

Abstract
What controls the length of life?
One of the greatest scientific mysteries, which has puzzled scientists for thousands of years, is what controls the length of life. At a time when human lifespan is increasing to previously unforeseen lengths, it is urgent that this ancient question is answered. Fortunately, recent progress suggests that the answers will soon be at hand.
Animal species exhibit great diversity in their maximum lifespan (Finch, 1990). Some, like the nematode Caenorhabditis elegans, have a lifespan measured in days—an average of 21 days for wild-type C. elegans strains grown in the laboratory. Others, such as giant tortoises, can live for centuries—specimens have been kept in zoos for more than 150 years. Mice have a lifespan of about three years, birds and bats have lifespans of decades and humans have a lifespan somewhat in excess of 100 years—the current reliably documented record is 122 years. There are even some species, such as the freshwater Hydra, for which no intrinsic senescence has been discerned and which may therefore have indefinite longevity (Martinez, 1998).
The main reason why it makes little sense to think in terms of clock-driven ageing is that ageing is hardly ever observable in natural populations
Because length of life is determined by survival, and survival is a trait on which Darwinian natural selection is expected to act with particular force, there is strong interest in trying to understand the factors that control length of life. There is also a biomedical and social urgency in doing so as soon as possible, in view of the remarkable increases in human life expectancy in recent decades. This increase began in industrialized nations during the early nineteenth century and was driven mainly by the decline in infectious disease mortality due to improvements in sanitation, housing and disinfection, and subsequently through vaccination and antibiotics. The previous high levels of fatal infection, especially among children, were dramatically reduced, and the predominant causes of mortality became degenerative conditions, such as heart failure, cancer and dementia, for which age is the single largest risk factor. As a result of this epidemiological transition, a large fraction of expenditure on medical and social care is now associated with older members of society, the number of which increases with each passing decade. This demographic shift towards the elderly is further accelerated by falling birth rates.
In addition, demographic trends over the past 2–3 decades have shown that, instead of reaching a plateau as most forecasters had predicted, human life expectancy has continued to increase and shows no sign of slowing. This has taken everyone by surprise, because the conventional view of ageing is that maximum life expectancy is somehow 'fixed'. Thus it was expected that when preventable early deaths were brought under control by medicine, we would simply see the ageing process more clearly revealed, with more and more of us living lifespans approaching the intrinsic limit. The rapid declines in old-age mortality over the past 50 years show that this is not happening, which has led us to re-examine the very nature of the ageing process.

© Getty
Fortunately, and perhaps not entirely coincidentally, these recent dramatic changes and the enormous challenges for society that they herald have come at a time when science is able, for the first time, to get to grips with the complex biology underlying the ageing process itself. This essay examines what we are learning about what controls human longevity by asking and attempting to answer a series of fundamental questions.
When pressed to explain what might determine a span of time, such as a lifespan, we naturally think in terms of some kind of clock. Manufactured clocks come in many forms, ranging from those that simply measure cyclical time, such as a sun dial or a wrist watch, to those that measure accumulated time and signal when a period has expired, such as an alarm clock or the fuse of a time bomb. Growing evidence reveals that biological clocks exist in a similarly diverse variety. The key questions with regard to ageing, however, are whether the ageing process is in fact clock-driven, and what evolutionary factors might have shaped the design of the clock.
In spite of a widespread belief that clocks drive the ageing process, progress—particularly in the evolutionary biology of ageing—tells us that this is highly unlikely. The main reason why it makes little sense to think in terms of clock-driven ageing is that ageing is hardly ever observable in natural populations. For most natural populations, extrinsic mortality—due to accidents, predation, starvation, disease, cold and so forth—is such that death occurs well before 'old age' (Fig 1). This means that whatever functional value is assigned to a clock for ageing, there is very little evidence that such value is actually realized. Also, it is extremely difficult to propose a satisfactory explanation for how such a clock might have evolved under natural selection, if animals in the wild do not normally survive to an age when the actions of the clock become apparent.
... if genes programme ageing, they do so only very loosely
Figure 1.
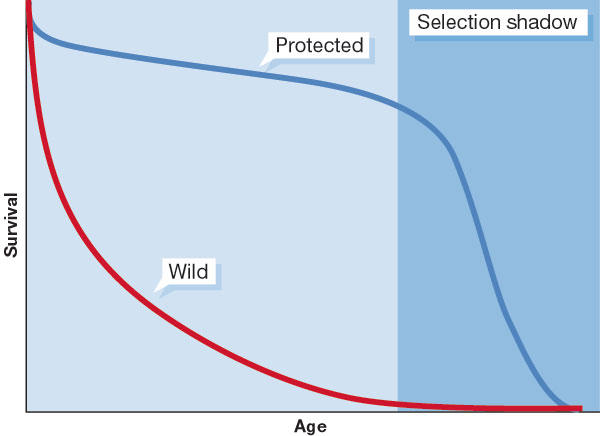
Survival curves for organisms in wild and protected environments. In the wild, the level of age-independent extrinsic mortality tends to be high, so that animals rarely reach an age where senescence makes its mark. For this reason, the ageing process has evolved mainly within the 'selection shadow' where natural selection is powerless to select for any kind of clock that might actively drive senescence.
Those who like to think in terms of clock-driven ageing sometimes follow the thinking pioneered by the great nineteenth century zoologist August Weismann (1891), who reasoned: “Worn out individuals are not only valueless to the species, but they are even harmful, for they take the place of those which are sound.” From this idea arises the notion that there might be clocks that induce senescence. However, Weismann's logic was circular. It supposed that older individuals are worn out, perhaps reproductively exhausted, when its very purpose was to explain why such age-related decline should occur. Furthermore, the idea would require the existence of an evolutionary process called 'group selection', since the benefit of such a clock is to the species or group rather than to the individual, for whom fitness would generally be enhanced by living and reproducing as long as possible, i.e. precisely by not having a clock for ageing.
The fact that survival rates in the wild are much lower than in the protected environments in which ageing can be observed means that the actions of any hypothetical clock must occur within what can be termed a 'selection shadow' (Fig 1). Peter Medawar (1952) recognized that the existence of such a shadow effectively counters the argument that ageing is actively programmed. Indeed, he went on to suggest that ageing might come about precisely because selection does not remove from the genome any late-acting deleterious mutations whose age of expression falls within the shadow. This idea, together with its subsequent elaboration by Williams (1957), provides the backbone for much of the current thinking about the evolutionary genetics of ageing. Our answer to the question “Is it a clock?” is thus a definite “No”.
Although the lack of support for an ageing clock means that we must be cautious about any idea that genes drive the ageing process, there is a clear heritable component in human longevity (Cournil & Kirkwood, 2001). In recent years large numbers of genes affecting longevity have been identified in organisms such as C. elegans. Indeed, the existence of single genes that have major effects on longevity in the nematode supports, at first sight, the idea of programmed ageing. But if genes programme ageing, they do so only very loosely. There are significant differences in ageing phenotypes between monozygotic human twins (Finch & Kirkwood, 2000). In C. elegans, even when genotype and environment are controlled, the variation in ageing phenotype, and in individual lifespan, is enormous (Herndon et al, 2002). This is in sharp contrast to the developmental process in this organism, which is so precisely regulated that each adult has just 959 somatic cells. Our answer to the question “Is it genes?” is thus “Kind of”.
Within the Western scientific tradition, Aristotle was the first to question what controls length of life. Among his thoughts, he advanced the belief that sex is responsible for the finitude of life and that each sex act has a direct life-shortening effect. This troubling concept enjoyed remarkable longevity, and was clearly expressed in the seventeenth century writings of Metaphysical poets such as John Donne: “...since each such act, they say, Diminisheth the length of life a day, ... I'll no more dote and run, To pursue things which had endamaged me.” It continues in present-day scientific studies on ageing, which reveal a trade-off between longevity and reproduction.
In the case of the fruit fly Drosophila melanogaster, there is particularly clear evidence for such trade-offs. Unmated flies live longer. An ingredient in the seminal fluid of males has been shown to have a direct life-shortening effect on female flies. Furthermore, successful artificial selection for longevity has repeatedly shown that long-lived populations tend to 'pay' for their longer lives through a decrease in fertility.
In the case of humans, a study by Westendorp and Kirkwood (1998) on the historical records of births, deaths and marriages of more than 33,000 British aristocrats revealed that females who died at the greatest ages tended to have increased levels of total infertility, delayed age at first childbirth and reduced family sizes, all of which suggest an inverse relationship between fertility and longevity. This finding has been supported in subsequent studies and a possible immunogenetic basis for such a trade-off has been suggested (Westendorp et al, 2001).
Reassuringly, however, and unlike Drosophila, there is no evidence that sex or reproduction have a direct life-shortening effect on humans. The trade-off between fertility and longevity is rather a predisposition, probably genetic in nature, that does not arise through the exercise of personal choice. Our answer to the question “Is it sex?” is thus “Not exactly”.
From a phenomenological perspective, there is much evidence that ageing is accompanied by an accumulation of 'wear and tear' in the form of molecular, cellular and tissue damage. Such an accumulation of damage cannot be inevitable, however, since there are organisms such as Hydra that do not age, and since the cell lineage that forms the germ line of all surviving species has continued through millions of years without succumbing to damage. Thus, the relationship between damage and ageing deserves close scrutiny.
Reassuringly, ... there is no evidence that sex or reproduction have a direct lifeshortening effect on humans
A concept that helps us to understand this relationship is the disposable soma theory (Kirkwood, 1977; Kirkwood & Holliday, 1979). It is based on the question of how an organism should optimally allocate its metabolic resources, chiefly energy, between the maintenance and repair of its soma and the other functions, which it must carry out in order to maximize its Darwinian fitness. The necessity for trade-off arises because resources that are allocated to one function are unavailable to another.
Somatic maintenance needs only to be good enough to keep the organism in sound physiological condition for as long as it has a reasonable chance of survival in the wild. For example, since more than 90% of wild mice die in their first year (Berry & Bronson, 1992), any investment of energy in survival beyond this age benefits, at most, 10% of the population. Nearly all of the mechanisms required for somatic maintenance and repair—such as DNA repair and antioxidant systems—require significant amounts of energy (ATP). Energy is scarce, as shown by the fact that the major cause of mortality for wild mice is cold, due to a failure to maintain thermogenesis (Berry & Bronson, 1992). The mouse will therefore benefit by investing any spare energy into thermogenesis or reproduction, rather than into somatic maintenance and repair, even though this means that damage will eventually accumulate to cause ageing. The three-year lifespan potential of the mouse is sufficient for its actual needs in the wild, and yet it is not excessive, given that some mice will survive into their second year and that age-related deterioration will become apparent before maximum lifespan potential is reached.
The disposable soma theory predicts that the primary cause of ageing is the accumulation of cellular and molecular damage, which arises because of evolved limitations in somatic maintenance and repair functions (Fig 2). The kinds of damage that have been associated with ageing include damage to most of the important macromolecular and cellular systems of the body. The immortality of Hydra is explained by the fact that this organism lacks a clear distinction between germ line and soma, and therefore has no soma to be disposable.
Figure 2.
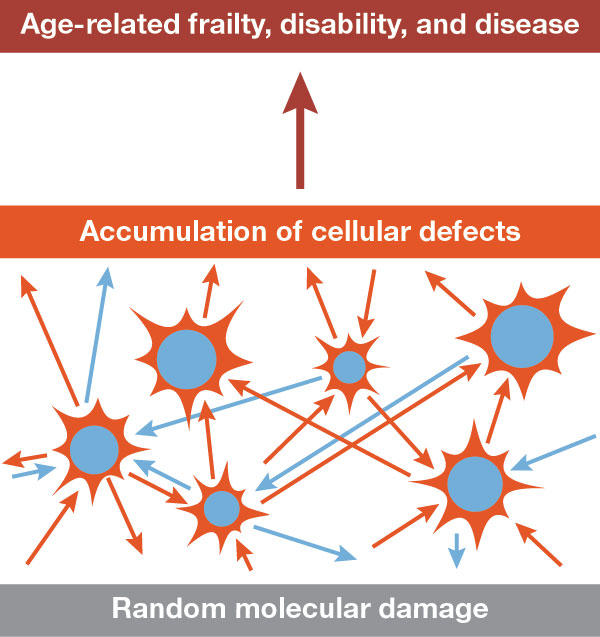
Underlying the ageing process is a progressive, gradual accumulation of molecular damage. Such damage is intrinsically random in nature but its rate of accumulation is regulated by genetic mechanisms for maintenance and repair. As cell defects accumulate, the effects on the body as a whole are eventually revealed as age-related frailty, disability and disease. This model accommodates the observed effects on length of life through environmental factors such as stress, nutrition, and exercise, which—in the case of adverse factors—can exacerbate the exposure to damage, and which—in the case of beneficial factors—can assist the body's maintenance and repair functions. The model therefore accommodates genetic, environmental and intrinsic chance effects on the ageing process.
Numerous studies have reported age-related increases in somatic mutation and other forms of DNA damage. In many human somatic tissues, a decline in cellular division capacity with age appears to be linked to the fact that the telomeres, which protect the ends of chromosomes, get progressively shorter as cells divide (Kim et al, 2002). This is due to the absence of the enzyme telomerase, which is normally expressed only in germ cells and in certain adult stem cells. Whereas the loss of telomeric DNA is commonly attributed to the so-called 'end-replication' problem—the inability of the normal DNA copying machinery to copy right to the very end of the strand in the absence of telomerase—it has been found that stress, especially oxidative stress, has a major effect on the rate of telomere loss, thus linking telomere erosion with damage (von Zglinicki, 2002).
...it seems likely that the genetic effects on longevity...are mediated through genes that regulate the levels of somatic maintenance and repair functions
Another important form of damage accumulation is seen in the build-up of mitochondrial DNA (mtDNA) mutations with age. Age-related increases in the frequency of cytochrome c oxidase (COX)-deficient cells, which are associated with mtDNA mutation, have been reported in human muscle (Brierley et al, 1998), brain (Cottrell et al, 2001) and gut (Taylor et al, 2003). Cells in which mtDNA mutations reach a high level are likely to suffer from impaired ATP production, which results in a decline in tissue bioenergenesis.
Much of the early, bewildering proliferation of ageing theories ... arose from a tendency to view the different ageing hypotheses as competing
Damage also accumulates in the protein constituents of cells, and contributes to a range of age-related disorders, including cataracts, Alzheimer's disease and Parkinson's disease. This accumulation appears to be reinforced by the age-related impairment of protein turnover due to functional declines in the activities of both proteasomes (Carrard et al, 2002) and chaperones (Soti & Csermely, 2003). These declines may be part of a more general failure of cellular 'waste-disposal' processes, which are an important means of maintaining homeostasis in the face of damage.
The age-related accumulation of cellular damage is likely to result in considerable tissue mosaicism, with damaged cells co-existing alongside relatively undamaged cells. Understanding how tissues respond to a growing burden of damaged cells, and what degree of burden can be tolerated before functional decline becomes apparent at the system level, are important current challenges (Kirkwood, 2005). Our answer to the question “Is it damage?” is clearly “Yes”.
In the preceding section, we considered how damage controls length of life. Of course, the rate at which damage accumulates is a function not only of the rate at which fresh damage occurs, but also of the rate at which it is repaired. Thus, the capacity for DNA repair will be an important determinant of the rate of ageing. This is particularly well illustrated by studies on the enzyme poly(ADP-ribose) polymerase-1 (PARP-1), a key player in the immediate cellular response to stress-induced DNA damage. In cross-species comparisons, higher poly-ADP ribosylation capacity is associated with longer lifespan (Grube & Bürkle, 1992), and there is some evidence that human centenarians have higher PARP-1 levels than the general population (Muiras et al, 1998). This suggests that innate differences in repair capacity may be part of the basis for heritability of human longevity. In general, it seems likely that the genetic effects on longevity noted earlier are mediated through genes that regulate the levels of somatic maintenance and repair functions. Our answer to the question “Is it repair?” is therefore also “Yes”.
It has been known for 70 years that underfeeding laboratory rodents extends their lifespan, apparently by slowing the intrinsic rate of ageing. Lifespan extension through food restriction is not confined only to rodents and has been reported in several other animal species, including fruit flies, nematodes, water fleas, spiders and fish. There is some indication that budding yeast also exhibits slower ageing when its nutrient supply is restricted. However, it is unclear whether food restriction affects length of life in primates or humans.

© Getty
In rodents, food restriction upregulates a wide range of somatic maintenance and repair functions, while simultaneously shutting down reproduction. The primary role of this response may be to shift resources away from reproduction during periods of famine, when the likelihood of reproductive success is small, and to invest as much as possible of the resultant savings into increased somatic maintenance. The potential benefit is that the animal gains an increased chance of survival, thereby allowing reproductive value to be preserved for when the famine is over (Shanley & Kirkwood, 2000).
In a broader context, it is clear from epidemiological studies that nutrition can affect human lifespan both adversely in the case of poor nutrition—a diet rich in sugars and saturated fats, for example—and positively in the case of good nutrition—a diet rich in fruits, vegetables, unsaturated fats and red wine. These positive and negative effects on ageing can be accommodated very readily within an understanding of ageing that is founded on damage and repair. Our answer to the question “Is it food?” is therefore “Partly”.
We have now reached a point at which we can see that whatever controls the length of life is primarily bound up in the dynamics of damage and repair. The simplicity offered by this view also makes it possible to better understand the phenomenological complexity of the ageing process. The logic of the disposable soma theory tells us that there are likely to be multiple kinds of damage that contribute to ageing, which are regulated by a complex network of maintenance and repair functions. It also tells us that the mechanisms of cellular and molecular ageing are inherently stochastic—strongly influenced by chance—which may explain the marked variability in lifespan even among genetically identical populations.
If we want to understand the underlying mechanisms satisfactorily, the inherent complexity of ageing cannot be ignored and reductionist science alone will not be enough. Much of the early, bewildering proliferation of ageing theories during the 1950s, 1960s and 1970s arose from a tendency to view the different ageing hypotheses as competing. However, the disposable soma theory suggests that multiple kinds of damage will accumulate in parallel within cells. This has led to recent initiatives to develop 'network' theories of ageing in which the contributions of various mechanisms are considered together, thereby allowing for interaction and synergism between different processes (Kirkwood et al, 2003). In order to make effective progress in unravelling the underlying complexity of ageing mechanisms, we are likely to become increasingly reliant on these kinds of systems biology approaches. Our answer to the question “Is it complicated?” is emphatically “Yes”.
We have seen that answers to the question “What controls the length of life?” are beginning to emerge. This is an area of science that—until relatively recently—was dismissed by many biologists as just too difficult for serious study. This picture is changing rather quickly. It is an exciting challenge to take our answers further. For the benefit of societies around the world, who will have to confront the big issues that surround population ageing, it is imperative that we should do so with all possible speed.

Acknowledgments
I thank the many colleagues and others whose work contributed to these ideas but could not be cited here due to space restrictions.
References
- Berry RJ, Bronson FH (1992) Life history and bioeconomy of the house mouse. Biol Rev Camb Philos Soc 67: 519–550 [DOI] [PubMed] [Google Scholar]
- Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM (1998) Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann Neurol 43: 217–223 [DOI] [PubMed] [Google Scholar]
- Carrard G, Bulteau AL, Petropoulos I, Friguet B (2002) Impairment of proteasome structure and function in aging. Int J Biochem Cell Biol 34: 1461–1474 [DOI] [PubMed] [Google Scholar]
- Cottrell DA, Blakely EL, Johnson MA, Ince PG, Borthwick GM, Turnbull DM (2001) Cytochrome c oxidase deficient cells accumulate in the hippocampus and choroid plexus with age. Neurobiol Aging 22: 265–272 [DOI] [PubMed] [Google Scholar]
- Cournil A, Kirkwood TB (2001) If you would live long, choose your parents well. Trends Genet 17: 233–235 [DOI] [PubMed] [Google Scholar]
- Finch CE (1990) Longevity, Senescence, and the Genome. Chicago, IL, USA: Chicago University Press [Google Scholar]
- Finch CE, Kirkwood TBL (2000) Chance, Development, and Aging. Oxford, UK: Oxford University Press [Google Scholar]
- Grube K, Bürkle A (1992) Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with speciesspecific life span. Proc Natl Acad Sci USA 89: 11759–11763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M (2002) Stochastic and genetic factors influence tissuespecific decline in ageing C. elegans. Nature 419: 808–814 [DOI] [PubMed] [Google Scholar]
- Kim S, Kaminker P, Campisi J (2002) Telomeres, aging and cancer: in search of a happy ending. Oncogene 21: 503–511 [DOI] [PubMed] [Google Scholar]
- Kirkwood TB (1977) Evolution of ageing. Nature 270: 301–304 [DOI] [PubMed] [Google Scholar]
- Kirkwood TB (2005) Understanding the odd science of aging. Cell 120: 437–447 [DOI] [PubMed] [Google Scholar]
- Kirkwood TB, Holliday R (1979) The evolution of ageing and longevity. Proc R Soc Lond B Biol Sci 205: 531–546 [DOI] [PubMed] [Google Scholar]
- Kirkwood TB, Boys RJ, Gillespie CS, Proctor CJ, Shanley DP, Wilkinson DJ (2003) Towards an e-biology of ageing: integrating theory and data. Nat Rev Mol Cell Biol 4: 243–249 [DOI] [PubMed] [Google Scholar]
- Martinez DE (1998) Mortality patterns suggest lack of senescence in hydra. Exp Gerontol 33: 217–225 [DOI] [PubMed] [Google Scholar]
- Medawar PB (1952) An Unsolved Problem of Biology. London, UK: H. K. Lewis [Google Scholar]
- Muiras ML, Müller M, Schächter F, Bürkle A (1998) Increased poly(ADP-ribose) polymerase activity in lymphoblastoid cell lines from centenarians. J Mol Med 76: 346–354 [DOI] [PubMed] [Google Scholar]
- Shanley DP, Kirkwood TB (2000) Calorie restriction and aging: a life history analysis. Evolution 54: 740–750 [DOI] [PubMed] [Google Scholar]
- Soti C, Csermely P (2003) Aging and molecular chaperones. Exp Gerontol 38: 1037–1040 [DOI] [PubMed] [Google Scholar]
- Taylor RW et al. (2003) Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest 112: 1351–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zglinicki T (2002) Oxidative stress shortens telomeres. Trends Biochem Sci 27: 339–344 [DOI] [PubMed] [Google Scholar]
- Weismann A (1891) Essays upon Heredity and Kindred Biological Problems. Oxford, UK: Clarendon [Google Scholar]
- Westendorp RG, Kirkwood TB (1998) Human longevity at the cost of reproductive success. Nature 396: 743–746 [DOI] [PubMed] [Google Scholar]
- Westendorp RG, van Dunne FM, Kirkwood TB, Helmerhorst FM, Huizinga TW (2001) Optimizing human fertility and survival. Nat Med 7: 873. [DOI] [PubMed] [Google Scholar]
- Williams GC (1957) Pleiotropy, natural selection, and the evolution of senescence. Evolution 11: 398–411 [Google Scholar]