

Abstract
Double-strand breaks (DSBs) elicit a DNA damage response, resulting in checkpoint-mediated cell-cycle delay and DNA repair. The Saccharomyces cerevisiae Sae2 protein is known to act together with the MRX complex in meiotic DSB processing, as well as in DNA damage response during the mitotic cell cycle. Here, we report that cells lacking Sae2 fail to turn off both Mec1- and Tel1-dependent checkpoints activated by a single irreparable DSB, and delay Mre11 foci disassembly at DNA breaks, indicating that Sae2 may negatively regulate checkpoint signalling by modulating MRX association at damaged DNA. Consistently, high levels of Sae2 prevent checkpoint activation and impair MRX foci formation in response to unrepaired DSBs. Mec1- and Tel1-dependent Sae2 phosphorylation is necessary for these Sae2 functions, suggesting that the two kinases, once activated, may regulate checkpoint switch off through Sae2-mediated inhibition of MRX signalling.
Keywords: Sae2, checkpoint, MRX, Mec1, Tel1
Introduction
Response to double-strand breaks (DSBs) in eukaryotes is governed by DNA damage checkpoint signal-transduction pathways, the key players of which belong to a protein kinase family including the mammalian ataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-related protein (ATR) and their Saccharomyces cerevisiae orthologues Tel1 and Mec1 (Longhese et al, 2003). An evolutionarily conserved protein complex, called MRX (Mre11–Rad50–Xrs2) in budding yeast, mediates DSB association of both ATM/Tel1 and ATR/Mec1 (Nakada et al, 2003, 2004).
The S. cerevisiae Sae2 protein is involved in meiotic and mitotic DSB processing and in subsets of recombination pathways together with the MRX complex (Keeney & Kleckner, 1995; Rattray et al, 2001; Lobachev et al, 2002; Neale et al, 2002; Clerici et al, 2005). Moreover, Sae2 undergoes Mec1- and Tel1-dependent phosphorylation that is important for its functions in DNA damage response (Baroni et al, 2004). Finally, the absence of Sae2 prolongs checkpoint-mediated cell-cycle arrest after UV irradiation (Baroni et al, 2004). This might be due to reduced efficiency in repairing DNA lesions or may reflect a more specific role of Sae2 in checkpoint switch off.
By examining the effects of lack of or excess Sae2 on checkpoint response to a single irreparable DSB, we now provide evidence that Sae2 negatively regulates signalling to Mec1- and Tel1-dependent pathways, possibly by modulating MRX association to DSBs.
Results and Discussion
Sae2 is required for DNA damage checkpoint switch off
Cells carrying a single irreparable DSB undergo checkpoint-mediated cell-cycle block, but then they adapt to the damage, decrease Rad53 checkpoint kinase activity and re-enter the cell cycle (Toczyski et al, 1997; Pellicioli et al, 2001). To test whether Sae2 has a role in turning off the checkpoint independently of DNA damage repair, we examined the checkpoint response to an unrepaired DSB in sae2Δ cells (Lee et al, 1998). A single DSB can be generated at the MAT locus of JKM139 derivative strains by expressing the site-specific HO endonuclease gene from a galactose-inducible promoter, and it cannot be repaired by homologous recombination, because the homologous donor sequences HML or HMR are deleted (Lee et al, 1998). As shown in Fig 1A, when G1-arrested cell cultures were spotted on galactose-containing plates, most wild-type JKM139 cells overrode the checkpoint-mediated cell-cycle arrest in 24–32 h, producing microcolonies with four or more cells, whereas most isogenic sae2Δ cells were still arrested at the two-cell dumbbell stage after 32 h. Moreover, when galactose was added to exponentially growing cell cultures of the same strains, Rad53 phosphorylation, which is required for its activation as a kinase, became detectable as electrophoretic mobility shift in both cell cultures about 2 h after HO induction (Fig 1B). Then, it persisted at least for a further 30 h in sae2Δ cells that did not re-enter the cell cycle, whereas it decreased in wild-type cells after 12–15 h, when most cells resumed cell-cycle progression (Fig 1A,B; data not shown). Thus, sae2Δ cells fail to override the DNA damage checkpoint response and to resume cell-cycle progression when DSB repair is prevented. As checkpoint inactivation in the presence of unrepaired DNA lesions occurs independently of repair pathways (Toczyski et al, 1997), the role of Sae2 in switching off the checkpoint response is probably independent of DSB repair.
Figure 1.
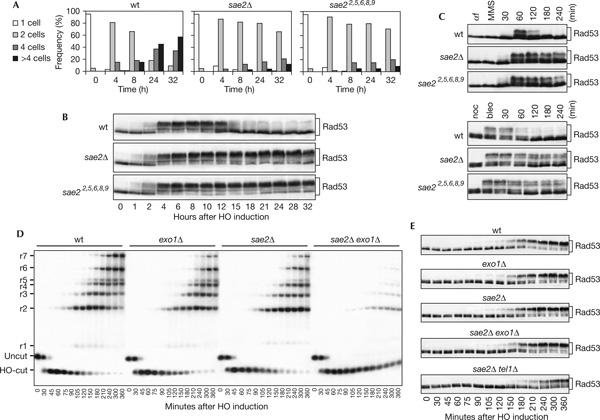
Response to a single irreparable double-strand break in sae2Δ cells. (A) YEP+raf G1-arrested cell cultures of wild-type (wt) JKM139 and isogenic sae2Δ and sae22,5,6,8,9 strains were spotted on galactose-containing plates incubated at 30°C (time zero). At the indicated time points, 200 cells for each strain were analysed to determine the frequency of single cells and of cells forming microcolonies of two, four or more than four cells. (B) Galactose was added at time zero to wild-type JKM139 and isogenic sae2Δ and sae22,5,6,8,9 cell cultures exponentially growing in YEP+raf. Protein extracts from aliquots withdrawn at the indicated times were analysed by western blot with anti-Rad53 antibodies. (C) Wild-type W303 and isogenic sae2Δ and sae22,5,6,8,9 cell cultures arrested in G1 with α-factor (αf) or in G2 with nocodazole (noc) were incubated for 15 min with methylmethane sulphonate (MMS, 0.02%) or bleomycin (bleo, 10 mU/ml), respectively, and then released in YEPD. Protein extracts from samples taken at the indicated times were analysed by western blot with anti-Rad53 antibodies. (D) YEP+raf nocodazole-arrested cell cultures of wild-type JKM139 and isogenic exo1Δ, sae2Δ, sae2Δ exo1Δ and sae2Δ tel1Δ strains were transferred to YEP+raf+gal in the presence of nocodazole at time zero. Genomic DNA prepared from aliquots taken at the indicated times was digested with SspI and separated on alkaline agarose gel. Gel blots were hybridized with a single-stranded RNA probe specific for the MAT locus, which shows HO-cut and uncut fragments of 0.9 and 1.1 kb, respectively. As depicted in supplementary Fig S2 online, 5′-to-3′ resection progressively eliminates SspI sites located 1.7, 3.5, 4.7, 5.9, 6.5, 8.9 and 15.8 kb centromere-distal from the HO-cut site, producing larger SspI fragments (r1–r7) detected by the probe. The kinetics of resection product accumulation in sae2Δ tel1Δ cell cultures (not shown) was undistinguishable from that of sae2Δ cells. (E) Protein extracts from samples taken at the indicated times during the experiment in (D) were analysed by western blot with anti-Rad53 antibodies.
The effects of Sae2 absence were not limited to the checkpoint triggered by an irreparable HO-induced DSB. In fact, Rad53 phosphorylation persisted much longer in sae2Δ cells treated with methylmethane sulphonate (MMS) or bleomycin than in wild-type cells (Fig 1C), and sae2Δ cells markedly delayed S-phase completion, compared with wild-type cells, after MMS treatment (supplementary Fig S1 online).
As single-stranded DNA (ssDNA) covered by RPA is one of the signals activating the checkpoint in response to DSBs (Lee et al, 1998; Zou & Elledge, 2003), we also monitored the kinetics of both HO-cut and 3′-ended ssDNA formation at the HO-cut site in galactose-induced cell cultures (Fig 1D) that were blocked in mitosis with nocodazole, to avoid specific cell-cycle effects on DSB processing. The sae2Δ JKM139 derivative strain showed some delay, compared with wild type, in the accumulation of 3′-ended resection products (r1–r7 in Fig 1D; see supplementary Fig S2 online for details), which then remained stable throughout the experiment (Fig 1D), because the HO break could not be repaired (data not shown). As Rad53 seems to be phosphorylated with similar kinetics in both wild-type and sae2Δ cells, even if the latter delayed DSB resection (Fig 1D,E), we asked whether checkpoint activation in the absence of Sae2 could occur independently of 3′-ended ssDNA generation. Therefore, we further slowed down accumulation of resection products in sae2Δ cells by deleting the EXO1 gene, encoding an exonuclease that contributes to DSB resection independently of MRX (Llorente & Symington, 2004; Nakada et al, 2004). As shown in Fig 1D,E, the appearance of ssDNA intermediates at the HO-cut site was markedly delayed, although not abolished, in nocodazole-arrested galactose-induced sae2Δ exo1Δ cells compared with that in sae2Δ and exo1Δ single mutants, whereas the kinetics of Rad53 phosphorylation was similar in the single and double mutants, indicating that signals other than ssDNA generation may activate the checkpoint in the absence of Sae2. These checkpoint signals may be unresected DSBs, as Rad53 phosphorylation after HO induction was delayed in sae2Δ cells lacking the Tel1 kinase (Fig 1E), the activation of which does not require ssDNA generation (Usui et al, 2001).
If Sae2 negatively regulates the checkpoint, as suggested by the above data, excess Sae2 might be expected to counteract checkpoint activation. Indeed, when G1-arrested cultures of JKM139 derivative strains were spotted on galactose-containing plates, cells carrying a galactose-inducible GAL-SAE2 construct formed microcolonies with more than four cells much faster than wild-type cells and similar to mec1Δ cells (Fig 2A). Moreover, when galactose was added to exponentially growing YEP+raf cell cultures, most wild-type cells were cell-cycle arrested with 2C DNA content and heavily phosphorylated Rad53 in 2–3 h after galactose addition, whereas SAE2-overexpressing cells were neither cell-cycle arrested (Fig 2E) nor underwent significant Rad53 phosphorylation (Fig 2B,F). This was not due to a failure to generate 3′-ssDNA at the DSB ends, as accumulation of the resection products in nocodazole-arrested galactose-induced GAL-SAE2 cells was similar to that in wild-type cells (Fig 2C).
Figure 2.
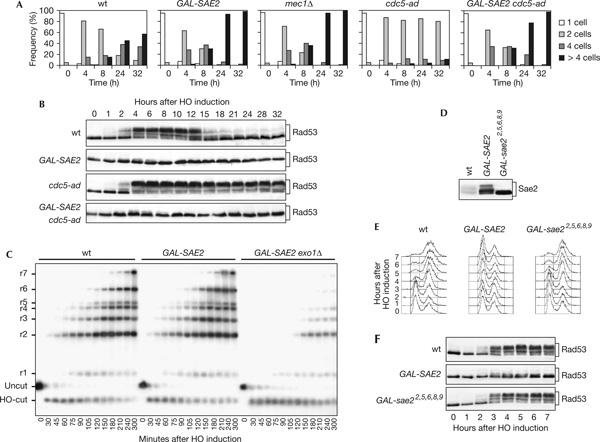
Response to a single irreparable double-strand break in SAE2-overexpressing cells. (A) YEP+raf G1-arrested cells of wild-type (wt) JKM139 and isogenic GAL-SAE2, cdc5-ad, GAL-SAE2 cdc5-ad and mec1Δ sml1Δ strains were spotted on galactose-containing plates incubated at 30°C (time zero). At the indicated times, 200 cells for each strain were analysed as in Fig 1A. (B) Galactose was added at time zero to wild-type JKM139 and isogenic GAL-SAE2, cdc5-ad and GAL-SAE2 cdc5-ad cell cultures exponentially growing in YEP+raf. Protein extracts from samples taken at the indicated times were subjected to western blot analysis with anti-Rad53 antibodies. (C) YEP+raf nocodazole-arrested cell cultures of wild-type JKM139 and isogenic GAL-SAE2 and GAL-SAE2 exo1Δ strains were transferred to YEP+raf+gal in the presence of nocodazole at time zero. Genomic DNA from samples collected at the indicated times was analysed as described in Fig 1D. (D) Cell cultures of JKM139 derivative strains carrying either the SAE2-HA allele at the SAE2 chromosomal locus or the GAL-SAE2-HA and GAL-sae22,5,6,8,9-HA fusions at the URA3 locus were shifted to YEP+raf+gal for 1 h and protein extracts were analysed by western blot with anti-haemagglutinin antibodies. (E,F) Exponentially growing YEP+raf cell cultures of wild-type JKM139 and isogenic GAL-SAE2 and GAL-sae22,5,6,8,9 strains were transferred to YEP+raf+gal at time zero. Aliquots were taken at the indicated times for fluorescence-activated cell sorting analysis (E) or western analysis with anti-Rad53 antibodies (F).
A similar impairment of checkpoint activation was also observed when SAE2 was overexpressed in the presence of the cdc5-ad allele, which prevents adaptation to a single unrepaired DSB (Toczyski et al, 1997). In fact, galactose-induced GAL-SAE2 cdc5-ad JKM139 derivative cells progressed through cell cycle with unphosphorylated Rad53, whereas otherwise isogenic cdc5-ad cells did not re-enter the cell cycle for at least 32 h after galactose addition (Fig 2A,B).
Therefore, Sae2 seems to negatively regulate the checkpoint independently of the ability of the cell to repair the break or to adapt to an unrepaired DSB.
Conversely, DNA-damage-induced Mec1- and Tel1-dependent Sae2 phosphorylation, besides being essential for other Sae2 functions (Baroni et al, 2004), is required for Sae2-mediated checkpoint switch off. In fact, replacement of Sae2 in JKM139 with the Sae22,5,6,8,9 variant, in which alanine substitutions of serines and threonines in all its [S/T]Q motifs abolish Sae2 phosphorylation without affecting protein level (Baroni et al, 2004; Fig 2D), caused permanent arrest at the two-cell dumbbell stage and persistent Rad53 phosphorylation after HO induction and MMS or bleomycin treatment (Fig 1A–C). Moreover, unlike GAL-SAE2, GAL-sae22,5,6,8,9 induction in the JKM139 strain (Fig 2D) did not alter the ability of the cell to arrest with 2C DNA content and phosphorylated Rad53 after HO induction (Fig 2E,F). Thus, DNA-damage-activated Mec1 and Tel1 might themselves trigger checkpoint switch off through Sae2 phosphorylation.
Sae2 is required for timely Mre11 removal from DSBs
One of the first proteins recruited to DNA-damaged sites is the MRX complex (Lisby et al, 2004), which is also necessary to trigger Rad53 phosphorylation in response to an HO-induced DSB even in the absence of Sae2 (Ira et al, 2004; Fig 3C). As Sae2 acts together with MRX in meiotic and mitotic DSB resection (Keeney & Kleckner, 1995; Neale et al, 2002; Clerici et al, 2005) and belongs to the MRX epistasis group with respect to HO-induced DSB end resection (supplementary Fig S3 online), Sae2 may regulate the checkpoint by modulating MRX signalling activity. Consistent with this hypothesis, the hypomorphic rad50s mutant, which shows the same meiotic and mitotic recombination defects as sae2Δ cells and which has been proposed to be impaired in Rad50–Sae2 interaction (Keeney & Kleckner, 1995; Rattray et al, 2001; Clerici et al, 2005), showed persistent Rad53 phosphorylation (Fig 3C) and failed to override the cell-cycle arrest after an HO-induced DSB (data not shown).
Figure 3.
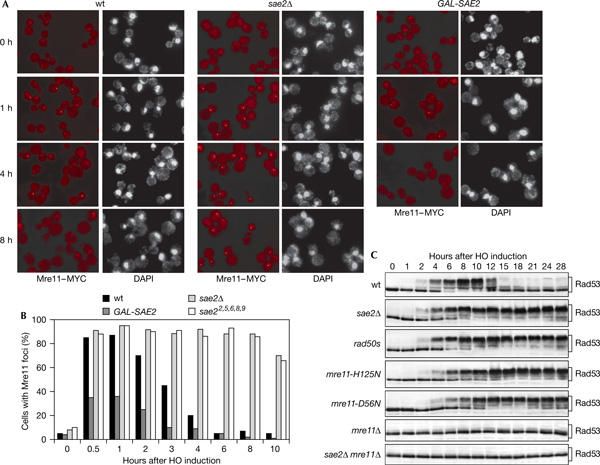
Mre11 localization at HO breaks in the absence or excess of Sae2. YEP+raf nocodazole-arrested cell cultures of wild-type (wt) JKM139 and isogenic sae2Δ and GAL-SAE2 strains, all carrying an MRE11-MYC-tagged allele at the MRE11 locus, were transferred to YEP+raf+gal in the presence of nocodazole (time zero). Cell samples taken at the indicated times were processed for staining with 4,6-diamidino-2-phenylindole (DAPI) and anti-Myc antibody indirect immunofluorescence. (A) Representative fields were photographed at the indicated times. (B) Kinetics of Mre11 foci formation were determined by scoring 200 cells for each strain at each time points. (C) Wild-type JKM139 and isogenic sae2Δ, rad50s, mre11-H125N, mre11-D56N, mre11Δ and mre11Δ sae2Δ strains, exponentially growing in YEP+raf, were transferred to YEP+raf+gal. Protein extracts from samples taken at the indicated times were analysed by western blot with anti-Rad53 antibodies.
As Sae2 lack has been shown to prolong Mre11 foci persistence after γ-irradiation or DSBs induced by the I-SceI endonuclease (Lisby et al, 2004), we asked whether the effects caused by the lack of Sae2 on checkpoint switch off correlated with alterations in the pattern of Mre11 localization at HO-induced DSBs. To this purpose, we analysed Mre11 localization after galactose addition in nocodazole-arrested wild-type and sae2Δ JKM139 derivative strains expressing a fully functional MRE11-MYC-tagged allele from the MRE11 promoter. As shown in Fig 3A,B, Mre11 localization detected by immunofluorescence was diffusely nucleoplasmic in all cell types growing in raffinose, as confirmed by nuclear 4,6-diamidino-2-phenylindole (DAPI) staining, whereas 85% of wild-type and sae2Δ nuclei contained one Mre11 focus already 30 min after galactose addition. Then, the Mre11 signal started to decrease in wild-type cells concomitantly with the appearance of fully phosphorylated Rad53 (Fig 3C), such that only 20% of them contained one Mre11 focus after 4 h. Conversely, Mre11 foci in sae2Δ cells were not only of higher intensity than those in wild–type cells, but also mostly persisted, together with Rad53 phosphorylation, for at least 10 h after HO induction (Fig 3).
MRX persistence at the HO-cut site in sae2Δ cells may constitutively signal to the checkpoint. If this was the case, SAE2 overexpression, which inhibits checkpoint activation, might impair Mre11 association to HO-induced DNA breaks. Strikingly, less than 40% of SAE2-overexpressing cells showed Mre11 foci 30 min after HO induction, and most foci disappeared in 4 h in these cells (Fig 3A,B). Consistent with defective Mre11 association to HO-induced DSBs in Sae2-overproducing cells, 3′-ended ssDNA accumulated much more slowly in GAL-SAE2 exo1Δ cells than in isogenic exo1Δ, sae2Δ or mre11Δ cells (compare Figs 1D and 2C; supplementary Fig S3 online). Moreover, the amount of 3′-ended ssDNA was reduced to the same extent in both GAL-SAE2 exo1Δ and sae2Δ exo1Δ cells compared with that in GAL-SAE2 cells (compare Figs 1D and 2C), indicating that Exo1 is mainly responsible for DSB resection in galactose-induced GAL-SAE2 cells.
As MRX has a nuclease activity (Paull & Gellert, 1998) and Mre11 foci persistence after γ-rays in the nuclease-deficient mre11-D56N and mre11-H125N mutants is similar to that found in sae2Δ cells (Lisby et al, 2004), the lack of Sae2 may freeze Mre11 at sites of DNA damage by altering its nuclease activity. In agreement with this hypothesis, we found that Rad53 phosphorylation persisted for at least 28 h after HO induction in the nuclease-deficient mre11-D56N and mre11-H125N cells (Fig 3C) that did not re-enter the cell cycle (data not shown), whereas it decreased in wild-type cells after 12–15 h (Fig 3C). Thus, the lack of Mre11 nuclease activity might constitutively signal to the checkpoint by causing MRX persistence at HO-induced DSBs.
Sae2 modulates Mec1- and Tel1-dependent checkpoints
The lack of Sae2 has been shown to trigger activation of a Tel1-dependent DNA damage checkpoint in mec1Δ cells (Usui et al, 2001). However, as the MRX complex is necessary for Mec1-dependent Rad53 phosphorylation in response to an HO-induced DSB (Ira et al, 2004) and for association of both Tel1 and Mec1 to DSBs (Nakada et al, 2003, 2004), Sae2-mediated inhibition of MRX signalling activity should influence both Mec1- and Tel1-dependent pathways.
To test this hypothesis, we used YMV80 derivative strains, in which a single HO cut can be generated on chromosome III by expressing a galactose-inducible GAL-HO fusion (Vaze et al, 2002). As shown in Fig 4A,B, Rad53 phosphorylation was below the detection level in mec1Δ cells throughout the experiment, whereas it became detectable 2 h after galactose addition and persisted until the end of the experiment in sae2Δ mec1Δ cells (Fig 4B), although its amount was not sufficient to slow down cell-cycle progression (Fig 4A). Moreover, persistent Rad53 phosphorylation was observed in both sae2Δ and sae2Δ tel1Δ cells, which remained arrested with 2C DNA content (Fig 4A) and undivided nuclei (data not shown) for at least 12 h, whereas it started to decrease in wild-type and tel1Δ cells when they re-entered the cell cycle about 9 h after HO induction (Fig 4A,B). As DNA-damage-induced Rad53 phosphorylation is completely abolished in sae2Δ mec1Δ tel1Δ cells (Usui et al, 2001; our unpublished observation), this indicates that the lack of Sae2 impairs both Tel1 and Mec1 inactivation in the presence of a DSB.
Figure 4.
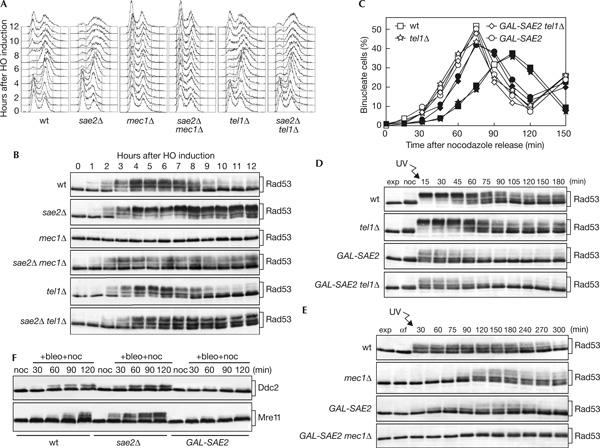
Mec1- and Tel1-dependent checkpoints in sae2Δ and SAE2-overexpressing cells. (A,B) Exponentially growing YEP+raf cell cultures of wild-type YMV80 and isogenic sae2Δ, mec1Δ sml1Δ, sae2Δ mec1Δ sml1Δ, tel1Δ and sae2Δ tel1Δ strains were transferred to YEP+raf+gal to induce HO expression (time zero). Samples withdrawn at the indicated times were used for fluorescence-activated cell sorting analysis of DNA contents (A) and western blot analysis of protein extracts with anti-Rad53 antibodies (B). (C,D) Exponentially growing YEP+raf cell cultures of wild-type W303 and isogenic tel1Δ, GAL-SAE2 and GAL-SAE2 tel1Δ strains were arrested with nocodazole in the presence of galactose for 2 h (noc), UV-irradiated (45 J/m2) and released into cell cycle in YEP+raf+gal. Samples were collected at the indicated times to determine the percentage of binucleate cells in unirradiated (open symbols) and UV-irradiated (closed symbols) cultures (C) and to monitor Rad53 phosphorylation from UV-irradiated cell cultures by western analysis (D). (E) Exponentially growing YEP+raf cell cultures of wild-type W303 and isogenic mec1Δ sml1Δ, GAL-SAE2 and GAL-SAE2 mec1Δ sml1Δ strains were arrested in G1 with α factor in the presence of galactose for 2 h (αf), UV-irradiated (30 J/m2) and released into cell cycle in YEP+raf+gal. Protein extracts prepared at the indicated times were analysed by western blot with anti-Rad53 antibodies. (F) YEP+raf nocodazole-arrested cell cultures of wild-type W303 and isogenic sae2Δ and GAL-SAE2 strains, all expressing the DDC2-HA3- or MRE11-HA3-tagged alleles from the corresponding endogenous promoters, were transferred in YEP+raf+gal containing 10 mU/ml bleomycin and 15 μg/ml nocodazole (+bleo+noc). Protein extracts prepared at the indicated times were subjected to western analysis with anti-haemagglutinin antibodies.
Because of the poor Rad53 phosphorylation in response to an HO-induced DSB in SAE2-overexpressing cells, we investigated the effects of high levels of Sae2 on Rad53 phosphorylation after UV irradiation of W303 derivative strains (Fig 4). Both GAL-SAE2 and GAL-SAE2 tel1Δ cells released into cell cycle after UV irradiation in G2 divided nuclei much faster (Fig 4C) and phosphorylated Rad53 much less efficiently (Fig 4D) than similarly treated tel1Δ cells. Moreover, Rad53 phosphorylation after UV irradiation in G1 and release into cell cycle (Fig 4E) was reduced in GAL-SAE2 cells compared with that in wild-type cells, whereas it could be detected in mec1Δ cells 90 min after release and was below the detection level in GAL-SAE2 mec1Δ cells. Thus, also SAE2 overexpression affects both Mec1- and Tel1-dependent pathways.
In agreement with the above findings, bleomycin-induced phosphorylation of the Mre11 and Ddc2 proteins, which specifically depends on Tel1 and Mec1, respectively (Longhese et al, 2003), was more efficient in sae2Δ cells than in wild-type cells, whereas it was below the detection level in SAE2-overexpressing cells (Fig 4F).
Altogether, our data indicate that Sae2 negatively regulates both Mec1- and Tel1-dependent checkpoint responses, possibly by modulating MRX association to damaged sites. As Mec1- and Tel1-dependent Sae2 phosphorylation is necessary for Sae2-mediated checkpoint switch off, it is tempting to speculate that these kinases, once activated, may limit MRX-dependent checkpoint signalling by phosphorylating Sae2.
Methods
Yeast strains JKM139 (MATa hmlΔ hmrΔ ade1 lys5 leu2-3,112 trp1∷hisG ura3-52 ho ade3∷GAL-HO; Lee et al, 1998), YMV80 and W303 (MATa or MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3, rad5-535) were used to disrupt the SAE2 gene, to replace the SAE2 chromosomal copy with the sae22,5,6,8,9 allele, or to integrate the GAL-SAE2 and GAL-sae22,5,6,8,9 fusions at the URA3 locus. YMV80 is isogenic to YFP17 (MATΔ hmlΔ hmrΔ ade1 lys5 ura3-52 trp1 ho ade3∷GAL-HO leu2∷cs), except for the presence of a LEU2 fragment inserted 25 kb centromere-distal to leu2∷cs (Vaze et al, 2002). Strains JKM139 and YMV80 were kindly provided by J. Haber (Brandeis University, Waltham, MA, USA).
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/7400593-s1.pdf).
Supplementary Material
Supplementary Information
Acknowledgments
We thank J. Haber, J. Diffley, N. Kleckner, L. Symington, D. Toczyski and K. Nasmyth for providing yeast strains and antibodies. This work was supported by grants from Associazione Italiana Ricerca sul Cancro, Cofinanziamento Ministero dell'lstruzione, dell'Università e della Ricerca/Università Milano-Bicocca and European Community's Human Potential Programme HPRN-CT-2002-00238 to M.P.L. and Fondo Investimenti Ricerca di Base to G.L.
References
- Baroni E, Viscardi V, Cartagena-Lirola H, Lucchini G, Longhese MP (2004) The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol Cell Biol 24: 4151–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Mantiero D, Lucchini G, Longhese MP (2005) The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double-strand break ends. J Biol Chem 280: 38631–38638 [DOI] [PubMed] [Google Scholar]
- Ira G et al. (2004) DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431: 1011–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Kleckner N (1995) Covalent protein–DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci USA 92: 11274–11278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE (1998) Saccharomyces Ku70, Mre11/Rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94: 399–409 [DOI] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, Rothstein R (2004) Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118: 699–713 [DOI] [PubMed] [Google Scholar]
- Llorente B, Symington LS (2004) The Mre11 nuclease is not required for 5′ to 3′ resection at multiple HO-induced double-strand breaks. Mol Cell Biol 24: 9682–9694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobachev KS, Gordenin DA, Resnick MA (2002) The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 108: 183–193 [DOI] [PubMed] [Google Scholar]
- Longhese MP, Clerici M, Lucchini G (2003) The S-phase checkpoint and its regulation in Saccharomyces cerevisiae. Mutat Res 532: 41–58 [DOI] [PubMed] [Google Scholar]
- Nakada D, Matsumoto K, Sugimoto K (2003) ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev 16: 1957–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Hirano Y, Sugimoto K (2004) Requirement of the Mre11 complex and exonuclease 1 for activation of the Mec1 signaling pathway. Mol Cell Biol 24: 10016–10025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale MJ, Ramachandran M, Trelles-Sticken E, Scherthan H, Goldman AS (2002) Wild-type levels of Spo11-induced DSBs are required for normal single-strand resection during meiosis. Mol Cell 9: 835–846 [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M (1998) The 3′ to 5′ exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol Cell 7: 969–979 [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE (2001) Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell 7: 293–300 [DOI] [PubMed] [Google Scholar]
- Rattray AJ, McGill CB, Shafer BK, Strathern JN (2001) Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: a role for SAE2/COM1. Genetics 158: 109–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toczyski DP, Galgoczy DJ, Hartwell LH (1997) CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90: 1097–1106 [DOI] [PubMed] [Google Scholar]
- Usui T, Ogawa H, Petrini JHJ (2001) A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 7: 1255–1266 [DOI] [PubMed] [Google Scholar]
- Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, Foiani M, Haber JE (2002) Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10: 373–385 [DOI] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information