results on the lack of pRb acetylation under ultraviolet light treatment (Fig 1), ultraviolet light treatment failed to alter or enhance the nuclear immunostaining pattern (data not shown). Importantly, the immunostaining pattern was specific, as the staining observed with SK37 was competed by the acetylated K873/874 peptide but not by the non-acetylated peptide (Fig 2B), and SK37 failed to stain the nuclei of cells that lacked wild-type pRb (supplementary Fig 2 online). The SK37 nuclear immunostaining pattern did not seem to be similar to that of several DNA-damage-responsive proteins, including E2F-1, H2AX, proliferating-cell nuclear antigen (PCNA), PML, BRCA1 or nucleophosmin (data not shown).
Figure 2.
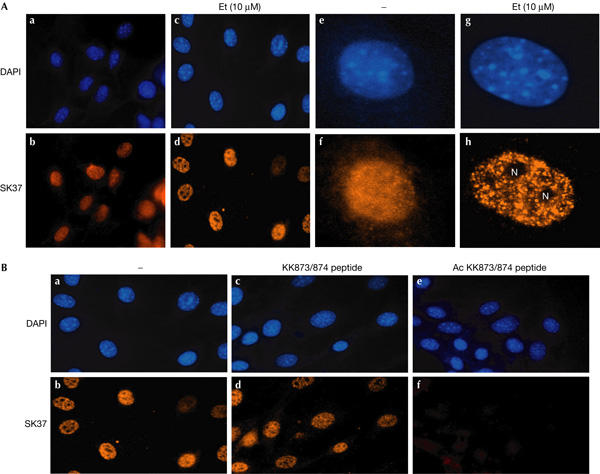
Altered distribution of acetylated pRb following DNA damage. (A) NIH3T3 cells either untreated (a,b) or treated (10 μM etoposide (Et) for 8 h; c,d,g,h) followed by 4,6-diamidino-2-phenylindole (DAPI) treatment (a,c,d,e) or immunostaining with anti-acetylated pRb antibody (b,d,f,h). The nucleoli (N) are indicated. Increased magnifications of typical cells are shown in (e,f,g,h). (B) NIH3T3 cells were treated with 10 μM etoposide for 8 h either in the absence (a,b) or presence of the unacetylated (5 μg; c,d) or acetylated (5 μg; e,f) peptide and immunostained with the anti-acetylated pRb antibody. Panels (a,c,e) represent DAPI treatment.
Acetylation of K873/874 governs binding to E2F-1
We reasoned that the increased acetylation at K873/874 may affect the binding of pRb to certain cellular proteins, and considered E2F-1 as a possibility. It was previously reported that, in addition to the pocket-dependent interaction with E2F, a second interaction site located entirely within the C-terminal domain and outside the pocket region of pRb is specific for E2F-1 (Dick & Dyson, 2003). It was also suggested that this interaction was regulated under DNA damage conditions, although the mechanism involved and the residues responsible were not defined.
We assessed the binding of E2F-1 to the pRb C-terminal region encompassed between residues 763 and 928 and to the same region of pRb in which the K residues were altered to either RR873/874 or QQ873/874 (Fig 3A), where the R substitution provides a similar basic residue as K, and Q residue mimics the acetylation of a K residue (Chan et al, 2001). In extracts prepared from 293 cells, the binding of E2F-1 to RR873/874 was marginally reduced relative to the interaction with the wild-type pRb domain; in contrast, the binding to QQ873/874 was significantly less (Fig 3B). As a control for the integrity of the pRb mutant derivatives, we measured the binding of pRb to Mdm2, because Mdm2 interacts with the C-terminal region of pRb (Xiao et al, 1995). Both RR873/874 and QQ873/874 bound equally well as the wild-type pRb domain to Mdm2 (Fig 3B). These results imply that the interaction of E2F-1 with the C-terminal domain of pRb is controlled through the acetylation status of KK873/874.
Figure 3.
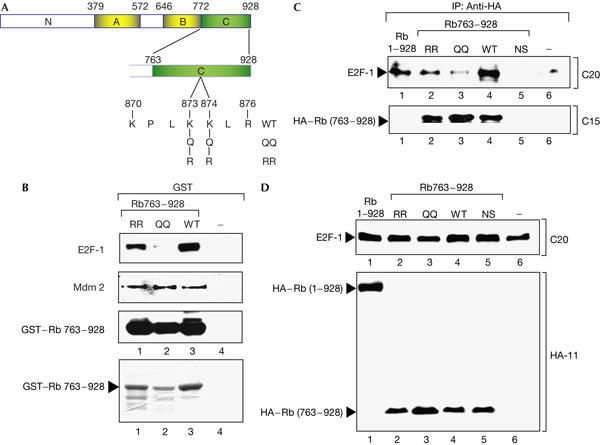
Acetylation of pRb KK873/874 regulates the specific interaction with E2F-1. (A) Diagram of pRb, indicating the mutant derivatives RR873/874 and QQ873/874 in the context of the carboxy-terminal domain from residues 763 to 928. (B) Binding of glutathione S-transferase (GST)–pRb763–928, RR873/874 and QQ873/874 (1 μg) to E2F-1 and Mdm2 in 293 cell extracts assayed by immunoblotting with anti-E2F-1 and anti-Mdm2. The level of input GST–pRb763–928 is shown in the lower panel after blotting with anti-pRb. (C) Expression vectors encoding haemagglutinin (HA)-tagged pRb763–928, RR873–874 and QQ873/874 together with E2F-1 and pCMV-β-gal (1 μg throughout) were transfected into 293 cells as described. Extracts were normalized and immunoprecipitated (IP) with anti-HA followed by immunoblotting with anti-E2F-1 and anti-Rb. The level of input E2F-1 and pRb is shown in (D).
To establish whether these results reflect similar interactions in cells, we assessed the association of pRb763–928 with E2F-1 in 293 cells. Similar to the behaviour of the proteins in the in vitro binding assay, we found that pRb763–928 bound to E2F-1 and that altering KK873/874 to RR873/874 had a marginal effect on binding efficiency (Fig 3C). In contrast, pRb QQ873/874 was significantly reduced in its interaction with E2F-1 (Fig 3C). The interaction of pRb763–928 with E2F-1 and the influence of QQ873/874 observed in vitro is therefore recapitulated in cells.
Further, we assessed the properties of the mutant derivatives in regulating E2F activity. We studied the effect on different E2F-responsive promoters and present data for cyclin E, a known E2F target gene (Botz et al, 1996). As expected from previous studies, the effect of wild-type pRb was to downregulate E2F-1 activity (Fig 4A). We found that QQ873/874 showed a reproducibly reduced ability to downregulate E2F-1 activity (Fig 4A). In contrast, RR873/874 more efficiently repressed E2F-1 than QQ873/874, but showed less repression activity than wild-type pRb in this assay (Fig 4A). These effects were not due to differences in expression levels because wild-type pRb, QQ873/874 and RR873/874 were expressed at similar levels (Fig 4B). Furthermore, both full-length and 763–928 pRb derivatives underwent nuclear accumulation with similar efficiency (Fig 4C), although QQ873/874 in the context of full-length pRb showed a marginal increase in cytoplasmic localization. However, altering KK873/874 to QQ873/874 did not have a major influence on nuclear localization. We conclude therefore that the modification of residues 873/874 influences the interaction between the C-terminal pRb-binding domain and E2F-1 and that this has functional consequences on the regulation of E2F-1 activity.
Figure 4.
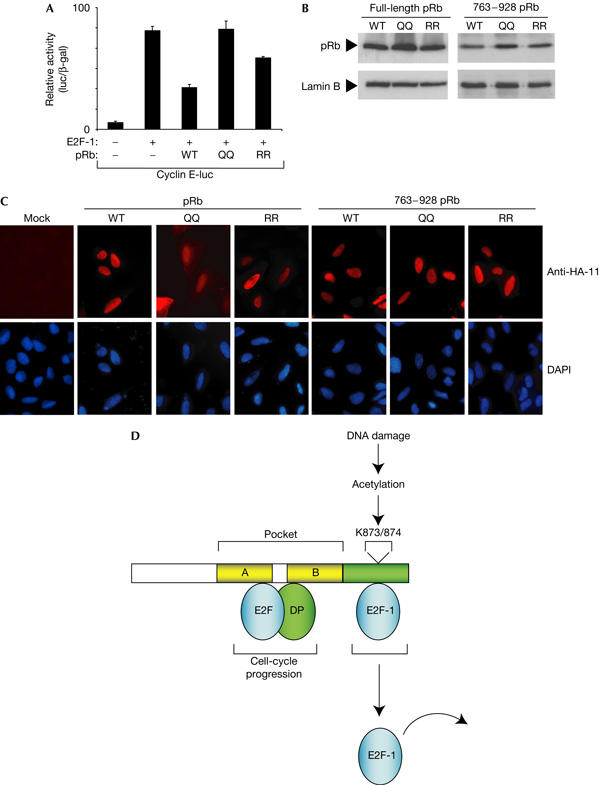
Functional properties of pRb. (A) Functional consequences of pRb acetylation on E2F-1 activity. Expression vectors encoding wild-type (WT) pRb, pRb RR873/874 or pRb QQ873/874 (1 μg) were coexpressed with E2F-1 (200 ng), and the effect on cyclin E-luciferase (200 ng) was measured. Transfection into U2OS cells was carried out as described previously (Chan et al, 2001). (B) Levels of wild-type pRb and 763–928 derivatives in U2OS cells transfected as described in (A). (C) Intracellular location of pRb and 763–928 derivatives in U2OS cells (transfected with 1 μg expression vector and treated as described). The 4,6-diamidino-2-phenylindole (DAPI) staining shows the location of nuclei. (D) Model for the influence of DNA-damage-dependent pRb acetylation on E2F-1 activity. The results presented in this study suggest that residues KK873/874 influence the interaction between E2F-1 and the pRb carboxy-terminal E2F-1-specific binding domain. Acetylation of KK873/874 activated through the DNA damage response liberates E2F-1 and theoretically allows E2F-1 activity to be regulated independently of generic E2F binding to the pRb pocket domain.
Discussion
pRb acetylation is DNA damage responsive
Using an antibody that selectively recognizes acetylated KK873/874, we have shown that pRb acetylation is physiologically upregulated during the DNA damage response. The functional consequence of acetylation at KK873/874 in the context of pRb control was implied from previous studies, which suggested that residues KK873/874 are located within a cyclin/CDK docking site (Adams et al, 1996; Chan et al, 2001). Thus, we reasoned and subsequently gained substantial evidence in support of acetylation as a modification that blocked pRb phosphorylation, mediated through hindering the interaction between cyclin/CDK complexes and pRb (Chan et al, 2001).
This model is consistent with the observations reported here. During the DNA damage response, pRb is maintained in an active growth-suppressing state, through the inhibition of cyclin/CDK activity by inhibitors, such as p21 (Harrington et al, 1998; Knudsen et al, 2000). The p21 gene is a direct target for p53, which, in turn, is activated by the DNA damage signalling pathway (Sherr, 2000). The acetylation of K873/874 provides a mechanism that may ensure and perhaps reinforce the block to pRb phosphorylation through a process that physically hinders the interaction with cyclin/CDK complexes.
Acetylation at KK873/874 regulates binding to E2F-1
In addition to the well-characterized interaction between the pRb pocket and E2F, the C-terminal region of pRb harbours another E2F-1-specific binding domain (Dick & Dyson, 2003). Previous results indicated a reduced interaction in cells treated with DNA-damaging agents, although the mechanism involved in regulating the C-terminal interaction with E2F-1 was not clarified (Dick & Dyson, 2003). Our results suggest that the different interactions between pRb and E2F, namely within the pocket and C-terminal region, may be separately regulated. Specifically, KK873/874 are key residues that influence the binding activity of E2F-1 to the C-terminal specific domain. The acetylation of KK873/874 thus results in the release of E2F-1 from pRb, implying that the DNA-damage-dependent acetylation of KK873/874 impedes binding to E2F-1. Another effect of KK873/874 acetylation is to retain pRb in a hypophosphorylated state and E2F binding to the pRb pocket (Chan et al, 2001). Thus, pRb pocket and C-terminal binding to E2F-1 can, theoretically, be independently regulated.
With respect to these results, it is interesting to note that a considerable amount of evidence has connected E2F-1-dependent apoptosis with regulation through DNA damage response pathway (Stevens & La Thangue, 2004). Specifically, E2F-1 is inducible by DNA damage where it follows similar kinetics to the induction of p53 (Stevens et al, 2003). Thus, following release from pRb, it is possible that E2F-1 is further regulated by DNA damage signals, such as phosphorylation by Chk2 (Stevens et al, 2003), and thereafter becomes stabilized and primed to induce apoptosis. The results described here on the DNA-damage-dependent acetylation of KK873/874 add further detail to this emerging pathway by defining an acetylation signal that releases E2F-1 from the C-terminal pRb domain.
Overall, these results show that the DNA damage regulation of pRb KK873/874 acetylation is intimately connected with E2F-1 activity. The pathway allows E2F-1 to be liberated from pRb in a manner that, theoretically, occurs independently of E2F binding to the pRb pocket (Fig 4D). Thus, pRb acetylation provides a mechanism whereby E2F-1 activity can be independently regulated from other E2F subunits and directly integrated with the DNA damage response.
Methods
Plasmids. pGEX-Rb, pGEX-Rb(873/874RR), pGEX-Rb(873/874QQ), pGEX-Rb(763–928), pCMV-E2F-1, pCMV-DP-1, Flag-p3001135–2414 and pCycE-luc (Botz et al, 1996) were previously described (Bandara et al, 1991; Shikama et al, 1999; Chan et al, 2001). pGEX-Rb RR873/874 and pGEX-RB KK873/874 were generated by site-directed mutagenesis.
Tissue culture and transfections. NIH3T3 fibroblasts, 293, SAOS2 and U2OS cells were cultured in Dulbecco's modified Eagle's medium (GIBCO, Paisley, UK) supplemented with 10% fetal calf serum (GIBCO) at 37°C in 5% CO2. To induce DNA damage, cells were treated by adding etoposide directly to the growth medium or removing the medium and exposing the cells to UV in a UV crosslinker (Amersham, Little Chalfont, Buckinghamshire, UK). SAOS2 cells were transfected by calcium phosphate precipitation.
Antibodies. The rabbit polyclonal anti-peptide antiserum SK37 directed against Rb was acetylated at K873/874 using a 16-amino-acid peptide taken from human pRb (residues 863–878) and chemically acetylated on residues 873 and 874 (AEGSNPPKPLK(Ac)K(Ac)LRFC). Crude antiserum raised against the acetylated peptide was passed through an acetylated KK873/874 peptide column and the bound material passed through a second column containing the non-acetylated peptide as described (Stevens et al, 2003).
In vitro protein acetylation. In vitro protein acetylation was performed as described previously (Chan et al, 2001) using 1 μg of GST-purified Rb or RR/QQ Rb mutants, 1 μg of Flag-purified p3001135–2414 and 90 pmol of acetyl-CoA incubated at 30°C for 30 min in 5 × HAT assay buffer (250 mM Tris–HCl (pH 8.0), 25% glycerol, 0.5 M EDTA (pH 8.5), 250 mM KCl and 10 mM sodium butyrate). The reactions were stopped by the addition of SDS loading buffer (150 mM Tris–HCl (pH 6.8), 6% SDS, 30% glycerol, 0.3% bromophenol blue and 3% β-mercaptoethanol) and analysed by SDS–polyacrylamide gel electrophoresis followed by immunoblotting.
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/7400591-s1.pdf).
Supplementary Material
Supplementary Information
Acknowledgments
We thank R. Williams for help in preparing the manuscript. This work was supported by Cancer Research UK, the Medical Research Council, the European Union and the Association of International Cancer Research.
References
- Adams PD, Sellers WR, Sharma SK, Wu AD, Nalin CM, Kaelin WG (1996) Identification of a cyclin–cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol Cell Biol 16: 6623–6633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandara LR, Adamczewski JP, Hunt T, La Thangue NB (1991) Cyclin-A and the retinoblastoma gene-product complex with a common transcription factor. Nature 352: 249–251 [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 [DOI] [PubMed] [Google Scholar]
- Botz J, Zerfass-Thome K, Spitkovaky D, Delius H, Vogt B, Eilers M, Hatzigeorgiou A, Jansen-Dürr P (1996) Cell cycle regulation of the murine cyclin E gene depends on an E2F binding site in the promoter. Mol Cell Biol 16: 3401–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB (2001) Acetylation control of the retinoblastoma tumour-suppressor protein. Nat Cell Biol 3: 667–674 [DOI] [PubMed] [Google Scholar]
- Darbon JM, Penary M, Escalas N, Casagrande F, Goubin-Gramatica F, Baudouin C, Ducommun B (2000) Distinct Chk2 activation pathways are triggered by genistein and DNA-damaging agents in human melanoma cells. J Biol Chem 275: 15363–15369 [DOI] [PubMed] [Google Scholar]
- Dick FA, Dyson N (2003) pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol Cell 12: 639–649 [DOI] [PubMed] [Google Scholar]
- Dyson N (1998) The regulation of E2F by pRB-family proteins. Genes Dev 12: 2245–2262 [DOI] [PubMed] [Google Scholar]
- Harrington EA, Bruce JL, Harlow E, Dyson N (1998) pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci USA 95: 11945–11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen KE, Booth D, Naderi S, Sever-Chroneos Z, Fribourg AF, Hunton IC, Feramisco JR, Wang JYJ, Knudsen ES (2000) RB-dependent S-phase response to DNA damage. Mol Cell Biol 20: 7751–7763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laj A, Lee JM, Yang WM, DeCaprio JA, Kaelin WG, Seto E, Branton PE (1999) RBP1 recruits both histone deacetylase-dependent and -independent repression activities to retinoblastoma family proteins. Mol Cell Biol 19: 6632–6641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya R et al. (2001) ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev 15: 1067–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DX, Baglia LA, Huang SM, Baker CM, McCance DJ (2004) Acetylation regulates the differentiation-specific functions of the retinoblastoma protein. EMBO J 23: 1609–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu ZQ, Shaw AS, PiwnicaWorms H (1997) Mitotic and G(2) checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 277: 1501–1505 [DOI] [PubMed] [Google Scholar]
- Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP (2000) DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet 25: 338–342 [DOI] [PubMed] [Google Scholar]
- Sherr CJ (2000) The Pezcoller lecture: cancer cell cycles revisited. Cancer Res 60: 3689–3695 [PubMed] [Google Scholar]
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C (2000) The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 14: 289–300 [PMC free article] [PubMed] [Google Scholar]
- Shikama N, Lee CW, France S, Delavaine L, Lyon J, Krstic-Demonacos M, La Thangue NB (1999) A novel cofactor for p300 that regulates the p53 response. Mol Cell 4: 365–376 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Stevens C, La Thangue NB (2003) E2F and cell cycle control: a double-edged sword. Arch Biochem Biophys 412: 157–169 [DOI] [PubMed] [Google Scholar]
- Stevens C, La Thangue NB (2004) The emerging role of E2F-1 in the DNA damage response and checkpoint control. DNA Repair 3: 1071–1079 [DOI] [PubMed] [Google Scholar]
- Stevens C, Smith L, La Thangue NB (2003) Chk2 activates E2F-1 in response to DNA damage. Nat Cell Biol 5: 401–409 [DOI] [PubMed] [Google Scholar]
- Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D (2001) Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol Cell Biol 21: 6484–6494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA (1995) The retinoblastoma protein and cell-cycle control. Cell 81: 323–330 [DOI] [PubMed] [Google Scholar]
- Xiao ZX, Chen JD, Levine AJ, Modjtahedi N, Xing J, Sellers WR, Livingston DM (1995) Interaction between the retinoblastoma protein and the oncoprotein Mdm2. Nature 375: 694–698 [DOI] [PubMed] [Google Scholar]
- Yang ST, Kuo C, Bisi JE, Kim MK (2002) PML-dependent apoptosis after DNA damage is regulated by the checkpoint kinase hCds1/Chk2. Nat Cell Biol 4: 865–870 [DOI] [PubMed] [Google Scholar]
- Zhang JR, Willers H, Feng ZH, Ghosh JC, Kim S, Weaver DT, Chung JH, Powell SN, Xia F (2004) Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol 24: 708–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information