

Abstract
When it comes to studying ageing and the means to slow it down, mice are not just small humans
All mammalian cells use similar molecular mechanisms to regulate growth, replication, differentiation and death. Even specialized cells, such as fibroblasts or secretory epithelial cells, are often quite similar in structure and function between species. Mice and humans are good examples of this metabolic homogeneity—they have the same organs and systemic physiology, and they also show great similarities in disease pathogenesis. For example, mouse tumours have similar histological features to comparable human tumours. In addition, mice acquire mutations in an equivalent spectrum of proto-oncogenes and tumour suppressor genes (Balmain & Harris, 2000). These similarities constitute one of the main reasons why mouse models are used to study human disease pathogenesis and ageing processes. They have also been the driving force behind efforts to understand the effects on humans of caloric restriction, which is the only experimental manipulation so far shown to reduce disease incidence and to significantly increase both mean and maximum lifespan in mammals (Weindruch & Walford, 1988). In view of the similarities between mice and human cells and physiology, will caloric restriction have an impact on humans similar to that observed in mice? Furthermore, how appropriate is the mouse as a model to study ageing in humans?
Answering these questions requires an understanding of the general mechanisms that drive the ageing process. Ageing, in its broadest sense, is the continuous and irreversible decline in the efficiency of various physiological processes once the reproductive phase of life is over (Sohal & Weindruch, 1996). Species that belong to the same phyletic lineage and have similar rates of senescence would therefore have similar molecular mechanisms driving the ageing process. Consequently, caloric restriction in such species should have equivalent effects on their longevity. However, species with divergent rates of senescence would typically have dissimilar molecular mechanisms. When caloric restriction is imposed on such species, the outcome on longevity may be quite different.
Thus, to evaluate the significance of mouse models in studies of caloric restriction in particular, and in the analysis of ageing in general, one question needs to be addressed. Is the ageing process in mice and humans characterized by equivalent rates of senescence and concomitantly homologous molecular mechanisms, or are the systems distinguished by dissimilar rates of senescence with correspondingly divergent underlying mechanisms of ageing? In this article, I review various physiological, demographic and evolutionary arguments to show that mice and humans, despite a certain congruence in systemic physiology and a similarity in age-associated disease pathogenesis, have demonstrative differences in their rates of senescence. Consequently, the mechanisms underlying the ageing process in the two species are likely to be distinct. This review is organized to a large extent around the concept of metabolic stability, which was introduced to explain the large variation in lifespan observed between species (Demetrius, 2004). Metabolic stability, roughly speaking, describes the capacity of cellular regulatory networks to maintain metabolic homeostasis in response to stress. The term 'stress' in this context is defined as any agent of endogenous origin that disrupts the rates of intracellular metabolic conversions.
This analysis predicts that the impact of caloric restriction on non-obese human populations will result in a relatively small increase in mean lifespan and no change in maximum lifespan potential
I use the metabolic stability concept to elucidate two aspects of the phenomenon of senescence. The first one pertains to the proposal of a molecular mechanism for ageing—the progressive and irreversible loss of function accompanied by increasing mortality. The mechanism I postulate is derived from the metabolic stability–longevity principle (Demetrius, 2004), which relates metabolic stability to maximal lifespan potential, and asserts that metabolic stability is the prime determinant of ageing and is positively correlated with longevity. Accordingly, strongly stable networks will be defined by slow rates of ageing, whereas weakly stable networks will be defined by rapid rates of ageing.
The second aspect explores the evolutionary rationale for different rates of senescence between species. This argument is developed in terms of directionality theory, which is an analytical model based on the concept of evolutionary entropy as a measure of Darwinian fitness (Demetrius, 1997). It considers the ecological constraints that impinge on a population and their impact on the dynamics of mutation and natural selection. Directionality theory predicts that mice, which have evolved under ecological conditions that are defined by highly variable resource constraints, are characterized by weakly stable metabolic networks and hence fast rates of ageing. By contrast, humans have been subjected throughout most of their evolutionary history to ecological constraints defined by limited but relatively constant resources, and are characterized by highly stable metabolic networks and hence slow rates of ageing.
The metabolic stability–longevity principle has important implications for research on caloric restriction and ageing. Empirical studies support the hypothesis that caloric restriction increases lifespan by increasing the stability of metabolic networks (Dhahbi et al, 2001; Cao et al, 2001). I invoke this principle to predict that the large increases in mean and maximum lifespan observed in mice under caloric restriction will not appear in humans. This analysis predicts that the impact of caloric restriction on non-obese human populations will result in a relatively small increase in mean lifespan and no change in maximum lifespan potential.
Simple extrapolation from mouse models to human systems in studies of the ageing process may ... be invalid
Although mice share genes, organ systems and systemic physiology with humans, the two species differ significantly in terms of morphometry, physiology and life history. Humans are about 3,000 times larger than mice, and this size difference imposes constraints on physiology and life history with significant effects on the species' ability to adapt to environmental conditions. In the following, I discuss the effects of the differences in metabolic activity, disease pathogenesis and life history.
As mice are significantly smaller than humans, their basal metabolic rate—the rate of energy production over a set period of time under constant conditions—is much less than that of man, because there is simply less body mass and less total energy production. However, the basal metabolic rate per gram of body weight—the mass-specific rate—is seven times greater in mice than in humans. This difference in metabolic rate will have contrasting effects on the senescence process in the two species.
Reactive oxygen species (ROS) are a by-product of energy metabolism in the mitochondria (Beckman & Ames, 1998). They mediate their effects on lifespan through two distinct mechanisms. According to Finkel & Holbrook (2000), increased ROS production may cause random damage to proteins, lipids and DNA with detrimental effects that lead to cell death. Decreased ROS production may cause the deactivation of redoxsensitive signalling pathways with a concomitant deleterious effect on cell-cycle regulation. Consequently, the generation of ROS, and the capacity of the cell to maintain ROS production within certain bounds, will be the strongest correlate with overall longevity.
ROS production does not necessarily increase in proportion to mitochondrial oxygen consumption because the free-radical leak in the respiratory chain is not constant. Consequently, the relationship between the metabolic rate itself with longevity will be quite complex, as the studies described in Speakman et al (2004) indicate. However, analysis of model systems (Demetrius, 2004) suggests that organisms with a small massspecific metabolic rate will typically be characterized by low ROS production and small deviations of ROS levels from the normal state. In such organisms, the capacity to maintain ROS levels within certain limits is relatively strong. Organisms with large mass-specific metabolic rates will typically be characterized by high ROS production and relatively large deviations from their prescribed values. They will have a weak capacity to maintain homeostasis. This difference in homeostatic capacities has important implications for comparing the rate of senescence in mice and humans. Mice, which have a higher mass-specific metabolic rate, have a weak capacity to maintain cellular homeostasis and consequently age relatively fast. Humans have a lower mass-specific metabolic rate, a strong capacity to maintain cellular homeostasis, and accordingly, a slower rate of ageing (Fig 1).
... mice and humans are different beasts when it comes to cancer—a disease typical of old age
Figure 1.
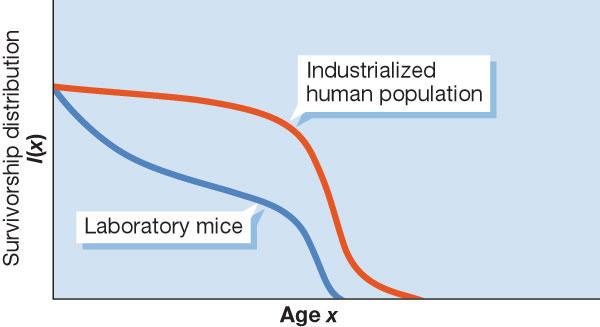
Survivorship distribution for laboratory mice and an industrialized human population.
We can therefore conclude that mice and humans, in spite of their similarities at the molecular, cellular and physiological level, show decisive dissimilarities in the rates at which they age. These divergences in the rate of senescence underscore the fact, succinctly expressed by Rangarajan & Weinberg (2003), that mice are not small people. Simple extrapolation from mouse models to human systems in studies of the ageing process may therefore be invalid.
In addition to the differences in the metabolic rate of the two species, there are striking variations in disease pathogenesis, for example, in terms of cancer susceptibility. Assuming that tumour progression from benign to malignant is similar in both rodent and human cancers (Fearon & Vogelstein, 1990; Dragan & Pitot, 1992), we would infer that humans have higher rates of cancer incidence, given that they live, on average, 30–50 times longer than mice. Epidemiological studies, however, show that this is not the case. Furthermore, the dynamics of disease pathogenesis differ significantly between the two species. In mice, the incidence of invasive cancer increases exponentially with age. In humans, the pattern is more complex (Piantanelli, 1988; DePinho, 2000): an exponential increase of cancer incidence begins at 40 years old and ceases at age 80. Beyond this age, cancer incidence ultimately levels out. Whereas cancer afflicts 30% of laboratory rodents at the end of their 2–3-year lifespan, a comparable percentage is observed in humans only beyond 70 years of age (Holliday, 1996). Cancer susceptibility studies also show that mice develop malignant tumours with multiple genetic alterations within a 6–18-month period, whereas malignancy in humans usually takes many years to impact on lifespan (Balmain & Harris, 2000). As the incidence of disease pathogenesis indicates, mice and humans are different beasts when it comes to cancer—a disease typical of old age.
The term 'life history' describes the age-specific fecundity and mortality of individuals in a given population. Certain species concentrate their reproductive activity over a very narrow period of their total lifespan, whereas others distribute their reproduction over the greater part of their lifespan. The life history of a population can be described by a unique function if certain demographic constraints are imposed on life-history patterns (Demetrius, 1997). This function is called 'evolutionary entropy' (see sidebar), owing to its formal similarity to the entropy concept in physics. Evolutionary entropy can be illustrated using plant populations: annuals are a low-entropy species that concentrate their reproduction at a single stage in their life, whereas high-entropy perennials allocate their reproduction to several distinct points in their life cycle. As Demetrius (2004) showed, evolutionary entropy can be expressed in terms of three demographic components: age of sexual maturity, litter size and reproductive span. Mice, a low-entropy species, become sexually mature at 35–50 days, produce a litter of 8–10 pups and have a reproductive span of two years. Humans, a high-entropy species, become sexually mature at 13 years of age, give birth to one offspring at a time and have a reproductive span of 40 years (Figs 2,3). Mice and humans thus occupy different positions on the entropy scale.
Evolutionary entropy: an analytical characterization.
The arguments in this article revolve around the concept of 'evolutionary entropy', which is a function of the age-specific fecundity and survival rates of the individuals in a given population. Evolutionary entropy is analytically characterized by the expression:
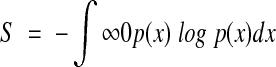 |
where p(x)dx is the probability that the mother of a randomly chosen newborn belongs to the age category (x,x+dx).
The function p(x) can be expressed in terms of the survivorship distribution l(x) (the proportion of newborns surviving to age x) and the fecundity distribution m(x) (the mean number of offspring produced by an individual of age x):
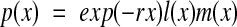 |
where r is the population growth rate.
Figure 2.
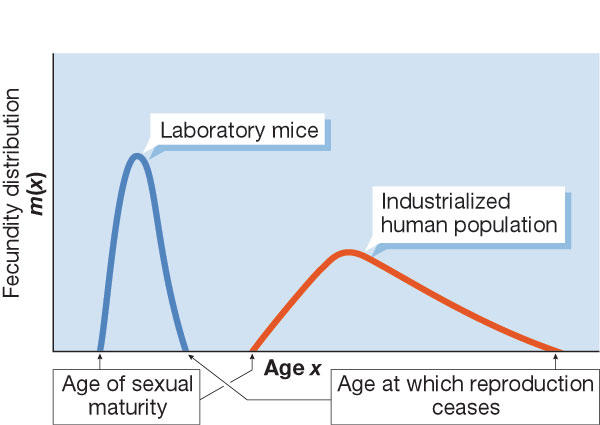
Fecundity distribution for laboratory mice and an industrialized human population.
Figure 3.
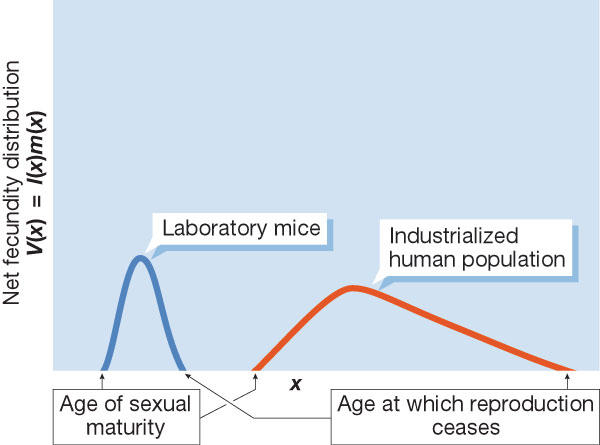
The net fecundity distribution V(x) = l(x)m(x) for laboratory mice and humans. The curve for mice describes a population with early age of sexual maturity, large litter size and narrow reproductive span (low entropy) whereas that for humans shows the effect of late age of sexual maturity, small litter size and broad reproductive span (large entropy).
The significance of the evolutionary entropy concept for ageing studies arises from the observation that entropy is positively correlated with lifespan potential. This relationship, which was originally derived from model systems (Demetrius, 2004), is supported by various studies of ageing in mammalian lineages (Austad, 1997). These empirical studies delineate the following relationships between demographic components and lifespan: late age of sexual maturity, small litter size and broad reproductive span are positively correlated with long maximum lifespan, whereas early age of sexual maturity, large litter size and narrow reproductive span are positively correlated with short maximum lifespan. In view of the characterization of entropy in terms of the three demographic variables, we can conclude that entropy and maximum lifespan are positively correlated.
The large divergences in physiological properties, such as metabolic rate and disease pathogenesis, and life-history properties, such as evolutionary entropy, suggest that mice and humans, despite congruencies at the molecular and cellular level, are significantly different in their rate of ageing. With this in mind, new questions arise. What are the molecular mechanisms that underlie these divergences? Can these divergences be explained at the cellular level? These issues, as Demetrius (2004) showed, can be resolved in terms of the concept of metabolic stability—a measure of the robustness of the regulatory networks that control cell metabolism.
The random nature of dysregulation ... suggests that metabolic stability is not simply correlated with lifespan, but is a critical determinant of longevity
The concept of metabolic stability refers to the ability of cells to maintain steady state values of redox couples in the face of random perturbations in the rates of enzymatic reactions. According to the stability–longevity model, which forms the cornerstone of the theory of ageing proposed in Demetrius (2004), the loss of function that comes with older age is therefore due to the dysregulation of this steady state, which causes fluctuations in the concentrations of various metabolites. The effect of these random perturbations accumulates over time and ultimately leads to the impairment of cellular signalling, the dysregulation of ontogenetic events and cell death. Accordingly, senescence is the result of spontaneous and cumulative changes in cell metabolism during normal development.
Metabolic dysregulation derives from random molecular perturbations and is thus an intrinsic property of all metabolic processes. The point at which dysregulation occurs in a metabolic network depends on the ability of the system to maintain cellular homeostasis in the face of stress. This can be enhanced by factors, such as DNA repair mechanisms, which significantly prolong the point at which metabolic dysregulation results in cell death. Organisms with high metabolic stability have a strong capacity to maintain homeostatic regulation when under stress, whereas organisms with low metabolic stability are defined by a weak capacity to maintain homeostasis. These observations imply that maximal lifespan potential is positively correlated with metabolic stability. The random nature of dysregulation of the steady state condition that describes the metabolic work further suggests that metabolic stability is not simply correlated with lifespan, but is a critical determinant of longevity.
Two important implications follow immediately from this principle: the metabolic stability of an organism decreases with age and it is positively correlated with maximum potential lifespan. The latter is supported by various cross-species comparisons of metabolic stability and lifespan. Cutler (1984) studied the effects on cells of endogenous mutagenic agents that are known to destabilize homeostasis, and showed that cells from longer-lived species are intrinsically more insensitive to mutagenic agents. Kapahi and colleagues (1999) comprehensively studied stress resistance in primary cultures of mammalian skin fibroblasts and noted that cell stress resistance is positively correlated with species lifespan. Consequently, species with highly stable metabolic networks, such as humans, have a strong capacity for maintaining the steadystate values of critical metabolites under random perturbation, and will therefore be long-lived. Species with weakly stable metabolic networks have a weak capacity to maintain their homeostatic condition and live only short lives.
So can the metabolic stability–longevity principle be explained in an evolutionary context? This problem can be resolved by appealing to directionality theory (Demetrius, 1997), a model of the evolutionary process that is based on the concept of evolutionary entropy as a measure of Darwinian fitness. Directionality theory distinguishes between two classes of population in terms of their response to resource limits. Equilibrium species spend the main part of their evolutionary history in the stationary growth phase or at a fairly constant population size. Humans are a canonical example of an equilibrium species. Their population has increased in size as they moved from the hunter/gatherer state to the agricultural and then the industrialized state. Human population growth rate was fairly slow during the hunter/gatherer phase—between 0.007 and 0.0015 per thousand per year—a period that represents 99% of human history. The large increase in growth rate (to 0.36 per 1000) when humans turned to agriculture 10,000 years ago, and the recent explosion in growth rate (0.56 per 1000) since 1750, represent only 1% of human evolutionary history. By contrast, opportunistic species are subject to episodes of rapid and exponential population growth when resources are ample, which are followed by sharp declines as soon as resources become depleted. Wild mice are a typical example of an opportunistic species.
Directionality theory revolves around the idea that Darwinian fitness—the demographic measure that determines the outcome of a competition between a mutant allele and its wild type—is characterized by the robustness or demographic stability of the population. This notion of stability refers to the capacity of a population to maintain its population size in the face of chance perturbations in birth and death rates. It is positively correlated with the life-history parameter, evolutionary entropy (Demetrius, 1997).
Accordingly, directionality theory predicts that mutations in equilibrium species that increase demographic stability will have a selective advantage, as they increase the capacity of the population to exploit limited but constant resources in their environment. In opportunistic species, however, mutations that decrease demographic stability will have a selective advantage as they enhance the ability of the population to quickly exploit large variations in resource availability. It thus follows that equilibrium species will typically be characterized by strong demographic stability, large entropy and, consequently, long maximum lifespan potential. Opportunistic species are typically described by weak demographic stability, small entropy and a correspondingly short maximum lifespan potential.
Directionality theory also predicts the evolutionary dynamics of metabolic stability. Evolutionary changes in demographic stability are positively correlated with changes in metabolic stability (Demetrius, 2004). Accordingly, equilibrium species have metabolic networks that are highly resistant to random perturbations of endogenous origin, whereas opportunistic species' metabolic networks are highly sensitive to random fluctuations. These properties also entail that equilibrium and opportunistic species will be largely divergent in their rates of senescence and in their incidence of age-related diseases. By appealing to the relationship between metabolic stability and longevity, and the fact that equilibrium and opportunistic species are described by highly and weakly stable metabolic networks, respectively, I predict that in equilibrium species, mortality rates increase exponentially with age but abate at advanced age (a mortality plateau), whereas they increase exponentially with age (a Gompertzian distribution) in opportunistic species (Table 1).
Table 1. Physiological and demographic properties for equilibrium and opportunistic species.
| Physiological and demographic properties | Equilibrium species (humans) | Opportunistic species (mice) |
|---|---|---|
| Metabolic stability | Strong | Weak |
| Demographic stability | Large | Small |
| Rate of senescence | Slow | Fast |
| Survivorship curve | Non-Gompertzian (mortality plateau) | Gompertzian |
| Maximum lifespan potential | Long | Short |
This analysis accounts for the observation that humans, a typical equilibrium species, consist of cells that are highly stress resistant, whereas mice, a typical opportunistic species, are composed of cells with metabolic dynamics that are highly sensitive to random perturbations. The large differences in metabolic stability or the robustness of the metabolic network in humans and mice have their origin in the different ecological constraints that these species have endured over their evolutionary history.
This conceptual framework has important implications for understanding the impact of caloric restriction on mammalian longevity. The hypothesis that caloric restriction acts by increasing metabolic stability, and the observation that equilibrium and opportunistic species differ significantly in terms of this feature, suggest that the response to caloric restriction is different for each species. Homo sapiens has been subject to ecological constraints that ensured stationary or very slow population growth throughout most of its evolutionary history and has therefore evolved a life-history schedule defined by high entropy: late sexual maturity, small litter size and a maximum lifespan potential of 120 years. The high entropy condition implies that metabolic networks in humans are highly resistant to change. Caloric restriction will therefore have only negligible effects on metabolic stability and hence no effect on maximum lifespan. However, it may have an effect on mean lifespan by reducing the incidence of diseases such as diabetes, atherosclerosis and hypertension. But these changes, as demographic studies described in Keyfitz (1985) indicated, will result in only a 3–5-year increase in lifespan—a moderate effect. Because caloric restriction has a negligible effect on metabolic stability, it exerts no effect on the rate of ageing and hence induces no changes to the senescence process.
Mice are classical examples of opportunistic species and are therefore defined by low entropy: early sexual maturity, large litter size and a maximum lifespan potential of four years. The low entropy condition suggests that the stability of their metabolic networks is close to the minimal condition. Because caloric restriction increases longevity by increasing metabolic stability, caloric restriction can be expected to influence the rate of ageing and thereby induce changes in both the maximum and mean lifespan. Studies on mice are consistent with this prediction (Weindruch & Walford, 1988).
...any efforts to exploit mouse systems to elucidate human ageing or disease must take into account the vast differences in the metabolic stability of the cells within these animals
I should point out here the ongoing studies of caloric restriction on lifespan in rhesus monkeys that indicate lower disease risk and less incidence of age-related diseases in caloric restriction monkeys compared with controls (Roth et al, 2004). Rhesus monkeys share 92% gene homology and many biological similarities with humans in the profile of ageing. These similarities have suggested to many researchers that caloric restriction in humans may result in increases in mean and maximum lifespan that are comparable with the increases observed in rhesus monkeys. I contend, however, that although rhesus monkeys and humans are both equilibrium species and should therefore have comparable ageing patterns, the effect of caloric restriction on mean and maximum lifespan in the two species will be quantitatively distinct because they show significant differences in life history. In rhesus monkeys, sexual maturity occurs at 3–5 years of age, mean lifespan is 25 years and maximum lifespan is estimated to be 40 years. Hence, rhesus monkeys have lower evolutionary entropy, and consequently a weaker metabolic stability, than humans. This analysis thus predicts that caloric restriction will induce changes in longevity in rhesus monkeys that are less pronounced than in mice, but significantly more pronounced than any changes that may occur in humans.
Mouse models of human senescence have become a central element in many areas of biomedical research. The significance of this system derives from its experimental accessibility and the fact that mice share organ systems, systemic physiology and genes with humans. However, any efforts to exploit mouse systems to elucidate human ageing or disease must take into account the vast differences in the metabolic stability of the cells within these animals. These differences derive from the contrasting evolutionary history of the species as determined by their environmental conditions and the resource constraints that these conditions induce. An understanding of this history, and its signature at the cellular level—the metabolic stability, or robustness of the cellular regulatory network—are thus crucial in elucidating human ageing and disease pathogenesis from mouse models.

References
- Austad SN (1997) Comparative aging and life histories in mammals. Exp Gerontol 32: 22–38 [DOI] [PubMed] [Google Scholar]
- Balmain A, Harris CC (2000) Carcinogenesis in mouse and human cells: parallels and paradoxes. Carcinogenesis 21: 371–377 [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN (1998) The free radical theory of aging matures. Physiol Rev 78: 547–581 [DOI] [PubMed] [Google Scholar]
- Cao SX, Dhahbi JM, Mote PL, Spindler SR (2001) Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice. Proc Natl Acad Sci USA 98: 10630–10635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG (1984) Antioxidants, aging and longevity. In Pryor WA (ed) Free Radicals in Biology (vol 6) pp371–428. New York, NY, USA: Academic [Google Scholar]
- Demetrius L (1997) Directionality principles in thermodynamics and evolution. Proc Natl Acad Sci USA 94: 3491–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetrius L (2004) Caloric restriction, metabolic rate, and entropy. J Gerontol A Biol Sci Med Sci 59: B902–B915 [DOI] [PubMed] [Google Scholar]
- DePinho RA (2000) The age of cancer. Nature 408: 248–254 [DOI] [PubMed] [Google Scholar]
- Dhahbi JM, Mote PL, Wingo J, Rowley BC, Cao SX, Walford RL, Spindler SR (2001) Caloric restriction alters the feeding response of key metabolic enzyme genes. Mech Ageing Dev 122: 1033–1048 [DOI] [PubMed] [Google Scholar]
- Dragan YP, Pitot HC (1992) Multistage hepatocarcinogenesis in the rat: insights into risk estimation. Prog Clin Biol Res 374: 261–279 [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61: 759–767 [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408: 239–247 [DOI] [PubMed] [Google Scholar]
- Holliday R (1996) Neoplastic transformation: the contrasting stability of human and mouse cells. Cancer Surv 28: 103–115 [PubMed] [Google Scholar]
- Kapahi P, Boulton ME, Kirkwood TB (1999) Positive correlation between mammalian life span and cellular resistance to stress. Free Radic Biol Med 26: 495–500 [DOI] [PubMed] [Google Scholar]
- Keyfitz N (1985) Applied Mathematical Demography. New York, NY, USA: Springer [Google Scholar]
- Piantanelli L (1988) Cancer and aging: from the kinetics of biological parameters to the kinetics of cancer incidence and mortality. Ann NY Acad Sci 521: 99–109 [DOI] [PubMed] [Google Scholar]
- Rangarajan A, Weinberg RA (2003) Comparative biology of mouse versus human cells: modelling human cancer in mice. Nat Rev Cancer 3: 952–959 [DOI] [PubMed] [Google Scholar]
- Roth GS, Mattison JA, Ottinger MA, Chachich ME, Lane MA, Ingram DK (2004) Aging in rhesus monkeys: relevance to human health interventions. Science 305: 1423–1426 [DOI] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R (1996) Oxidative stress, caloric restriction, and aging. Science 273: 59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speakman JR, Talbot DA, Selman C, Snart S, McLaren JS, Redman P, Krol E, Jackson DM, Johnson MS, Brand MD (2004) Uncoupled and surviving: individual mice with high metabolism have greater mitochondrial uncoupling and live longer. Aging Cell 3: 87–95 [DOI] [PubMed] [Google Scholar]
- Weindruch R, Walford RL (1988) The Retardation of Aging and Disease by Dietary Restriction. Springfield, IL, USA: CC Thomas [Google Scholar]