

Abstract
Focal adhesion kinase (FAK) was first identified as a viral Src (v-Src) substrate, but the role of FAK in Src transformation events remains undefined. We show that stable expression of the FAK C-terminal domain (termed FRNK) in v-Src-transformed NIH 3T3 fibroblasts inhibited cell invasion through Matrigel and blocked experimental metastases in nude mice without effects on cell motility. FRNK inhibitory activity was dependent upon its focal contact localization. FRNK expression disrupted the formation of a v-Src-FAK signaling complex, inhibited p130Cas tyrosine phosphorylation, and attenuated v-Src-stimulated ERK and JNK kinase activation. However, FRNK did not affect v-Src-stimulated Akt activation, cell growth in soft agar, or subcutaneous tumor formation in nude mice. FRNK-expressing cells exhibited decreased matrix metalloproteinase-2 (MMP-2) mRNA levels and MMP-2 secretion. Transient FRNK expression in human 293 cells inhibited exogenous MMP-2 promoter activity and overexpression of wild-type but not catalytically-inactive (Ala-404) MMP-2 rescued v-Src-stimulated Matrigel invasion in the presence of FRNK. Our findings show the importance of FAK in Src-stimulated cell invasion and support a role for Src-FAK signaling associated with elevated tumor cell metastases.
Keywords: cell invasion/FAK/matrix/metalloproteinase/metastasis/Src
Introduction
FAK is a non-receptor protein-tyrosine kinase (PTK) that facilitates integrin signaling, regulating cell cycle progression, cell survival and cell migration (reviewed in Parsons et al., 2000; Zhao and Guan, 2000; Schaller, 2001). FAK is a major tyrosine-phosphorylated protein in adherent cells, and the transient recruitment of Src-family PTKs to FAK is one of the best measures of integrin-stimulated FAK activation. Studies with human tumor tissues and tumor-derived cell lines have shown that enhanced tumorigenicity in vivo is correlated with elevated FAK expression, tyrosine phosphorylation and c-Src association with FAK (Cance et al., 2000; Hecker et al., 2002).
In Rous sarcoma virus (v-Src)-transformed chicken fibroblasts, FAK was identified as a highly tyrosine-phosphorylated protein that directly associated with v-Src (Schaller et al., 1992). Both Src homology-2 (SH2)- and SH3-binding interactions stabilize the v-Src–FAK signaling complex (Hauck et al., 2001a). Although v-Src is a potent oncogene (reviewed in Martin, 2001; Frame et al., 2002) and FAK-associated proteins such as p130Cas are important mediators of Src-stimulated cell growth (Honda et al., 1998), the role of a v-Src–FAK complex remains undefined, as studies with FAK-null cells showed that FAK was not essential for enhanced v-Src-stimulated cell growth (Roy et al., 2002).
A number of studies have shown that FAK localization with integrins at cellular focal contact sites is required for maximal FAK activation. FAK focal contact localization is mediated by FAK C-terminal domain interactions with integrin-associated proteins (Tachibana et al., 1995; Klingbeil et al., 2001). The FAK C-terminal domain is also expressed as a separate mRNA transcript termed FRNK in chicken fibroblasts (Schaller et al., 1993; Nolan et al., 1999) and in smooth muscle cells after vascular injury (Taylor et al., 2001). FRNK functions as a potent dominant-negative inhibitor of FAK activity by competitively preventing FAK localization to focal contacts (Richardson and Parsons, 1996). In this manner, FRNK exerts inhibitory effects on cell cycle progression (Zhao and Guan, 2000), cell migration (Hauck et al., 2001b; Taylor et al., 2001) and cell survival (Xu et al., 2000).
Here, we test the importance of FAK in v-Src- stimulated cell transformation through the dominant-negative effects of FRNK expression. We found that stable FRNK expression prevented v-Src-stimulated cell invasion in vitro and experimental metastases in vivo. FRNK localization to focal contacts disrupted the v-Src– FAK signaling complex, inhibited v-Src-stimulated ERK2 and JNK activation and reduced the gene transcription of targets such as matrix metalloproteinase-2 (MMP-2). Our studies support the hypothesis that increased Src–FAK signaling in transformed cells is connected to the generation of an invasive cell phenotype.
Results
FRNK inhibits v-Src-stimulated cell invasion but not cell motility
To determine the significance of v-Src–FAK association with respect to cell transformation, FRNK was stably expressed as a dominant-negative inhibitor of FAK function in NIH-3T3 fibroblasts transformed by the Schmidt–Ruppin D v-src expression plasmid psrc11 (v-Src3T3s). Since pooled populations of v-Src3T3s were heterogeneous for FRNK expression (data not shown), cell clones were obtained and expanded, and three that expressed equivalent levels of FRNK (termed R2, A2 and O4) were selected for further analyses (Figure 1A). Stable FRNK expression levels are approximately equal to endogenous FAK levels in v-Src3T3s (data not shown). The ability of FRNK to act as a dominant-negative inhibitor of FAK depends upon its localization to focal contact sites, and this can be inactivated by a point mutation (Leu1034 to serine in FAK) that de-stabilizes the focal adhesion targeting domain and disrupts paxillin binding (Sieg et al., 1999; Hayashi et al., 2002). Since FRNK S-1034 is a stable cytoplasmically distributed protein, v-Src3T3s expressing FRNK S-1034 were used as a control for FRNK (Figure 1A).

Fig. 1. Stable FRNK expression in v-Src3T3s does not inhibit cell motility. (A) Equal protein lysates of either NIH-3T3 or v-Src3T3 fibroblasts were analyzed by blotting with a v-Src-specific mAb (top panel) and re-probed with an HA tag mAb to visualize FRNK (clones R2, A2 and O4) or FRNK S-1034 expression (lower panel). (B) c-Src and v-Src were immunoprecipitated (mAb 2-17) from lysates of serum-starved cells and were analyzed for associated in vitro kinase (IVK) activity using GST–FAK-CT as a substrate. (C) Haptotaxis motility was assayed with immobilized FN and the indicated cells. Random cell motility was measured in the presence of BSA. Values are means ± SD from four experiments. (D) Chemotaxis motility was assayed on collagen-coated membranes with the indicated cells using a serum stimulus added to the lower chamber. Random cell motility was measured in the presence of BSA. Values are means ± SD from three experiments. (E) Wound-healing scratch motility assays were performed with the indicated cells plated onto Matrigel-coated glass slides in the presence of serum. Cell migration was assessed by 1 mm grid comparisons of four image sets at 0 and 24 h; representative images are shown.
Compared with mock-transfected v-Src3T3s, equivalent v-Src expression and in vitro kinase activities were present in FRNK clones R2 and A2 (Figure 1A and B). Lower levels of v-Src expression and in vitro kinase activity were detected in FRNK clone O4 compared with v-Src3T3s (Figure 1B). Analyses of fibronectin (FN)-stimulated haptotaxis motility as measured by Boyden chamber assays revealed that v-Src3T3s exhibited reduced migration compared with normal NIH-3T3 fibroblasts (Figure 1C). This result may be related to the weakening of cell contact sites either through v-Src-enhanced β1-integrin cytoplasmic domain phosphorylation (Sakai et al., 2001) or effects of enhanced protease secretion from v-Src-transformed cells (Datta et al., 2001). Importantly, FRNK expression in v-Src3T3s did not inhibit haptotaxis motility (Figure 1C), serum-stimulated chemotaxis motility (Figure 1D) or two-dimensional motility on top of Matrigel as performed in scratch-type assays (Figure 1E) compared with control v-Src3T3s. However, when analyzed for three-dimensional cell invasion activity through a polymerized Matrigel or collagen type I barrier (Figure 2), FRNK inhibited v-Src3T3 cell invasion in all clones analyzed. Notably, cell invasion was stimulated by v-Src as NIH-3T3 fibroblasts were motile (Figure 1), but these cells displayed only low invasive activity (Figure 2). FRNK S-1034 did not inhibit v-Src- stimulated cell invasion through either Matrigel or collagen type I barriers (Figure 2). Together, these results support the conclusion that FRNK inhibition of cell invasion occurs through a mechanism that is independent of effects on cell motility within a v-Src-transformed cell background.

Fig. 2. FRNK inhibits v-Src-stimulated three-dimensional cell invasion. (A) Matrigel (30 µg) invasion assays were performed with the indicated cells for 24 h using a serum stimulus in the lower chamber. Values are means ± SD from four experiments. No cell invasion was detected in the presence of BSA. (B) Representative images (60×) of the lower porous membrane surface from Matrigel invasion assays. Crystal violet-stained cells can be distinguished from the 8 µm membrane pores. (C) Cell invasion through polymerized collagen type I was assayed with the indicated cells for 24 h using a serum stimulus in the lower chamber. Values are means ± SD from two independent experiments. Random invasion activity was assayed in the presence of BSA.
Disruption of the v-Src–FAK signaling complex by FRNK
When expressed at high levels in chicken or rat fibroblasts, v-Src promotes the loss of focal contacts and actin stress fibers with the concomitant formation of actin-rich ventral contact sites termed podosomes (Tarone et al., 1985; Meijne et al., 1997). v-Src localizes to both podosomes and perimeter focal contact sites (Fincham et al., 1996; Hauck et al., 2002). v-Src3T3s form podosomes when plated onto FN in the absence of serum, whereas they form actin stress fibers and focal contacts when plated onto FN in the presence of serum (data not shown). FRNK-expressing v-Src3T3s exhibit a fusiform cell shape and form podosomes or focal contacts on FN in a manner identical to v-Src3T3s (Figure 3A). FRNK expression did not affect v-Src distribution in v-Src3T3s (data not shown). However, FRNK expression in many cell systems promotes FAK dephosphorylation potentially by displacement of FAK from focal contact sites (Parsons et al., 2000). To evaluate the effect of FRNK on FAK tyrosine phosphorylation, N-terminally directed antibodies were used to isolate endogenous FAK and comparative analyses were performed between FRNK clone R2 (herein termed v-Src FRNK) and v-Src FRNK S-1034 cells, as they are equal for v-Src expression and activity (see Figure 1A and B). Lower levels of both FAK-associated in vitro kinase activity and v-Src co-immunoprecipitation (IP) were detected in lysates from v-Src FRNK compared with both v-Src3T3s and v-Src FRNK S-1034 cells (Figure 3B). Since equal levels of FAK were isolated in the IP analyses, these results support the conclusion that FRNK expression disrupts the signaling complex formed between v-Src and FAK.

Fig. 3. FRNK inhibits the formation of a v-Src–FAK signaling complex, promotes FAK and p130Cas dephosphorylation, but does not affect cell morphology. (A) v-Src3T3 FRNK cells plated onto FN in the presence or absence of serum visualized by phase, actin staining with FITC–phalloidin or indirect staining for focal contact-associated vinculin. The scale bar is 30 µm. (B) FAK was immunoprecipitated (IP) from the indicated serum-starved cells and analyzed for 32P-associated in vitro kinase (IVK) activity. The FAK IPs were resolved by SDS–PAGE, transferred to a PVDF membrane and exposed to film. Equal levels of FAK and the co-immunoprecipitation of v-Src were visualized by blotting. (C) FAK IPs from the indicated serum-starved cells were analyzed sequentially with anti-phosphotyrosine (pTyr) or with phospho-specific antibodies to FAK Tyr397 (pY397), Tyr861 (pY861) or Tyr925 (pY925). FAK levels were determined by FAK blotting, and these analyses were performed on two sets of IPs. (D) p130Cas IPs from the indicated cells were analyzed sequentially by anti-P.Tyr and p130Cas blotting.
As shown previously (Schlaepfer and Hunter, 1996) and evaluated through the use of phospho-specific antibodies to FAK, FAK Tyr397 can be highly phosphorylated in starved NIH-3T3 cells (Figure 3C) under conditions where FAK-associated kinase activity is very low (Figure 3B). Therefore, the analysis of FAK Tyr397 phosphorylation is not always an accurate measure of FAK activation. However, v-Src transformation of NIH-3T3 fibroblasts results in elevated FAK tyrosine phosphorylation at sites in the C-terminal domain such as Tyr861 (Calalb et al., 1996) and the Grb2 SH2-binding site at Tyr925 (Schlaepfer et al., 1994). FRNK expression in v-Src3T3s inhibited FAK phosphorylation at Tyr861 and Tyr925, but not FAK phosphorylation at Tyr397 compared with v-Src3T3s (Figure 3C). Notably, FRNK also inhibited the tyrosine phosphorylation of FAK-associated proteins such as p130Cas (Figure 3D). Importantly, FRNK S-1034 expression did not significantly decrease v-Src-enhanced FAK or p130Cas tyrosine phosphorylation (Figure 3C and D). These results indicate that FRNK disrupts the v-Src–FAK signaling complex and promotes the site-specific inhibition of v-Src-mediated FAK and p130Cas tyrosine phosphorylation.
FRNK inhibits v-Src-stimulated JNK1 and ERK2 but not Akt activation
As FRNK expression in v-Src3T3s selectively reduced p130Cas tyrosine phosphorylation and FAK phosphorylation at Tyr925, and as these events have been connected to JNK/SAP kinase and ERK/MAP kinase activation, respectively (Dolfi et al., 1998; Schlaepfer et al., 1998), blotting comparisons of whole-cell lysates were made with phospho-specific antibodies to activated ERKs, JNKs and Akt (Figure 4A–C). Under serum-starved conditions, v-Src promoted the strong activation of ERK2, JNK1 and Akt kinases in v-Src3T3s compared with the lack of signaling pathway activation in NIH-3T3s (Figure 4A–C, lanes 1 and 2). In v-Src FRNK cells, the level of activated ERK2 and JNK1 detected by blotting was reduced compared with v-Src3T3s and v-Src FRNK S-1034 cells (Figure 4A and B, lanes 2–4). ERK2 in vitro kinase activity was reduced ∼2-fold, whereas JNK kinase activity was reduced ∼3-fold in cells expressing FRNK compared with v-Src3T3 controls (Figure 4A and B). However, FRNK was not acting as a general inhibitor of v-Src-stimulated signaling events, as the level of activated Akt detected by blotting and in vitro kinase assays was equivalent in FRNK, FRNK S-1034 and v-Src3T3 cells (Figure 4C).

Fig. 4. FRNK inhibits v-Src-stimulated ERK2 and JNK1 but not Akt activation. FRNK does not affect v-Src-stimulated cell growth in vitro. (A–C) Lysates were made from the indicated serum-starved cells. Blotting analyses were performed on whole-cell lysates with phospho-specific antibodies to either activated ERK (pERK), JNK (pJNK) or Akt (pAkt). The corresponding blots were stripped and reprobed with polyclonal antibodies to ERK2, JNK1 and Akt, respectively. (A) ERK2 IPs from the indicated cells were analyzed for in vitro kinase activity by the phosphorylation of myelin basic protein (MBP). (B) IPs using polyclonal antibodies to JNK1 and JNK2 from the indicated cell lysates were analyzed for in vitro kinase activity using GST–ATF-2 as a substrate. (C) Akt IPs from the indicated cells were analyzed for in vitro kinase activity by the phosphorylation of GST–GSK-3. Values in (A–C) are means ± SD from two experiments. (D) Growth of the indicated cells on gelatin-coated dishes in 0.5% serum. Cell counts were performed every 24 h in triplicate. Values are means ± SD from two independent experiments. NIH-3T3 (diamonds), v-Src3T3 (open squares), v-Src FRNK (open triangles) and v-Src FRNK S-1034 (filled circles). (E) Anchorage-independent cell growth in soft agar was evaluated by plating the indicated cells in 10% serum within 0.3% agar on a solidified base. Images show typical fields at 60×.
FRNK does not block v-Src-stimulated cell growth or tumor formation
To determine whether FRNK-mediated inhibition of v-Src-stimulated ERK or JNK activity affected cell growth or survival, analyses measuring serum-independent and anchorage-independent growth of v-Src3T3s were performed (Figure 4D and E). Under 0.5% serum conditions, v-Src3T3s but not normal NIH-3T3 cells continued to proliferate and form foci (Figure 4D). No differences in either cell growth or foci formation after 12 days were observed for the v-Src3T3s, v-Src FRNK or v-Src FRNK S-1034 cells in 0.5% serum (Figure 4D) or in normal 10% serum conditions (data not shown).
Although JNK (Turkson et al., 1999) and ERK2 activation (Penuel and Martin, 1999) have been implicated in promoting v-Src-stimulated cell growth, v-Src FRNK cells exhibited robust anchorage-independent growth in soft agar (Figure 4E). No differences in either colony number or size were observed between v-Src FRNK, v-Src FRNK S-1034 or v-Src3T3 cells. Since in vitro cell proliferation does not always correlate with cell growth in vivo, solid tumor formation was analyzed by subcutaneous injection of cells into athymic nude mice (Figure 5A). v-Src FRNK cells grew equally well as a solid tumor mass compared with both v-Src FRNK S-1034 and v-Src3T3s, whereas NIH-3T3 cells did not form tumors. Blotting analyses of tumor tissue revealed that equivalent levels of v-Src, FRNK and FRNK S-1034 expression were maintained in vivo (Figure 5B). Together, these results support the conclusion that FRNK selectively inhibited v-Src-stimulated cell invasion through a molecular mechanism that was independent of growth-promoting signaling pathways.

Fig. 5. FRNK inhibits experimental metastasis but not v-Src-stimulated solid tumor growth. (A) Athymic nude mice were injected subcutaneously with the indicated cells and, after 10 days, visible tumor size was measured with calipers. Values are means ± SD from two experiments with eight animals per group. NIH-3T3 (diamonds), v-Src3T3 (open squares), v-Src FRNK (open triangles) and v-Src FRNK S-1034 (filled circles). (B) Protein lysates from the indicated subcutaneously grown tumor tissue were analyzed by FAK (top), v-Src (middle) or HA tag blotting (lower). (C) Representative Bouin’s-fixed lungs from mice 4 weeks after injection with the indicated cells. Multiple tumor nodules on the lung surface can be seen in v-Src3T3- and v-Src FRNK S-1034-injected animals. The scale bar is 1 cm. (D) Representative H&E-stained mouse lung sections from animals injected with the indicated cells. Metastatic tumor cells (arrows) and blood vessels (B) are indicated. (E) Blotting analyses of lung tissue after 4 weeks from animals injected with the indicated cells. FAK blotting was performed on protein lysates (top), v-Src was detected by immunoprecipitation and blotting (middle), and FRNK was detected by HA tag immunoprecipitation and blotting (lower). (F) HA tag blotting analysis was performed on protein lysates of lung tumor nodules from either v-Src FRNK- or v-Src FRNK S-1034-injected mice. The M number is used to identify mice.
FRNK expression prevents v-Src-stimulated experimental metastasis
Since the in vitro Matrigel invasion assays do not fully reconstitute the multiple steps of tumor cell metastasis in vivo (Curran and Murray, 2000), experimental metastases assays were also performed. Solid tumor growth assays were stopped after 19 days due to skin ulceration and, at this point, no lung or organ metastases were detected in animals (data not shown). Therefore, cells were tail vein injected and experimental lung metastases were evaluated after 4 weeks. A total of 75% (12/16) of v-Src3T3- and 84% (16/19) of v-Src FRNK S-1034-injected animals exhibited multiple and large tumor nodules on the lung surface (Figure 5C; Table I). Thin sectioning and staining of lungs revealed tumor cell growth in areas close to blood vessels for v-Src3T3- and v-Src FRNK S-1034- but not for v-Src FRNK- or NIH-3T3 cell-injected animals (Figure 5D).
Table I. Subcutaneous tumor growth and experimental lung metastases in athymic nude mice.
| Cell line | Subcutaneous growth |
Lung metastases |
||||
|---|---|---|---|---|---|---|
| Mice with tumors/total mice | Average diameter (mm) | Mice with visible nodules | (%) | Average No. of surface nodules | FRNK expression in nodules (%) | |
| NIH-3T3 | 0/4 | 0 | 0/12 | 0 | 0 | N/A |
| v-Src3T3 | 16/16 | 15.5 ± 0.6 | 12/16 | 75 | 6.2 | N/A |
| v-Src FRNK | 16/16 | 15.6 ± 2.0 | 3/16 | 19 | 1 | 0 |
| v-Src FRNK S-1034 | 16/16 | 14.2 ± 2.1 | 16/19 | 84 | 11 | 100 |
The average diameter of subcutaneous tumors was determined 19 days after injection. Visible lung tumor nodules (1–10 mm in size) were counted under a stereomicroscope 4 weeks after tail vein injection. HA tag blotting of excised tumor nodule tissue was evaluated for FRNK expression. N/A, not applicable.
Notably, only 19% (3/16) of v-Src FRNK-injected animals exhibited macroscopic metastatic tumor nodules on the lung surface (Table I) and the average number of surface tumor nodules per lung lobe in v-Src FRNK-injected animals was 10-fold less than in mice injected with v-Src FRNK S-1034 cells (Figure 5C; Table I). As a more sensitive means to detect microscopic metastases, protein extracts from lung tissue were analyzed by either v-Src or hemagglutinin (HA) tag blotting analyses (Figure 5E). Equivalent endogenous FAK expression was used as a loading control. Importantly, v-Src expression was detected in the lung tissue from v-Src3T3- and v-Src FRNK S-1034-injected animals, and HA tag blotting also revealed strong FRNK S-1034 expression (Figure 5E). However, neither v-Src nor FRNK were detected in the lung tissue from v-Src FRNK-injected animals (Figure 5E).
Since FRNK expression was maintained in the cells grown as subcutaneous tumors (Figure 5B), HA tag blotting analyses were also performed with protein extracts from two individual lung surface tumor nodules isolated from either v-Src FRNK- or v-Src FRNK S-1034-injected animals (Figure 5F). Whereas v-Src was detected equally in the tumor nodules (data not shown), surprisingly, FRNK expression was not detected (Figure 5F). This result contrasts with the 100% frequency of detectable FRNK S-1034 expression in multiple lung tumor nodules analyzed (Figure 5F; Table I). Since experimental metastases are believed to arise from the colonization of single cells, we speculate that the low frequency of metastases observed after v-Src FRNK cell injection may represent cells within the clonal population that ceased to express FRNK.
FRNK inhibits MMP-2 secretion and expression
For cell invasion and experimental metastasis, cells must be able to proliferate and migrate, and be capable of degrading and moving through extracellular matrix barriers (Curran and Murray, 2000). v-Src can promote increased expression of gelatinases (Hamaguchi et al., 1995) and collagenases (Vincenti et al., 1998) involved in breaking down tissue barriers. By gelatin zymography of conditioned media, there is increased gelatinase A (or MMP-2) and gelatinase B or (MMP-9) secretion from equal numbers of v-Src3T3 compared with NIH-3T3 cells (Figure 6A). FRNK-expressing clones R2, A2 and O4 showed reduced secretion of MMP-2 compared with v-Src3T3s, as determined by both zymography and blot ting of conditioned media (Figure 6B). In contrast, no differences in MMP-2 secretion were observed for v-Src FRNK S-1034 compared with v-Src3T3s (Figure 6B). Proteolytic activation of 72 kDa proMMP-2 occurs in part through the actions of a membrane-associated MMP, termed MT1-MMP (Nagase and Woessner, 1999). v-Src promotes increased MT1-MMP expression that cleaves MMP-2 to an ∼64 kDa active form (Kadono et al., 1998). Whereas low levels of active MMP-2 were detected by zymography of FRNK R2-conditioned media (Figure 6B), no differences in MT1-MMP expression were detected bet ween v-Src3T3s and FRNK-expressing clones (Figure 6B).

Fig. 6. FRNK inhibits v-Src-stimulated MMP-2 secretion. (A) Gelatin zymography of conditioned media from equal numbers of NIH-3T3 or v-Src3T3 fibroblasts. The positions of proMMP9, proMMP-2 and active MMP-2 are indicated. (B) Conditioned medium from the indicated cells was analyzed by gelatin zymography and MMP-2 blotting. Whole-cell lysates from the indicated cells were analyzed by MT1-MMP blotting. (C) Conditioned medium from the indicated cells was analyzed for soluble gelatinase activity. Values are expressed as a percentage of an activated MMP-2 standard. (D) Whole-cell lysates from the indicated cells were analyzed for soluble gelatinase activity. (E) Matrigel invasion assays were performed with v-Src3T3s in the presence of recombinant human TIMP-2 at the concentration indicated in the top chamber. Values are means ± SD from two independent experiments.
Since MMP activity is also regulated by TIMP binding (Nagase and Woessner, 1999) and the dissociation of TIMP–MMP complexes during gel electrophoresis prior to zymography assays acts to enhance the apparent activity of proMMP isoforms, soluble gelatinase activity in the cell-conditioned media samples was assayed using a peptide substrate (Figure 6C). Both v-Src3T3 and v-Src FRNK S-1034 cells contained high levels of soluble gelatinase activity, whereas NIH-3T3 and the various v-Src FRNK cell clones had ∼4-fold lower levels of gelatinase activity secreted from the same number of cells (Figure 6C). Analysis of whole-cell lysates also revealed that FRNK expression resulted in lower levels of cell-associated gelatinase activity (Figure 6D). Although blotting analyses did not reveal significant changes in TIMP expression (data not shown), addition of recombinant TIMP-2 to v-Src3T3s inhibited Matrigel invasion activity in a dose-dependent manner (Figure 6E). Taken together, these results support the hypothesis that FRNK may block v-Src-stimulated cell invasion through the inhibition of MMP-2 secretion and/or production.
To determine whether FRNK inhibition of MMP-2 secretion was associated with lower levels of MMP-2 expression, semi-quantitative RT–PCRs were performed with total RNA samples isolated from v-Src3T3s and FRNK-expressing cells (Figure 7A). Compared with v-Src3T3s and normalized to β-actin and GAPDH levels (values set to 1), MMP-2 mRNA levels were 2.5- to 3.3-fold lower in FRNK-expressing cells. The v-Src FRNK S-1034 cells contained MMP-2 mRNA levels equivalent to those of v-Src3T3s, and no significant differences were observed for MMP-9 mRNA levels in the cells analyzed (Figure 7A). To evaluate whether FRNK expression directly inhibited MMP-2 mRNA production, transient transfection assays in human 293T cells were performed with a 1686 bp fragment of the rat MMP-2 promoter region fused to a luciferase reporter (Figure 7B). This pT4-Luc-1686 construct exhibited high basal activity (Harendza et al., 1995), and v-Src co-transfection did not result in enhanced luciferase production (data not shown). However, co-transfection of FRNK inhibited MMP-2 promoter activity ∼4 fold in a dose-dependent manner (Figure 7B), and increased FRNK expression promoted endogenous FAK dephosphorylation in 293T cells (data not shown). Co-transfection of FRNK S-1034 did not significantly (P = 0.09) inhibit MMP-2 promoter activity. Since MMP-2 mRNA differences as determined by RT–PCR (Figure 7A) parallel the amount of both MMP activity and secreted protein detected in cell-conditioned media (Figure 6B) and also correlate with the invasive activity of v-Src3T3s (Figure 2), our combined results support the hypothesis that FRNK expression blocks v-Src3T3 cell invasion through the inhibition of MMP-2 gene expression.

Fig. 7. FRNK inhibits MMP-2 gene expresion. (A) Semi-quantitative RT–PCR analysis was performed with RNA isolated from the indicated cells using primer pairs to amplify MMP-2 (449 bp), MMP-9 (433 bp), β-actin (302 bp) or GAPDH (265 bp). The intensity of either the MMP-2 or MMP-9 bands was normalized to both β-actin and GAPDH signals. Values for v-Src3T3s were set to 1. (B) Luciferase activity from a 1686 bp region of the MMP-2 promoter (pT4-Luc-1686) or the empty luciferase vector (pGL2-Basic). Constructs were transiently transfected into human 293T cells along with increasing concentrations of FRNK or FRNK S-1034. Results are expressed as the ratio of luciferase versus Renilla activity. Values are means ± SD from two experiments.
Inhibition of JNK blocks v-Src-stimulated cell invasion and MMP-2 secretion, but not cell motility
The regulation of MMP gene expression and secretion involves the convergence of multiple signaling pathways (Nagase and Woessner, 1999). Since FRNK expression inhibited both v-Src-stimulated ERK2 and JNK activity, pharmacological inhibitors of these signaling pathways were tested for effects on v-Src3T3 cell invasion (Figure 8A). Treatment of cells with 25 µM PD98059 MEK1 inhibitor reduced v-Src-stimulated ERK2 in vitro kinase activity ∼2-fold and blocked ERK2 phosphorylation as detected by an activation-specific monoclonal antibody (mAb) (Figure 8B). However, treatment of v-Src3T3s with 25 µM PD98059 did not alter v-Src3T3 cell invasion through Matrigel (Figure 8A) and did not inhibit cell motility in scratch assays (data not shown). However, treatment of v-Src3T3s with the SP600125 JNK inhibitor reduced v-Src3T3 cell invasion in a dose-dependent manner (Figure 8A). At 25 µM, SP600125 reduced v-Src3T3 cell invasion 4-fold (Figure 8A), inhibited JNK in vitro kinase activity ∼2.5-fold (Figure 8C) and blocked JNK1 phosphorylation as detected by an activation-specific mAb (Figure 8C). Importantly, treatment of cells with 25 µM of either the SP600125 or PD98059 inhibitors did not cross-inhibit either v-Src-stimulated ERK2 or JNK activity, respectively (Figure 8B and C). As treatment of v-Src3T3s with SP600125 also did not inhibit cell motility in scratch assays performed on Matrigel-coated slides (data not shown), analyses of MMP activity in v-Src3T3-conditioned media showed that incubation of cells with 25 µM SP600125 for 18 h resulted in reduced MMP-2 secretion (Figure 8D). These results support the hypothesis that v-Src–FAK signaling through JNK is an important pathway regulating MMP-2 secretion and cell invasion.
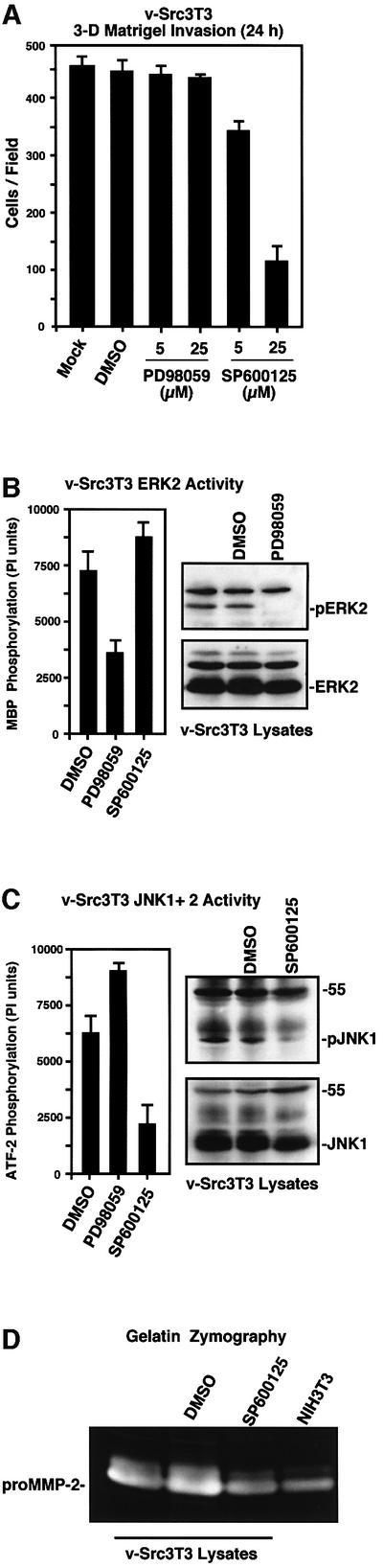
Fig. 8. JNK activity is important for v-Src-stimulated cell invasion and MMP-2 secretion. (A) Matrigel invasion assays were performed with v-Src3T3s in the presence of DMSO or the indicated concentrations of MEK1 (PD98059) or JNK (SP600125) inhibitors added to both chambers. Values are means ± SD from two experiments. (B and C) v-Src3T3s were mock treated with DMSO or incubated in the presence of 25 µM of either PD98059 or SP600125 for 18 h in starved conditions. Protein lysates were analyzed for (B) ERK2 in vitro kinase activity or (C) JNK in vitro kinase activity. Values are means ± SD from two experiments. Whole-cell lysates were blotted for either activated ERK (pERK) or activated JNK (pJNK1) and re-probed for either total ERK2 or JNK1 protein expression, respectively. (D) Conditioned media from either serum-starved, mock-treated (DMSO) or 25 µM SP600125-treated (18 h) v-Src3T3s were analyzed by gelatin zymography. A conditioned medium sample from the same number of serum-starved NIH-3T3 fibroblasts is shown for comparison.
MMP-2 overexpression rescues FRNK blockage to cell invasion
Since MMP-2 is probably one of several v-Src-stimulated targets that are regulated in part by a FAK–JNK signaling pathway, epitope-tagged human MMP-2 or a catalytically inactive (Ala404) point mutant (Morgunova et al., 1999) were stably overexpressed to test whether increased MMP-2 expression could reverse the FRNK blockage of cell invasion (Figure 9). Pooled populations of v-Src3T3s and v-Src FRNK cells showed equal levels of exogenous secreted His-tagged MMP-2 and His-tagged Ala404 MMP-2 by blotting analyses of conditioned media (Figure 9A). No changes in either v-Src or FRNK expression were detected in cells overexpressing MMP-2 (Figure 9A). Importantly, incubation of the 72 kDa pro-form of His-MMP-2 with the catalytic domain of MT1-MMP showed that this recombinant MMP-2 protein could be cleaved and activated efficiently, as detected by both blotting and zymography analyses (Figure 9B).

Fig. 9. MMP-2 overexpression rescues the FRNK-mediated blockage of cell invasion. (A) Conditioned media samples from the indicated cells stably overexpressing and secreting either MMP-2 or catalytically inactive MMP-2 (Ala404) were analyzed by His tag blotting (top). v-Src (middle) or HA tag (lower) blotting of whole-cell lysates from the indicated cells. (B) In vitro cleavage and activation of His-tagged MMP-2 by incubation with the catalytic domain of MT1-MMP as visualized by MMP-2 blotting (top) and gelatin zymography (lower). (C) Conditioned media samples from the indicated cells were analyzed by gelatin zymography. The positions of proMMP-9, proMMP-2 and active MMP-2 are indicated. (D) v-Src was immunoprecipitated from the indicated serum-starved cell lysates, and associated in vitro kinase activity (IVK) was measured by GST–FAK-CT phosphorylation. (E) Matrigel (30 µg) invasion assays were performed with the indicated cells for 24 h. Values are means ± SD from two independent experiments. No cell invasion was detected in the presence of BSA.
Notably, the amount of proMMP-2 secreted and active MMP-2 generated from both v-Src3T3s and v-Src FRNK cells was enhanced by wild-type but not Ala404 MMP-2 expression (Figure 9C). This result indicates that FRNK did not block the increased secretion and proteolytic activation of overexpressed His-tagged proMMP-2. When analyzed in Matrigel invasion assays, MMP-2 overexpression in v-Src FRNK cells promoted a >3-fold increase in cell invasion through Matrigel compared with v-Src FRNK cells (Figure 9E). This corresponded to an ∼70% rescue in v-Src FRNK cell invasion compared with v-Src3T3s. Importantly, Ala404 MMP-2 expression did not increase v-Src FRNK cell invasion and no significant invasion differences were observed in v-Src3T3 cells expressing either wild-type or Ala404 MMP-2 (Figure 9E). Since no changes in v-Src in vitro kinase activity were detected in cells overexpressing MMP-2 (Figure 9D), these results support the conclusion that the inhibitory effects of FRNK on v-Src-stimulated cell invasion were due in large part to reduced MMP-2 expression.
Discussion
Through the stable expression of the FAK C-terminal domain, termed FRNK, in v-Src3T3s, we show that FRNK functioned as a specific inhibitor of v-Src-stimulated cell invasion in vitro and experimental metastasis in vivo. Quite surprisingly, FRNK expression did not inhibit cell motility detectably in v-Src transformed cells. Addition ally, FRNK was not acting as a general inhibitor of v-Src-mediated cell transformation events as there were no detectable effects of FRNK on v-Src kinase activity, v-Src-stimulated cell growth in low serum, anchorage-independent growth in soft agar or solid tumor formation in nude mice. The inhibitory effects of FRNK were connected to the disruption of an activated v-Src–FAK signaling complex, as FRNK expression inhibited the site-specific phosphorylation of FAK at Tyr861 and Tyr925 as well as v-Src-stimulated p130Cas tyrosine phosphorylation. Importantly, we show that the inhibitory effects of FRNK could be inactivated by a point mutation (Leu1034 to serine) that prevents FRNK localization to focal contacts and the stable expression of which did not inhibit FAK or p130Cas tyrosine phosphorylation.
Studies have shown that the v-Src SH3 domain is important for targeting v-Src to focal contacts (Frame et al., 2002). Gain-of-function point mutations within the v-Src SH3 RT loop also promote the binding to target proteins containing a PXXPXXΦ motif (where Φ is a hydrophobic residue) in addition to normal c-Src type I or type II SH3-binding motifs (Hauck et al., 2001a). FAK contains three PXXPXXΦ motifs, and comparisons of Src–/– cells transformed by various v-Src isoforms showed that gain-of-function mutations in the v-Src SH3 domain acted to stabilize the v-Src–FAK signaling complex and enhance Matrigel invasion activity (Hauck et al., 2002). Notably, in Src–/– cells transformed by a v-Src isoform lacking gain-of-function SH3 mutations responsible for enhanced FAK association, Matrigel invasion activity was severely attenuated. However, adenoviral-mediated FAK overexpression facilitated the formation of a Src–FAK signaling complex, enhanced invadopodia projections and increased Matrigel invasion 9-fold in these cells (Hauck et al., 2002). Importantly, adenoviral- mediated FAK overexpression did not promote an invasive phenotype of normal Src–/– cells. Taken together with our current results using FRNK as a dominant-negative inhibitor of FAK within v-Src3T3s, our combined studies support the conclusion that Src–FAK signaling is connected to the generation of an invasive cell phenotype in transformed cells.
FRNK did not inhibit the motility or proliferation of v-Src3T3s
Previous studies have shown that FRNK expression in normal fibroblast (Sieg et al., 1999, 2000), smooth muscle (Hauck et al., 2000) or human carcinoma cells (Hauck et al., 2001b; Slack et al., 2001) blocked both integrin- and growth factor-stimulated cell motility. However, our studies showed that FRNK did not inhibit v-Src3T3 motility as measured by both Boyden chamber and wound-healing assays. We speculate that this surprising result may be associated with the maintenance of FAK Tyr397 phosphorylation in v-Src3T3 cells expressing FRNK at levels equivalent to endogenous FAK expression. Notably, high levels of adenoviral-mediated FRNK expression promote FAK Tyr397 dephosphorylation and inhibit cell motility (Hauck et al., 2001b; Slack et al., 2001). Since FAK Tyr397 phosphorylation is required for promoting cell motility and a number of different SH2 domain-containing proteins bind to this site (Schlaepfer et al., 1999), it is likely that FAK function was not completely inhibited by the level of FRNK expression in v-Src3T3s. This conclusion is supported by the findings that adenoviral-mediated FRNK overexpression in v-Src- transformed primary mouse fibroblasts inhibited cell motility (D.Hsia, unpublished results).
Although FRNK expression has been connected to the inhibition of cell proliferation in carcinoma cells (Aguirre Ghiso, 2002), other cell lines such as immortalized smooth muscle cells (Hauck et al., 2000), chicken embryo fibroblasts (Schaller et al., 1993) or N1E-115 neuroblastoma cells (Zhou and Song, 2001) either endogenously express or can tolerate exogenous FRNK expression without effects on cell proliferation. We found that FRNK did not inhibit v-Src3T3 cell growth in low serum, soft agar or as a tumor mass in nude mice. This result is consistent with the finding that FAK is not essential for Src-enhanced cell growth as determined using FAK–/– cells (Roy et al., 2002). Although FRNK expression inhibited v-Src-stimulated ERK2/MAP kinase and JNK/SAP kinase activation in v-Src3T3s, studies have shown that multiple signaling pathways contribute to v-Src-stimulated cell proliferation (Odajima et al., 2000). In particular, as v-Src can also bind directly to the p85 subunit of phosphatidylinositol (PI) 3′-kinase and stimulate PI 3′-kinase activity (Liu et al., 1993), it is possible that the strong activation of downstream targets of PI 3′-kinase such as Akt contributes to the anchorage-independent growth of v-Src3T3 cells expressing FRNK.
FAK signaling regulates MMP expression
FRNK disruption of the v-Src–FAK signaling complex inhibited the v-Src-mediated phosphorylation of FAK Tyr861 and FAK Tyr925, and the tyrosine phosphorylation of p130Cas. FAK Tyr861 phosphorylation is important for FAK association with integrins (Eliceiri et al., 2002), FAK Tyr925 is important for FAK signaling to ERK2/MAP kinase (Schlaepfer et al., 1998), whereas p130Cas tyrosine phosphorylation has been connected to JNK/SAP kinase activation (Dolfi et al., 1998). Pharmacological inhibition of JNK prevented v-Src3T3 cell invasion through Matrigel but not cell motility, whereas pharmacological inhibition of MEK1 upstream of ERK2 did not block v-Src3T3 invasion. FRNK inhibited v-Src-stimulated JNK activity, and this pathway can regulate MMP expression (Han et al., 2001). Both FRNK expression and JNK inhibition resulted in reduced MMP-2 secretion from v-Src3T3s. To test the importance of MMP-2 secretion to v-Src3T3 cell invasion, we found that overexpression of wild-type but not catalytically inactive MMP-2 resulted in an ∼70% rescue of cell invasion activity in v-Src3T3 cells expressing FRNK.
Previous studies have shown that FRNK overexpression can inhibit MMP-9 and MMP-2 secretion from human carcinoma cells (Hauck et al., 2001b) and that FAK re-expression in FAK-null cells increases the secretion of these MMPs (Sein et al., 2000). The regulation of MMP-2 gene expression is complex, and studies have shown that JNK-responsive transcription factor-binding sites contribute to constitutive MMP-2 expression (Qin et al., 1999). As shown by RT–PCR, FRNK expression resulted in lower levels of MMP-2 but not MMP-9 mRNA in v-Src3T3s. Additionally, transient transfection of FRNK inhibited constitutive MMP-2 promoter activity in 293T cells under conditions promoting endogenous FAK dephosphorylation. Whereas previous studies have implicated FAK signaling in promoting elevated MMP-2 secretion (Zhang et al., 2002), our studies support a role for FAK in promoting increased MMP-2 gene expression leading to increased MMP-2 protein production and secretion, and enhanced cell invasion.
Role of FAK in tumorigenesis
In the absence of tumor immune responses to Rous sarcoma virus-associated antigens, v-Src can promote malignancy and rapid metastasis in chicken wing tissues (Stoker and Sieweke, 1989; Taylor et al., 1994). v-Src transformation of many different mammalian cell types has shown that this oncogene strongly promotes cell invasion in vitro and experimental metastases in vivo (Behrens et al., 1993; Aguirre-Ghiso et al., 1999). Whereas our current study was focused upon the molecular mechanism of the v-Src–FAK signaling in fibroblasts, our results also may portend a potential role for Src–FAK signaling in the context of tumorigenesis. Notably, FAK expression and tyrosine phosphorylation are elevated as a function of increased human tumor cell malignancy (Cance et al., 2000). Activating Src mutations are found in malignant progression of human colon cancer (Irby et al., 1999), Src activation contributes to the metastatic spread of carcinoma cells (Boyer et al., 2002), and Src–FAK signaling is implicated in malignant astrocytoma tumor growth (Hecker et al., 2002). Since FAK is a key regulator of normal cytotrophoblast- mediated invasion of the uterus during placental formation (Ilic et al., 2001), and FRNK expression potently inhibited both v-Src3T3 cell invasion activity in vitro and experimental metastasis formation in vivo, our findings support the conclusion that FAK signaling promotes cell invasion in both normal developmental and neoplastic cell settings. In conclusion, our studies warrant either the testing of FAK antisense treatments (Hauck et al., 2001b) or the development of inhibitors of FAK activity in the control of tumorigenesis.
Materials and methods
Cells, antibodies and reagents
v-Src3T3s transformed with the pSrc11 plasmid (Johnson et al., 1985) were maintained as described previously (Schlaepfer et al., 1994). Anti-P.Tyr (4G10) mAb and avian-specific mAb to v-Src (EC10) were from Upstate Biotechnology, anti-His tag mAb was from Qiagen, anti-HA-epitope tag mAb (16B12) was from Covance Research, and anti-p130Cas mAb was from BD/Transduction Laboratories. Anti-ERK2 mAb (B3B9), anti-c-Src mAb (2-17) and anti-HA epitope tag mAb (12CA5) ascites were used as described previously (Schlaepfer et al., 1998). Polyclonal antibodies to c-Src (Src-2), p130Cas (C20), MMP-2 (C19), MMP-9 (C20), JNK1 (C17), ERK2 (C14) and Akt1 (C20) were from Santa Cruz Biotechnology, and polyclonal antibodies to MT1-MMP (AB8102) were from Chemicon. Phospho-specific mAbs to activated ERK2 (E10 to pT202/pY204), activated JNK1 (G9 to pT183/pY185) and activated Akt (pS473) were from Cell Signaling Technology. Phospho-specific antibodies to FAK (FAK pY397, FAK pY861 and FAK pY925) were from BioSource International. Affinity-purified polyclonal antibodies to the N- and C-terminal domains of FAK were used as described previously (Sieg et al., 1999). Recombinant human TIMP-2 and the catalytic domain of MT1-MMP were purchased from Chemicon. Pharmacological inhibitors to MEK1 (PD98059) and JNK1 (SP600125) were purchased from Calbiochem.
DNA constructs and cell transfection
Triple HA-tagged constructs for FRNK and S-1034 FRNK in pCDNA3.1 (Hauck et al., 2001a) were transfected into v-Src3T3s using Effectene (Qiagen) and selected for growth in hygromycin B (250 µg/ml). Single cells were isolated by light scatter fluorescence-activated cell sorting (FACS) into 96-well culture dishes, and clonal cell lines were established. Human MMP-2 cDNA containing a secretion signal sequence followed by an internal His6 tag was cloned into pCEP-Puro. Catalytically inactive MMP-2 was generated by replacement of Glu404 with alanine as described previously (Morgunova et al., 1999). All constructs were confirmed by DNA sequence analyses.
Immunoprecipitation and immunoblotting
Cells were solubilized in modified RIPA lysis buffer (Schlaepfer et al., 1998) containing 1% Triton X-100, 1% sodium deoxycholate and 0.1% SDS. Antibodies (2.5 µg) were incubated with lysates for 3 h at 4°C and collected by binding to protein G-plus (Oncogene Research Products) or protein A (Repligen) agarose beads. Blotting and sequential membrane re-probing were performed as described previously (Schlaepfer et al., 1998).
In vitro kinase (IVK) assays
For FAK and Src, assays were initiated by addition of [γ-32P]ATP to immuno-isolated proteins and incubated at 32°C for 15 min in the presence (Src IVK) or absence (FAK IVK) of a Src substrate (GST–FAK-CT) (Schlaepfer et al., 1994). ERK2 IVK activity was measured using myelin basic protein, JNK IVK activity was measured using GST–ATF2, and Akt IVK activity was measured using GST–GSK-3 as described previously (Schlaepfer et al., 1998; Klingbeil et al., 2001). Following SDS–PAGE and electrophoretic transfer to PVDF membranes (Millipore), labeled proteins were visualized by autoradiography and the equal recovery of the immuno-isolated kinase was verified by blotting.
Migration and invasion assays
MilliCell (12 mm diameter with 8 µm pores) chambers (Millipore) were pre-coated on the membrane underside with 10 µg/ml FN (Sigma) for 2 h at room temperature for haptotaxis assays or the whole chamber was coated with 5 µg/ml rat tail collagen (Boehringer Mannheim) for 24 h at 4°C for chemotaxis assays. For invasion assays, growth factor-reduced Matrigel (BD Biosciences) was diluted (30 µg in 100 µl of H2O), added to the top chamber, allowed to gel for 1 h at 37°C, air-dried for 16 h, and the Matrigel barrier was reconstituted with 100 µl of Dulbecco’s modified Eagle’s medium (DMEM) for 2 h at 37°C prior to use. For collagen type I invasion, neutralized (240 µg in 100 µl) bovine dermal collagen (Vitrogen) was added to the top chamber, allowed to gel, dried and reconstituted as described above. For all assays, serum-starved cells were collected by limited trypsin treatment followed by the addition of soybean trypsin inhibitor as described previously (Sieg et al., 1999). Cells were added to the upper compartment (1 × 105 cells in 300 µl) in migration medium [DMEM with 0.5% bovine serum albumin (BSA)] and the chambers were placed into 24-well culture dishes containing 0.4 ml of migration medium for integrin-stimulated motility or with DMEM with 10% serum for chemotaxis motility and invasion assays, respectively. Haptotaxis controls were performed with BSA-coated membranes, whereas migration medium in the lower chamber served as the control for chemotaxis and invasion assays. After 4 (migration) or 24 h (invasion) at 37°C, migratory cells on the lower membrane surface were fixed by treatment with 2% formaldehyde/0.5% glutaraldehyde. Cells were stained (0.1% crystal violet, 0.1 M borate pH 9.0 and 2% ethanol) and migration values were determined either by dye elution (in 10% acetic acid) or by counting five high-power (40×) fields/chamber. Mean values were obtained from at least three individual chambers for each experimental point per assay.
For scratch motility assays, etched grid coverslips (Bellco) were pre-coated with 30 µg/ml Matrigel in phosphate-buffered saline (PBS) for 2 h at 37°C, rinsed, placed into a 35 mm dish, and 1 × 106 cells in 3 ml were allowed to adhere in migration medium for 6 h. Cells were wounded with a pipet tip, rinsed, photographed (0 h) and incubated in DMEM with 10% serum. After 24 h, cells were fixed in paraformaldehyde, rinsed and photographed using the same grid markers.
Gelatinase activity
A total of 5 × 106 cells were plated onto Matrigel-coated (30 µg/ml) 10 cm dishes in growth medium for 8 h, and then incubated in 5 ml of Opti-MEM (Gibco-BRL) in the absence of serum at 37°C for 18 h. The conditioned medium was collected, clarified by centrifugation, separated in non-reducing gels containing 0.1% (w/v) gelatin, and processed for zones of clearing activity by zymography as described previously (Hauck et al., 2001b). Purified human MMP-2 and murine MMP-9 (Chemicon) were used as migration standards. For blotting analyses, secreted proteins in the conditioned medium were precipitated by cold acetone and separated by SDS–PAGE. Both conditioned medium and whole-cell lysates were analyzed for soluble MMP-2 + MMP-9 activity using the Chemicon Gelatinase Activity kit (ECM700) as per the manufacturer’s instructions. Sample activity is expressed as a percentage of control (10 µl of p-aminophenylmercuric acetate-activated human MMP-2 as provided). In vitro activation of purified His-tagged MMP-2 was performed by incubation (1 h at 37°C) with the recombinant catalytic domain of MT1-MMP (MT1:MMP-2 ratio 1:10) as described previously (English et al., 2001).
RT–PCR
Cells were incubated in Opti-MEM overnight and total RNA was isolated using an RNeasy kit (Qiagen). Reverse transcription was carried out using 5 µg of RNA, oligo(dT) primers and the SuperScript first strand cDNA synthesis kit (Gibco-BRL). The method of primer dropping PCR was used as a semi-quantitative means to analyze MMP RNA levels. Briefly, primer pairs (R&D systems) were used to amplify MMP-2 (449 bp), MMP-9 (433 bp), β-actin (302 bp) and GAPDH (265 bp) PCR products. Range-finding preliminary PCR experiments were used to determine the subsaturating levels of PCR amplification for each primer pair so that the products remain proportional to the amount of target mRNA. Twenty-three cycles were used to amplify the GAPDH and β-actin products, MMP-2 was amplified using 30 cycles, and 35 cycles were used to amplify MMP-9. Co-amplification of MMP-2 or MMP-9 with β-actin and GAPDH was carried out by adding β-actin and GAPDH primer pairs after PCR cycle 7 or cycle 12, respectively. Digitized images of the PCR products were quantified using ImageQuant (Molecular Dynamics).
MMP-2 promoter activity
We used 1686 bp of the 5′ rat MMP-2 promoter region cloned into the promoterless luciferase pGL2-basic vector (pT4-Luc-1686) as described previously (Harendza et al., 1995). Human 293 T cells were transiently transfected with either 0.5 µg of pGL-2-basic or pT4-Luc-1686 with either 0.5 µg pCDNA3.1 or increasing amounts of pCDNA3.1 FRNK or pCDNA3.1 FRNK S-1034 up to 0.5 µg using Lipofectamine Plus (Gibco-BRL). A 20 ng aliquot of pTK-Renilla luciferase (Promega) was included in all transfections. Cells were harvested after 36 h and luciferase activity was assessed using the dual-luciferase assay kit (Promega). Final values were corrected for transfection efficiency as determined by Renilla activity.
Cell growth and soft agar assays
Cells were serum starved overnight and 1 × 104 cells were plated onto gelatin-coated (0.1% in PBS) dishes in DMEM containing 0.5 or 10% serum. Every 24 h, cells were collected after trypsin treatment and counted. For measurement of anchorage-independent growth, 4 × 104 cells were suspended in 0.3% agar (Difco), seeded onto a solidified base of growth medium containing 0.6% agar and overlaid with 1 ml of growth medium. Colonies were scored after 3 weeks, and all values were determined in triplicate.
Tumor growth and experimental metastasis
Athymic WeHi nude mice (6–8 weeks old) were injected subcutaneously with 5 × 105 cells in 200 µl of DMEM and, after 7 days, tumor growth was monitored every other day by caliper measurement. After 19 days, the tumors were removed, weighed, measured and snap frozen. For experimental metastases assays, 6 × 105 cells in 200 µl of DMEM were injected into the tail vein and, after 4 weeks, the lungs were removed, analyzed for macroscopic tumor nodule formation and either snap frozen or fixed in Bouin’s solution. For histochemical analyses, lungs were paraffin embedded, thin sectioned and mounted onto glass slides, and microscopic metastases were visualized after hematoxylin and eosin (H&E) staining. Detection of v-Src or FRNK protein expression in subcutaneous tumors or in lung tissue was by western blotting after protein extraction in modified RIPA buffer containing 0.5% SDS.
Statistical analysis
Ordinary one-way analysis of variance (ANOVA) was used to determine significance within data groups.
Acknowledgments
Acknowledgements
We thank Michael Rühlmann for his advice on animal experiments, Sigrid Harendza and David Lovett for the MMP-2-luciferase reporter construct, Vito Quaranta for critical reading of the manuscript, and Amanda Moore for administrative assistance. Care for the mice, surgery and injection protocols were carried out according to institutional and National Institutes of Health guidelines. C.H. was supported in part by a fellowship from the Deutsche Forschungsgemeinschaft (HA-2856/1-1) and X.P. was supported by a long-term fellowship from the Human Frontier Science Program. This work was supported by grants from the National Cancer Institute to D.A.C. (CA50286, CA45726 and CA78045) and to D.D.S. (CA75240 and CA87038). This work was initiated with support from the American Cancer Society (RPG-98-109-TBE) to D.D.S. This is manuscript number 14601-IMM from The Scripps Research Institute.
References
- Aguirre-Ghiso J.A. (2002) Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene, 21, 2513–2524. [DOI] [PubMed] [Google Scholar]
- Aguirre-Ghiso J.A., Frankel,P., Farias,E.F., Lu,Z., Jiang,H., Olsen,A., Feig,L.A., de Kier Joffe,E.B. and Foster,D.A. (1999) RalA requirement for v-Src- and v-Ras-induced tumorigenicity and overproduction of urokinase-type plasminogen activator: involvement of metallo proteases. Oncogene, 18, 4718–4725. [DOI] [PubMed] [Google Scholar]
- Behrens J., Vakaet,L., Friis,R., Winterhager,E., Van Roy,F., Mareel,M.M. and Birchmeier,W. (1993) Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/β-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J. Cell Biol., 120, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer B., Bourgeois,Y. and Poupon,M.F. (2002) Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene, 21, 2347–2356. [DOI] [PubMed] [Google Scholar]
- Calalb M.B., Zhang,X., Polte,T.R. and Hanks,S.K. (1996) Focal adhesion kinase tyrosine-861 is a major site of phosphorylation by Src. Biochem. Biophys. Res. Commun., 228, 662–668. [DOI] [PubMed] [Google Scholar]
- Cance W.G., Harris,J.E., Iacocca,M.V., Roche,E., Yang,X., Chang,J., Simkins,S. and Xu,L. (2000) Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin. Cancer Res., 6, 2417–2423. [PubMed] [Google Scholar]
- Curran S. and Murray,G.I. (2000) Matrix metalloproteinases: molecular aspects of their roles in tumour invasion and metastasis. Eur. J. Cancer, 36, 1621–1630. [DOI] [PubMed] [Google Scholar]
- Datta A., Shi,Q. and Boettiger,D.E. (2001) Transformation of chicken embryo fibroblasts by v-src uncouples β1 integrin-mediated outside-in but not inside-out signaling. Mol. Cell. Biol., 21, 7295–7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolfi F., Garcia-Guzman,M., Ojaniemi,M., Nakamura,H., Matsuda,M. and Vuori,K. (1998) The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc. Natl Acad. Sci. USA, 95, 15394–15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliceiri B.P., Puente,X.S., Hood,J.D., Stupack,D.G., Schlaepfer,D.D., Huang,X.Z., Sheppard,D. and Cheresh,D.A. (2002) Src-mediated coupling of focal adhesion kinase to integrin α(v)β5 in vascular endothelial growth factor signaling. J. Cell Biol., 157, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English W.R., Holtz,B., Vogt,G., Knauper,V. and Murphy,G. (2001) Characterization of the role of the ‘MT-loop’: an eight-amino acid insertion specific to progelatinase A (MMP2) activating membrane-type matrix metalloproteinases. J. Biol. Chem., 276, 42018–42026. [DOI] [PubMed] [Google Scholar]
- Fincham V.J., Unlu,M., Brunton,V.G., Pitts,J.D., Wyke,J.A. and Frame,M.C. (1996) Translocation of Src kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. J. Cell Biol., 135, 1551–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame M.C., Fincham,V.J., Carragher,N.O. and Wyke,J.A. (2002) v-Src′s hold over actin and cell adhesions. Nat. Rev. Mol. Cell Biol., 3, 233–245. [DOI] [PubMed] [Google Scholar]
- Hamaguchi M., Yamagata,S., Thant,A.A., Xiao,H., Iwata,H., Mazaki,T. and Hanafusa,H. (1995) Augmentation of metalloproteinase (gelatinase) activity secreted from Rous sarcoma virus-infected cells correlates with transforming activity of src. Oncogene, 10, 1037–1043. [PubMed] [Google Scholar]
- Han Z., Boyle,D.L., Chang,L., Bennett,B., Karin,M., Yang,L., Manning,A.M. and Firestein,G.S. (2001) c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J. Clin. Invest., 108, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harendza S., Pollock,A.S., Mertens,P.R. and Lovett,D.H. (1995) Tissue-specific enhancer–promoter interactions regulate high level constitutive expression of matrix metalloproteinase 2 by glomerular mesangial cells. J. Biol. Chem., 270, 18786–18796. [DOI] [PubMed] [Google Scholar]
- Hauck C.R., Hsia,D.A. and Schlaepfer,D.D. (2000) FAK facilitates PDGF-BB stimulated ERK2 activation required for chemotaxis migration of vascular smooth muscle cells. J. Biol. Chem., 275, 41092–41099. [DOI] [PubMed] [Google Scholar]
- Hauck C.R., Hunter,T. and Schlaepfer,D.D. (2001a) The v-Src SH3 domain facilitates a cell adhesion-independent association with focal adhesion kinase. J. Biol. Chem., 276, 17653–17662. [DOI] [PubMed] [Google Scholar]
- Hauck C.R., Sieg,D.J., Hsia,D.A., Loftus,J.C., Gaarde,W.A., Monia,B.P. and Schlaepfer,D.D. (2001b) Inhibition of focal adhesion kinase expression or activity disrupts epidermal growth factor-stimulated signaling promoting the migration of invasive human carcinoma cells. Cancer Res., 61, 7079–7090. [PubMed] [Google Scholar]
- Hauck C.R., Hsia,D.A., Ilic,D. and Schlaepfer,D.D. (2002) v-Src SH3-enhanced interaction with focal adhesion kinase at β1 integrin-containing invadopodia promotes cell invasion. J. Biol. Chem., 277, 12487–12490. [DOI] [PubMed] [Google Scholar]
- Hayashi I., Vuori,K. and Liddington,R.C. (2002) The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat. Struct. Biol., 9, 101–106. [DOI] [PubMed] [Google Scholar]
- Hecker T.P., Grammer,J.R., Gillespie,G.Y., Stewart,J.,Jr and Gladson,C.L. (2002) Focal adhesion kinase enhances signaling through the Shc/extracellular signal-regulated kinase pathway in anaplastic astrocytoma tumor biopsy samples. Cancer Res., 62, 2699–2707. [PubMed] [Google Scholar]
- Honda H. et al. (1998) Cardiovascular anomaly, impaired actin bundling and resistance to Src-induced transformation in mice lacking p130Cas. Nat. Genet., 19, 361–365. [DOI] [PubMed] [Google Scholar]
- Ilic D., Genbacev,O., Jin,F., Caceres,E., Almeida,E.A.C., Bellingard-Dubouchaud,V., Schaefer,E.M., Damsky,C.H. and Fisher,S.J. (2001) Plasma membrane-associated pY397-FAK is a marker of cytotrophoblast invasion in vivo and in vitro. Am. J. Pathol., 159, 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irby R.B. et al. (1999) Activating SRC mutation in a subset of advanced human colon cancers. Nat. Genet., 21, 187–190. [DOI] [PubMed] [Google Scholar]
- Johnson P.J., Coussens,P.M., Danko,A.V. and Shalloway,D. (1985) Overexpressed pp60c-src can induce focus formation without complete transformation of NIH 3T3 cells. Mol. Cell. Biol., 5, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadono Y., Okada,Y., Namiki,M., Seiki,M. and Sato,H. (1998) Transformation of epithelial Madin–Darby canine kidney cells with p60(v-src) induces expression of membrane-type 1 matrix metalloproteinase and invasiveness. Cancer Res., 58, 2240–2244. [PubMed] [Google Scholar]
- Klingbeil C.K., Hauck,C.R., Hsia,D.A., Jones,K.C., Reider,S.R. and Schlaepfer,D.D. (2001) Targeting Pyk2 to β1-integrin-containing focal contacts rescues fibronectin-stimulated signaling and haptotactic motility defects of focal adhesion kinase-null cells. J. Cell Biol., 152, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Marengere,L.E., Koch,C.A. and Pawson,T. (1993) The v-Src SH3 domain binds phosphatidylinositol 3′-kinase. Mol. Cell. Biol., 13, 5225–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G.S. (2001) The hunting of the Src. Nat. Rev. Mol. Cell Biol., 2, 467–475. [DOI] [PubMed] [Google Scholar]
- Meijne A.M., Ruuls-Van Stalle,L., Feltkamp,C.A., McCarthy,J.B. and Roos,E. (1997) v-src-induced cell shape changes in rat fibroblasts require new gene transcription and precede loss of focal adhesions. Exp. Cell Res., 234, 477–485. [DOI] [PubMed] [Google Scholar]
- Morgunova E., Tuuttila,A., Bergmann,U., Isupov,M., Lindqvist,Y., Schneider,G. and Tryggvason,K. (1999) Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science, 284, 1667–1670. [DOI] [PubMed] [Google Scholar]
- Nagase H. and Woessner,J.F.,Jr (1999) Matrix metalloproteinases. J. Biol. Chem., 274, 21491–21494. [DOI] [PubMed] [Google Scholar]
- Nolan K., Lacoste,J. and Parsons,J.T. (1999) Regulated expression of focal adhesion kinase-related nonkinase, the autonomously expressed C-terminal domain of focal adhesion kinase. Mol. Cell. Biol., 19, 6120–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odajima J. et al. (2000) Full oncogenic activities of v-Src are mediated by multiple signaling pathways. Ras as an essential mediator for cell survival. J. Biol. Chem., 275, 24096–24105. [DOI] [PubMed] [Google Scholar]
- Parsons J.T., Martin,K.H., Slack,J.K., Taylor,J.M. and Weed,S.A. (2000) Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene, 19, 5606–5613. [DOI] [PubMed] [Google Scholar]
- Penuel E. and Martin,G.S. (1999) Transformation by v-Src: Ras–MAPK and PI3K-mTOR mediate parallel pathways. Mol. Biol. Cell, 10, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H., Sun,Y. and Benveniste,E.N. (1999) The transcription factors Sp1, Sp3 and AP-2 are required for constitutive matrix metalloproteinase-2 gene expression in astroglioma cells. J. Biol. Chem., 274, 29130–29137. [DOI] [PubMed] [Google Scholar]
- Richardson A. and Parsons,J.T. (1996) A mechanism for regulation of the adhesion-associated protein tyrosine kinase pp125FAK. Nature, 380, 538–540. [DOI] [PubMed] [Google Scholar]
- Roy S., Ruest,P.J. and Hanks,S.K. (2002) FAK regulates tyrosine phosphorylation of CAS, paxillin and PYK2 in cells expressing v-Src, but is not a critical determinant of v-Src transformation. J. Cell. Biochem., 84, 377–388. [DOI] [PubMed] [Google Scholar]
- Sakai T., Jove,R., Fassler,R. and Mosher,D.F. (2001) Role of the cytoplasmic tyrosines of β1A integrins in transformation by v-Src. Proc. Natl Acad. Sci. USA, 98, 3808–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M.D. (2001) Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim. Biophys. Acta, 1540, 1–21. [DOI] [PubMed] [Google Scholar]
- Schaller M.D., Borgman,C.A., Cobb,B.S., Vines,R.R., Reynolds,A.B. and Parsons,J.T. (1992) pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl Acad. Sci. USA, 89, 5192–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M.D., Borgman,C.A. and Parsons,J.T. (1993) Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol. Cell. Biol., 13, 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D.D. and Hunter,T. (1996) Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol. Cell. Biol., 16, 5623–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hanks,S.K., Hunter,T. and van der Geer,P. (1994) Integrin-mediated signal transduction linked to Ras pathway by Grb2 binding to focal adhesion kinase. Nature, 372, 786–791. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D., Jones,K.C. and Hunter,T. (1998) Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src and FAK-initiated tyrosine phosphorylation events. Mol. Cell. Biol., 18, 2571–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hauck,C.R. and Sieg,D.J. (1999) Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol., 71, 435–478. [DOI] [PubMed] [Google Scholar]
- Sein T.T., Thant,A.A., Hiraiwa,Y., Amin,A.R., Sohara,Y., Liu,Y., Matsuda,S., Yamamoto,T. and Hamaguchi,M. (2000) A role for FAK in the concanavalin A-dependent secretion of matrix metalloproteinase-2 and -9. Oncogene, 19, 5539–5542. [DOI] [PubMed] [Google Scholar]
- Sieg D.J., Hauck,C.R. and Schlaepfer,D.D. (1999) Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci., 112, 2677–2691. [DOI] [PubMed] [Google Scholar]
- Sieg D.J., Hauck,C.R., Ilic,D., Klingbeil,C.K., Schaefer,E., Damsky,C.H. and Schlaepfer,D.D. (2000) FAK integrates growth factor and integrin signals to promote cell migration. Nat. Cell Biol., 2, 249–256. [DOI] [PubMed] [Google Scholar]
- Slack J.K., Adams,R.B., Rovin,J.D., Bissonette,E.A., Stoker,C.E. and Parsons,J.T. (2001) Alterations in the focal adhesion kinase/Src signal transduction pathway correlate with increased migratory capacity of prostate carcinoma cells. Oncogene, 20, 1152–1163. [DOI] [PubMed] [Google Scholar]
- Stoker A.W. and Sieweke,M.H. (1989) v-src induces clonal sarcomas and rapid metastasis following transduction with a replication-defective retrovirus. Proc. Natl Acad. Sci. USA, 86, 10123–10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana K., Sato,T., D′Avirro,N. and Morimoto,C. (1995) Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J. Exp. Med., 182, 1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarone G., Cirillo,D., Giancotti,F.G., Comoglio,P.M. and Marchisio,P.C. (1985) Rous sarcoma virus-transformed fibroblasts adhere primarily at discrete protrusions of the ventral membrane called podosomes. Exp. Cell Res., 159, 141–157. [DOI] [PubMed] [Google Scholar]
- Taylor J.M., Mack,C.P., Nolan,K., Regan,C.P., Owens,G.K. and Parsons,J.T. (2001) Selective expression of an endogenous inhibitor of FAK regulates proliferation and migration of vascular smooth muscle cells. Mol. Cell. Biol., 21, 1565–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R.L. Jr, England,J.M., Kopen,G.C., Christou,A.A. and Halpern,M.S. (1994) Major histocompatibility (B) complex control of the formation of v-src-induced metastases. Virology, 205, 569–573. [DOI] [PubMed] [Google Scholar]
- Turkson J. et al. (1999) Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol. Cell. Biol., 19, 7519–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenti M.P., Schroen,D.J., Coon,C.I. and Brinckerhoff,C.E. (1998) v-src activation of the collagenase-1 (matrix metalloproteinase-1) promoter through PEA3 and STAT: requirement of extracellular signal-regulated kinases and inhibition by retinoic acid receptors. Mol. Carcinog., 21, 194–204. [PubMed] [Google Scholar]
- Xu L.H., Yang,X., Bradham,C.A., Brenner,D.A., Baldwin,A.S.,Jr, Craven,R.J. and Cance,W.G. (2000) The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. J. Biol. Chem., 275, 30597–30604. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Thant,A.A., Hiraiwa,Y., Naito,Y., Sein,T.T., Sohara,Y., Matsuda,S. and Hamaguchi,M. (2002) A role for focal adhesion kinase in hyluronan-dependent MMP-2 secretion in a human small-cell lung carcinoma cell line, QG90. Biochem. Biophys. Res. Commun., 290, 1123–1127. [DOI] [PubMed] [Google Scholar]
- Zhao J.H. and Guan,J.L. (2000) Role of focal adhesion kinase in signaling by the extracellular matrix. Prog. Mol. Subcell. Biol., 25, 37–55. [DOI] [PubMed] [Google Scholar]
- Zhou D. and Song,Z.H. (2001) CB1 cannabinoid receptor-mediated neurite remodeling in mouse neuroblastoma N1E-115 cells. J. Neurosci. Res., 65, 346–353. [DOI] [PubMed] [Google Scholar]