

Abstract
Arteri-, corona-, toro- and roniviruses are evolutionarily related positive-strand RNA viruses, united in the order Nidovirales. The best studied nidoviruses, the corona- and arteriviruses, employ a unique transcription mechanism, which involves discontinuous RNA synthesis, a process resembling similarity-assisted copy-choice RNA recombination. During infection, multiple subgenomic (sg) mRNAs are transcribed from a mirror set of sg negative-strand RNA templates. The sg mRNAs all possess a short 5′ common leader sequence, derived from the 5′ end of the genomic RNA. The joining of the non-contiguous ‘leader’ and ‘body’ sequences presumably occurs during minus-strand synthesis. To study whether toroviruses use a similar transcription mechanism, we characterized the 5′ termini of the genome and the four sg mRNAs of Berne virus (BEV). We show that BEV mRNAs 3–5 lack a leader sequence. Surprisingly, however, RNA 2 does contain a leader, identical to the 5′-terminal 18 residues of the genome. Apparently, BEV combines discontinuous and non-discontinous RNA synthesis to produce its sg mRNAs. Our findings have important implications for the understanding of the mechanism and evolution of nidovirus transcription.
Keywords: discontinuous transcription/nidovirus/RNA recombination/subgenomic mRNA synthesis/torovirus
Introduction
Positive (+)-strand RNA viruses have developed a wide range of strategies to express their genes. One recurring strategy, employed by many non-related viruses, entails the synthesis of subgenomic (sg) mRNAs (Miller and Koev, 2000). While the genome invariably serves as the mRNA for the viral replicase, the sgRNAs mediate the expression of structural and accessory proteins from cistrons, downstream of the replicase gene. These sg mRNAs are always 3′-coterminal with the viral genome but, with regard to their synthesis, important differences exist among the various virus groups. For some viruses, e.g. brome mosaic virus (Miller et al., 1985), transcription initiation of sg mRNAs occurs at internal promoter sequences on a genome-length, negative (–)-strand template. Alternatively, as proposed for red clover necrotic mosaic virus (Sit et al., 1998) and tomato bushy stunt virus (Choi et al., 2001), sg (–)strand RNAs, resulting from premature transcription–termination, may serve as templates. The Nidovirales, pathogens of vertebrates and invertebrates, are unique among (+)strand RNA viruses: the best studied nidoviruses, i.e. the corona- and arteriviruses, employ an unusual transcription mechanism, involving discontinuous RNA synthesis.
The Nidovirales, i.e. the toro-, corona-, arteri- and okaviruses, differ markedly in genome size, virion architecture and host range. Yet, their common ancestry is evident from sequence identity in their replicase proteins and from similarities in genome organization, gene order and replication strategy (Cowley et al., 2000; reviewed by Snijder et al., 1993; de Vries et al., 1997). Toro- and coronaviruses, because of their close evolutionary relationship (Snijder et al., 1990a), have been united taxonomically in the family Coronaviridae as separate genera (Cavanagh et al., 1993); the more distantly related arteri- and okaviruses (den Boon et al., 1991; Cowley et al., 2000) are assigned to the monogeneric families Arteriviridae and Roniviridae, respectively.
For all nidoviruses, the 5′-most two-thirds of the genome are taken up by the replicase polyprotein gene. Downstream, there are multiple smaller genes, which are expressed from a nested set of up to eight sg mRNAs. In the case of the arteri- and coronaviruses, these sg mRNAs are chimeric and both 3′- and 5′-coterminal with the genome: they all possess a 5′ common leader sequence, which is derived from the 5′ terminus of the genomic RNA and fused to the ‘body’ of the transcript (i.e. the 3′-terminal part, which carries the coding information). Coronavirus leaders are 55–92 nucleotides in length, whereas those of arteriviruses measure ∼200 nucleotides. The leader and mRNA body sequences are joined within a short conserved sequence motif, the transcription-regulating sequence (TRS), which precedes each transcription unit (van der Most and Spaan, 1995; de Vries et al., 1997; Lai and Cavanagh, 1997; Snijder and Meulenberg, 1998). A TRS is also found immediately downstream of the genomic leader sequence. Direct evidence that the TRSs are required for and determine the extent of sg mRNA synthesis was provided by mutagenesis studies (Makino et al., 1991; van der Most et al., 1994; van Marle et al., 1999).
Although there is general agreement that corona- and arteriviral mRNA synthesis involves discontinuous transcription, the models for the actual mechanism are still subject to debate. Initially, it was proposed that fusion of non-contiguous sequences occurs during (+)strand synthesis. Leader molecules, including the TRS, would be transcribed from the 3′ end of a genome-sized, negative-strand RNA, anneal internally to complementary (anti-) TRS sequences on this (–)strand template, and thus act as primers for transcription of mRNA body sequences (Lai et al., 1982b; Baric et al., 1983, 1985, 1987). The discovery of sg (–)strand RNAs (Sethna et al., 1989), which carry anti-leader sequences (Sethna et al., 1991) and serve as templates for transcription (Sawicki and Sawicki, 1990), has led to an alternative model, in which discontinuous transcription takes place during minus-strand synthesis (Sawicki and Sawicki, 1990, 1995, 1998). This latter view is gaining increasing experimental support (Schaad and Baric, 1994; Chang et al., 1996; Krishnan et al., 1996; Baric and Yount, 2000; Sawicki et al., 2001). In a series of elegant reverse genetics experiments, Snijder and co-workers (van Marle et al., 1999; Pasternak et al., 2000, 2001) recently provided persuasive evidence that, in arteriviruses, leader–body fusion indeed occurs during the synthesis of (–)strand RNA, presumably via a process resembling similarity-assisted copy-choice RNA recombination. Template switching apparently is guided by base pairing between the anti-TRS on the nascent sg (–)strand and the 5′-most genomic TRS.
It is tempting to assume that corona- and arteriviruses employ essentially the same transcription mechanism and that this mechanism applies for all nidoviruses. However, the mRNAs of gill-associated virus (GAV), the type Okavirus, were shown recently to be leader-less (Cowley et al., 2002). Moreover, there are indications that torovirus mRNAs may not have a common leader sequence (Snijder et al., 1990c, 1991).
The equine isolate Berne virus (BEV) so far is the only torovirus that can be propagated in tissue culture (Snijder and Horzinek, 1993). During infection, a genome-sized, 27 992 nucleotide mRNA 1 is produced and, in addition, four sg mRNA species (numbered 2–5) of 7, 2, 1.2 and 0.7 kb. The latter code for the spike protein (S), the membrane protein (M), a truncated hemagglutinin- esterase protein (HE) and the nucleocapsid protein (N), respectively (Figure 1; Snijder and Horzinek, 1993). The genes for M, HE and N are preceded by non-coding intergenic regions (IGRs), which contain the conserved sequence UCUUUAGA, the torovirus TRS equivalent. In contrast, the S gene overlaps with the replicase gene and there is no obvious TRS; a related motif, UGUUUAGU, was postulated to direct the synthesis of mRNA 2 (Snijder et al., 1990b,c).

Fig. 1. A schematic diagram of the BEV genome organization and expression. Indicated are the cap (black dot) and the poly(A) tail (An); boxes represent the genes for the replicase (ORFs 1a and 1b) and for the spike (S), membrane (M), hemagglutinin-esterase (H) and nucleocapsid proteins (N). Black arrows indicate transcription-regulating sequences. The nested set of the five BEV mRNAs (genome/mRNA 1 and sg mRNAs 2–5) is depicted below.
Snijder et al. (1990c) characterized the 5′ end of BEV mRNA 5 by primer extension and hybridization analysis. A comparison with the putative 5′ end of the BEV genome, as deduced by sequence analysis of defective interfering (DI) RNAs, led the authors to conclude that torovirus mRNAs do not possess large common leader sequences. However, the existence of a small leader (e.g. comprising the conserved sequence and some upstream nucleotides) could not be excluded rigorously. In particular, comparative primer extension analysis of BEV genomic and DI RNAs yielded conflicting results, raising doubts as to whether the DI sequences faithfully represented the 5′ terminus of the BEV genome (Snijder et al., 1991).
Here we have determined what we believe to be the true 5′ ends of the BEV genome and the sg mRNAs. We first ascertained that toroviral mRNAs contain a 5′ cap structure, and then employed an advanced RACE RT–PCR approach, which aims at the specific amplification of cDNA copies derived from capped transcripts (Maruyama and Sugano, 1994). The results were corroborated by primer extension analysis and RNA hybridization studies. Our data provide definitive proof that the BEV sg mRNAs 3–5 lack a common leader. Astoundingly, however, sg mRNA 2 contains a short non-contiguous leader, derived from the 5′ end of the BEV genome. BEV apparently utilizes both continuous and discontinuous RNA synthesis to produce its full set of sg mRNAs. These findings have important implications for our understanding of the mechanism and evolution of nidovirus transcription.
Results and discussion
Toroviral mRNAs possess a 5′ cap structure
Although most RNA viruses provide their (+)strand transcripts with a 5′ cap (Furuichi and Shatkin, 2000), there are conspicuous exceptions. Information with regard to the capping of nidoviral RNAs is limited (Brian, 2001): direct evidence for capping has been presented only for the genome and sg mRNAs of the coronavirus mouse hepatitis virus (Lai and Stohlman, 1981; Lai et al., 1982a) and for the genome of the arterivirus simian hemorrhagic fever virus (Sagripanti et al., 1986). In accordance with the latter observation, equine arteritis virus RNA transcribed in vitro from full-length cDNA templates is infectious only when provided with a cap (Glaser et al., 1999). To examine whether BEV mRNAs are capped, total cytoplasmic RNA was extracted from BEV-infected cells and subjected to immunoaffinity purification (IAP) using the cap-specific monoclonal antibody (mAb) H20 (Bochnig et al., 1987; Kabrane Lazizi et al., 1999). As a negative control, IAP with an isotype-matched mAb R78, directed against the T-cell receptor, was performed in parallel. Affinity-purified RNAs were separated by formaldehyde–agarose gel electrophoresis and subjected to in-gel hybridization with a 32P-labeled oligonucleotide probe 294. As this probe is designed after the 3′ end of the BEV genome (Table I), the complete nested set of viral mRNAs can be visualized. Torovirus mRNAs specifically bound to mAb H20 (Figure 2A, lanes ‘αCap +’). In short fluorographic exposures, only the smaller mRNAs 3–5 were seen; upon prolonged exposure, mRNA 2 could also be discerned. These results suggest that at least the sg mRNAs are capped. Most probably, however, full-length (intact) mRNAs 1 and 2 were selectively lost during IAP because of their large target size. In particular, RNA 1, with its length of 28 kb, is extremely vulnerable to mechanical shearing and enzymatic degradation.
Table I. Oligonucleotide primers.
| Oligo | Sequence | Polarity | Positiona | Gene/NTR |
|---|---|---|---|---|
| 294 | CTTACATGGAGACACTCAACCA | – | 27 940–27 941 | 3′-NTR |
| 1403 | AAGGATAAGGTACTGGCTCAC | – | 21 403–21 423 | S |
| 1404 | GATGGAAACATAGTACAAAGC | – | 26 235–26 255 | M |
| 1405 | CATTTGTAACAGTGTAGGGTG | – | 26 964–26 984 | HE |
| 1406 | ATTATTTTGAAGACGCTGCCG | – | 27 493–27 513 | N |
| 1407 | GTAAGTACTAAATGCACTCTC | – | 181–201 | 5′-NTR |
| 1553 | ATAAACTTCTAAAGATACGT | – | 1–20 | 5′-NTR |
| 1723 | CCGTGCGTACTTAGACATGC | – | NA | 18S rRNA |
| 1746 | GACGACAAAAACTTCTA | – | NA | mRNA 2 |
| 1748 | ACTTCTAAAGATACGT | – | 1–16 | 5′-NTR |
aPosition on the BEV genome.
NA, not applicable.
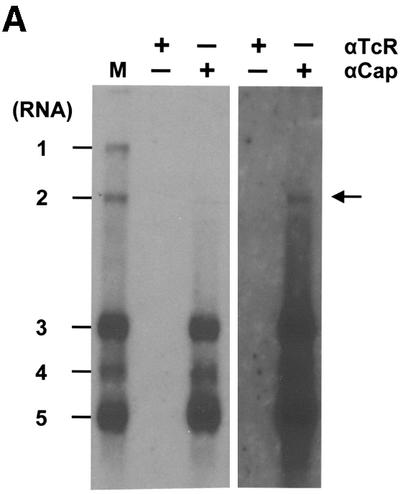

Fig. 2. (A) Immunoaffinity purification of BEV mRNAs with a cap-specific mAb. Total cytoplasmic RNA extracted from BEV-infected cells was incubated with protein G–Sepharose beads, coupled with either the m7G-cross-reactive mAb H20 (αCap) or, as a negative control, the isotype-matched mAb R78 (αTcR). RNA, bound to the mAb-coupled beads, was eluted, separated in denaturing 1% agarose gels and hybridized to a radiolabeled oligonucleotide probe (294; Table I) complimentary to the 3′ end of the genome. Untreated intracellular BEV RNAs served as a marker (M). Fluorographic exposures of 16 (left) and 192 h (right) are shown. (B) Xrn1p treatment of BEV RNAs. Total cytoplasmic RNA extracted from BEV-infected cells was incubated with purified Xrn1p at 37°C for 30 min, in either the presence (+) or absence (–) of EDTA. Samples were analyzed in parallel in non-denaturing ethidium bromide-stained 1% agarose gels (right hand panel), or in denaturing formaldehyde–1% agarose gels by hybridization to radiolabeled oligonucleotide 294 (left hand panel) or the equine 18S rRNA-specific oligonucleotide 1723 (middle panel). The locations of the BEV mRNAs and rRNAs are indicated.
To corroborate and extend the results of the IAP, total intracellular RNA of BEV-infected cells was subjected to digestion with RNase Xrn1p, a cytoplasmic 5′→3′ exoribonuclease, which exclusively degrades non-capped RNA (Boeck et al., 1998). Samples were analyzed in parallel in denaturing and in non-denaturing agarose gels (Figure 2B). Whereas the 28S and 18S rRNAs were degraded, the BEV sg mRNAs appeared fully resistant to digestion. Also, RNA 1 proved resistant to Xrn1p; as determined by β-scanning, 70% of this RNA species survived treatment versus only 7.5% of the 18S rRNA. Partial loss of mRNA 1, again, is most probably related to its huge target size. The combined data indicate that BEV (+)strand RNAs possess a 5′ cap structure.
BEV mRNAs 3 (M), 4 (HE) and 5 (N) lack a 5′ common leader sequence
The true 5′ termini of capped mRNAs can be determined by advanced RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) PCR (Maruyama and Sugano, 1994). This technique involves the specific attachment of a synthetic adaptor oligonucleotide to the 5′ ends of tobacco acid pyrophosphatase-treated, de-capped mRNAs. The chimeric adaptor–mRNA sequences can then be amplified readily by RT–PCR with a combination of RNA-specific and adaptor-specific primers.
RLM-RACE PCR was performed on total intracellular RNA of BEV-infected cells with primers located ∼160–220 nucleotides downstream of the TRSs. For BEV mRNAs 1, 2, 3 and 5, single, discrete PCR products were obtained; in the case of mRNA 4, two amplicons of 240 and 350 bp were generated (Figure 3A). All PCR products were inserted into cloning vector pGEM-T easy and subjected to sequence analysis. For each construct, 6–12 clones were characterized.

Fig. 3. RLM-RACE analysis of the BEV genome and sg mRNAs. (A) RLM-RACE products obtained for the purified viral genome (1) and for sg mRNAs 2–5 were separated in 2% agarose gels. (B) Purification of BEV genomic RNA. Tissue culture supernatant of BEV-infected Ederm cells was harvested and cleared by low speed centrifugation. Subsequently, virions were pelleted through a 10% (w/v) sucrose cushion. Genomic RNA was extracted and analyzed by RNA hybridization with oligonucleotide 294 (V); intracellular BEV mRNAs (IC) served as a marker.
Sequence analysis of mRNA 1-derived RT–PCR products identified adenosine as the 5′-terminal residue, with a cytidine at position 2, located in close proximity (four nucleotides) to a TRS consensus sequence (Figure 4). The 5′ end of mRNA 1, as predicted by RLM-RACE, is collinear with that of the BEV DI RNA DI-1000 (Snijder et al., 1991), but three nucleotides shorter. To confirm our observations, primer extension analysis was performed with oligonucleotide 1407 (Table I); 1407-primed dideoxy-sequencing samples of a DI-1000 cDNA clone served as molecular size marker. A predominant product of 197 nucleotides was detected, consistent with initiation at the AC-dinucleotide sequence identified by RLM-RACE PCR (Figure 4). In addition, a minor 198 nucleotide product was found. It is well established that reverse transcriptases, including Moloney murine leukemia virus (M-MuLV), exhibit terminal transferase activity (Patel and Preston, 1994; Kulpa et al., 1997). Most probably, the larger extension products seen by us and others (Snijder et al., 1991) result from the addition of non-templated nucleotides to the 3′ end of the full-length cDNA strand.

Fig. 4. Primer extension and RLM-RACE analysis of mRNA 1 and of genomic virion RNA. Left and middle panel: BEV genomic RNA, extracted from pelleted virions, and intracellular mRNA 1 were subjected to RLM-RACE. The resulting RT–PCR products were cloned and sequenced with oligonucleotide 1407 as a primer. Reaction mixtures were analyzed in 6% urea–polyacrylamide gels. Arrows indicate the 5′-most BEV nucleotide fused to the adaptor. The nucleotide sequence is presented, with adaptor-derived residues shaded in gray. Right panel: primer extension analysis of mRNA 1 was performed with oligonucleotide primer 1407. The 1407-primed dideoxy-sequencing samples of pDI-1000, a cloned full-length cDNA copy of the BEV DI-1000 RNA (Snijder et al., 1991), served as a molecular weight marker. The DI-1000 sequence, corresponding to the 5′ terminus of the BEV genome, is presented; the arrow indicates the run-off product.
Theoretically, the 5′ terminus of the intracellular mRNA 1 species may differ from that of the genome. To study this possibility, BEV genomic RNA was extracted from purified virus (Figure 3B) and subjected to RLM-RACE. Sequence analysis of the resulting RT–PCR product provided formal evidence that the 5′ termini of BEV mRNA 1 and genomic RNA are identical (Figure 4).
RLM-RACE and primer extension analysis of the 5′ termini of mRNAs 3–5 also identified adenosines as the 5′-most residues, with cytidines at positions 2. Again, the 5′ end of the RNAs was located in close proximity, three or four nucleotides, to a core TRS consensus sequence (Figures 5 and 6). Transcription initiation of mRNAs 3 and 4 was predicted to occur at A26 057 and A26 787, respect ively. Initiation of RNA 5 at A27 289, as predicted by RLM-RACE, is in perfect agreement with the results of primer extension analysis as reported by Snijder et al. (1990c).

Fig. 5. Primer extension and RLM-RACE analysis of BEV sg RNAs 2–5. RLM-RACE products obtained for mRNAs 2–5 (see Figure 3) were subjected to sequence analysis with oligonucleotide primers 1403, 1404, 1405 and 1406, respectively. In addition, primer extension analysis was performed with the respective oligonucleotides on mRNAs 2–4. Sequence reaction mixtures and primer extension products were run alongside in 6% urea–polyacrylamide gels. Arrows indicate the 5′-most residues of each mRNA as predicted by primer extension and/or RLM-RACE. The nucleotide sequence is presented, with adaptor- derived residues shaded in gray.

Fig. 6. Comparison of the 5′ termini of the BEV genome and mRNAs 3–5. Upper panel: alignment of the 5′ termini. Residues identical in three of the four sequences are shown in bold. Initiation codons of the M, HE and N genes are underlined. Lower panel: alignments of the 5′ termini from the BEV genome and from mRNAs 3–5 with the predicted 5′ end of DI-1000 (Snijder et al., 1991) and with the IGRs between the S and M genes (2/3 IGR), the M and HE genes (3/4 IGR) and the HE and N genes (4/5 IGR), respectively. The extended TRSs are shown in bold. The positions of the regions on the BEV genome (in nucleotides) are given on the right. Termination codons of the S and M genes are boxed; initiation codons of the genes for M, HE and N are underlined.
As mentioned, RLM-RACE PCR amplification of the 5′ terminus of mRNA 4 yielded, in addition to the expected 240 bp amplicon, a product of ∼350 bp (Figure 3A). This latter DNA resulted from mispriming by oligo 1405 at a 5′ CACCC 3′ motif within the BEV HE pseudogene, 111 residues downstream of the correct primer-binding site; the adaptor oligonucleotide, however, was fused to the 5′-terminal residue of mRNA 4, A26 787 (not shown).
The 5′ ends of sg RNAs 3–5 differ from each other and from the 5′ end of the viral genome, but they match in sequence the corresponding extended TRSs (AC-N2–3-UCUUUAGA) within the IGRs of the M, HE and N genes, respectively (Figure 6). These findings provide definitive evidence that BEV sg RNAs 3–5 lack a common leader sequence and are not produced via discontinuous transcription. Toroviruses thus differ in this respect from their closest relatives, the coronaviruses. Yet they are not unique among nidoviruses: GAV, a member of a recently recognized group of invertebrate nidoviruses (family Roniviridae, genus Okavirus) (Cowley et al., 2000), also specifies leaderless sg mRNAs (Cowley et al., 2002). Interestingly, transcription of the GAV mRNAs also initiates at AC-dinucleotides. Whether this similarity is fortuitous or reflects a common mechanism of transcription initiation in toro- and okaviruses remains to be determined.
Synthesis of BEV mRNA 2 (S) involves fusion of non-contiguous sequences
Snijder et al. (1990b) previously proposed that the sequence UGUUUAG (residues 21 257–21 263), which is located 50 nucleotides from the S initiation codon, represents the TRS of sg RNA 2. However, RLM-RACE amplification of the 5′-terminal sequences of RNA 2 with primer 1403 consistently yielded a 350 bp product (Figure 3A), suggesting that transcription initiation does not occur at the predicted site but some 150 nucleotides further upstream. Most surprisingly, the RLM-RACE product proved to be chimeric, comprising the 5′-most 15–18 nucleotides of the genome fused to open reading frame 1b (ORF1b) sequences (Snijder et al., 1990a), within the stretch G21 133–U21 136 (Figures 5 and 7). The chimeric product was not only found after RT–PCR with M-MuLV Superscript and Taq polymerase, but also under more stringent conditions with Thermus thermophilus (Tth) DNA polymerase, in one-tube reactions at temperatures ≥60°C. In accordance with the RLM-RACE data, primer extension analysis with oligonucleotide 1403 yielded one major run-off product of 303 nucleotides (Figure 5). A minor product of 314 nucleotides was also observed, the 3′ end of which would map to U21 108. There is no TRS-like sequence present at this location; the 314 nucleotide cDNA may well have arisen from premature termination during reverse transcription of mRNA 1 or from mispriming. The combined results of RLM-RACE and primer extension analysis would suggest that: (i) the proposed promoter motif UGUUUAG does not direct transcription; (ii) mRNA 2 extends far into ORF1b, at least up to U21 136; and (iii) perplexingly, mRNA 2 synthesis entails fusion of non-contiguous sequences.

Fig. 7. Hybridization analysis of the 5′ terminus of mRNA 2. Upper panel: total cytoplasmic RNA, extracted from BEV-infected cells, was separated in denaturing formaldehyde–1% agarose gels and hybridized to the indicated radiolabeled oligonucleotide probes. Hybridization with probes 294 and 1746 was performed at 5°C below the Tm. Hybridization with probe 1553 was performed at the indicated temperatures. Lower panel: nucleotide sequence comparison of the mRNA 2 RLM-RACE product, the corresponding ORF1b region and the 5′ terminus of the BEV genome. Asterisks indicate identical residues. Also given are the sequences of the oligonucleotide probes. Mismatches with the BEV 5′ terminus or the ORF1b sequence are italicized. Nucleotide positions on the viral genome are indicated.
To exclude in vitro recombination artifacts, which may have arisen during reverse transcription or subsequent PCR amplification, in-gel hybridization analysis of intracellular BEV RNAs was performed with a set of oligonucleotide probes (Figure 7). Probe 1748, designed after the 5′-most 16 residues of the BEV genome, detected both BEV RNAs 1 and 2. A second probe, 1553, was designed after the 5′-most 20 residues of the BEV genome, spanning the predicted mRNA 2 leader–body junction by two nucleotides. Thus, this oligonucleotide is fully complementary to the BEV RNA 1, but differs from the predicted chimeric RNA 2 sequence by a one-nucleotide mismatch. As anticipated, under stringent hybridization conditions at 44°C, only RNA 1 was detected. At 40°C, however, probe 1553 hybridized to both RNA 1 and 2. None of the other sg mRNAs was detected, not even when hybridization was performed at 36°C. Finally, probe 1746, which spans the mRNA 2 leader–body junction and contains three and four mismatches with the ORF1b sequence and the 5′ non-translated region (NTR) of the BEV genome, respectively, under stringent conditions preferentially bound to RNA 2.
To exclude the possibility that the chimeric RNA 2 species represented a DI RNA, BEV was plaque purified, and virus, taken from four separate plaques, was propagated at a low m.o.i. (≤0.05 p.f.u./cell). Under these conditions, DI RNAs are readily lost. However, in all cases, hybridization analysis of intracellular RNAs with primers 1553 and 1746 still detected RNA 2 (not shown). Based upon the combined data, we conclude that BEV RNA 2 is a genuine sg mRNA species, which, in contrast to the three other sg mRNAs, possesses a genome-derived 5′ leader sequence.
Fusion of mRNA 2 body and leader sequences: mediated by RNA secondary structure and duplex formation?
The BEV (–)strand RNAs have not yet been identified and characterized. Hence, we cannot formally exclude the possibility that toroviruses, for the synthesis of mRNAs 3–5, utilize an alphavirus-like transcription mechanism, in which the extended anti-TRSs on a genome-sized (–)strand RNA function as internal promoters. Most probably, however, toroviruses resemble the other nidoviruses in directing the synthesis of sg (–)strand templates (Sethna et al., 1989, 1991; Sawicki and Sawicki, 1990; den Boon et al., 1996). We propose that the toroviral promoters serve a dual function, acting as both (–)strand termination signals and transcription initiation signals for (+)strand synthesis (Figure 8B). As noted by Cowley et al. (2002), such a transcription mechanism would be reminiscent of that of the closteroviruses, (+)strand RNA viruses of plants (Gowda et al., 2001).


Fig. 8. (A) Nucleotide sequence comparison of the 5′ terminus of the BEV genome and the ORF1b fusion region (residues 21 118–21 161). The arrowhead indicates the mRNA 2 leader–body fusion site, comprising residues G21 133–U21 136 (italicized). As illustrated schematically, the residues upstream of the fusion site can adopt a hairpin structure. Asterisks indicate residues identical between ORF1b and the 5′ terminus of the BEV genome. Shown below is a comparison of the corresponding regions in PoTV. At present, only a partial sequence of the 5′ end of the PoTV genome is available. (B) Schematic hypothetical model for the discontinuous synthesis of BEV mRNA 2 and for the non-discontinuous transcription of mRNAs 3–5. For the latter RNAs, the TRSs on the (+)strand template (indicated in red) function as termination signals for (–)strand synthesis. The resulting sg (–)strands act in turn as templates for (+)strand sg RNA synthesis, with the anti-TRSs on the (–)strand template now functioning as transcription initiation signals. The viral RdRp is indicated as a gray circle. For the synthesis of mRNA 2, the stem–loop structure causes attenuation of (–)strand synthesis. Subsequently, a template switch occurs, during which the transcription complex is transferred to the 5′ end of the BEV genome, assisted by base pairing between ORF1b-derived sequences in the nascent (–) strand RNA and 5′-NTR sequences (indicated in blue). RNA synthesis resumes, resulting in the production of a transcription-competent sg RNA species, carrying a non-contiguous, genome-derived anti-TRS.
In contrast to the genes for M, HE and N, the BEV S gene seems to lack a TRS. Obviously, the absence of this cis-element would preclude the synthesis of a transcription-competent (–)strand template and, hence, expression of the S gene. We hypothesize that BEV solves this problem by producing (–)strand RNA 2 templates, which are provided with the genome anti-TRS via discontinuous transcription.
For discontinuous transcription in corona- and arteriviruses, current evidence supports a model in which (–)strand RNA synthesis is attenuated after copying a body TRS from the donor (+)strand template; pausing of the RdRp may be controlled by protein–TRS interaction and possibly local RNA secondary structure (Pasternak et al., 2000, 2001). The nascent (–)strand, presumably while still in complex with the RdRp, then detaches and switches to the leader TRS at the 5′ end of the genome, which serves as the acceptor template. Upon anti-TRS–TRS duplex formation, transcription resumes and synthesis of a chimeric, anti-leader-containing sg (–)strand RNA is completed. We believe that an analogous similarity-assisted, copy-choice RNA recombination event during (–)strand synthesis would best explain the chimeric structure of BEV sg RNA 2. Inspection of the mRNA 2 fusion region in ORF1b revealed the presence of a 17 nucleotide sequence (A21 118–U21 134), which can adopt a stem–loop structure (ΔG = –8.6 kcal/mol). Strikingly, this hairpin loop immediately precedes the leader–body junction site, followed by a stretch of 24 residues, with remarkable identity (79%) to residues 15–38 of the BEV genome (Figure 8A). Conceivably, this combination of sequence elements may create a recombinational hot spot, with the hairpin in the donor template causing attenuation of (–)strand synthesis and dissociation of the RdRp–RNA complex, after which the ORF1b-derived ‘guiding’ sequence mediates a template switch by base pairing to its complementary sequence at the 5′ end of the viral genome (Figure 8B). Such a mechanism would not be without precedent. Studies on tombusvirus RNA recombination showed that the 3′ base of a stem–loop structure in the donor template is a preferred site for similarity-assisted template switching (White and Morris, 1995). Torovirus discontinuous transcription would, however, differ from that of corona- and arteriviruses: the TRSs of the latter are not preceded by evident hairpins (A.O.Pasternak and E.J.Snijder, personal communication; R.J.de Groot, unpublished).
Our model for mRNA 2 synthesis is supported by phylogenetic data: the structural genes of porcine torovirus strain Markelo (Kroneman et al., 1998) display only 65% nucleotide identity with those of BEV. Yet, the BEV sg RNA promoter motif, 5′ AC-N2–3-UCUUUAGA 3′, is conserved in PoTV and is found immediately upstream of the PoTV genes for M, HE and N (S.L.Smits and R.J.de Groot, unpublished). However, as in BEV, the region upstream of the PoTV S gene does not contain an evident promoter motif. In fact, it bears a strong resemblance to the corresponding BEV sequence, and the potential stem–loop structure is conserved. Most notably, as compared with the BEV element, there are two substitutions in the stem of the PoTV hairpin, but base pairing is maintained (C–G→U–A; Figure 8A), reinforcing the notion that this RNA secondary structure may be relevant to discontintuous RNA 2 synthesis. Moreover, the similarities between BEV and PoTV suggest that the unusual combination of discontinuous and non-discontinuous transcription, displayed by BEV, does not result from tissue culture adaptation, but may also be employed by related naturally occurring toroviruses.
Discontinuous transcription in corona-, arteri- and toroviruses: divergent or convergent evolution?
It is commonly held that discontinuous transcription arose in a proto-nidovirus and that this mechanism was preserved in corona- and arteriviruses. This would imply that, in toroviruses, discontinuous transcription was replaced by a non-discontinuous transcription strategy for three of the four sg mRNAs, while in okaviruses, leader addition via discontinuous transcription was lost completely. However, it seems counterintuitive that a trait that survived the considerable evolutionary distance between corona- and arteriviruses would have been lost during the radiation of the corona-, toro- and okaviruses, which seems to have occurred more recently (A.E.Gorbalenya, personal communication). We would therefore like to offer an alternative and equally plausible explanation, namely that the proto-nidovirus utilized an okavirus/torovirus-like non-discontinuous transcription strategy and that discontinuous transcription arose independently in arteri-, corona- and toroviruses through convergent evolution. Indeed, there are remarkable differences between arteri- and coronaviruses with regard to the sizes of their leaders and TRSs. High frequency homologous RNA recombination appears to be a universal trait of nidoviruses (Brian and Spaan, 1997; Lai and Cavanagh, 1997; Molenkamp et al., 2000), which may have made them prone to developing analogous discontinuous transcription strategies. In arteri- and coronaviruses, the transcription initiation and termination signals, initially combined in the ancestral nidovirus TRS, may have become separated. In this view, leader addition would be refinement of a common nidoviral transcription scheme, in which the central phenomenon is not the fusion of non-contiguous sequences, but rather the attenuation/termination of (–)strand RNA synthesis at the TRS (Sawicki and Sawicki, 1995, 1998). Such a mechanism may have evolved to provide each of the sg mRNAs with an identical translational enhancer (i.e. a sequence element that increases translation efficiency).
This is the first report of an RNA virus that com bines two different strategies, discontinuous and non-discontinuous transcription, to produce its full set of sg mRNAs. Why toroviruses do so and how this situation arose is not known. If toroviruses evolved from an ancestor that relied on discontinuous mRNA synthesis, mRNA 2 transcription perhaps represents a relict. If, however, the torovirus ancestor utilized a non-discontinuous transcription strategy, the peculiar synthesis of mRNA 2 may be the consequence of a heterologous RNA recombination event. It is of note that the S gene overlaps with the replicase gene and, in contrast to the genes for M, HE and N, is not preceded by a non-coding IGR. A scenario can be envisaged in which the 5′ part of the S gene, including the IGR and TRS, was deleted or replaced by a foreign sequence and in which a new initiation codon was acquired by fusion to an internal (–1) ORF at the 3′ end of the replicase gene. If the resulting novel spike protein provided a strong selection advantage, even an initial inefficient expression may have allowed survival of the mutant virus. An infrequent, though consistent template switch in torovirus-infected cells, driven merely by chance sequence identity between ORF1b and the 5′ end of the BEV genome, may have given rise to a minor transcription-competent sg (–)strand RNA species. Subsequently, the discontinuous synthesis of mRNA 2 may have been perfected rapidly to optimize S expression. Be that as it may, torovirus transcription deserves further scrutiny.
Materials and methods
Cells and virus
BEV was grown in equine dermis (Ederm) cells (American Type Tissue Culture Collection). A BEV stock, devoid of DI viruses, was used in all experiments. This stock was prepared by performing single round plaque purification, followed by two rounds of end-point dilution. The purified virus was propagated at an m.o.i. of 0.05 TCID50 units/cell.
Isolation of genomic and intracellular BEV RNA
For the isolation of genomic RNA, 2 × 108 Ederm cells were infected with BEV at an m.o.i. of 0.05 TCID50 units/cell. Tissue culture supernatant, harvested at 16 h post-infection, was pre-cleared by centrifugation at 2400 g for 10 min at 4°C. Virus was pelleted through a 10% (w/v) sucrose cushion by centrifugation in a Beckman SW28 rotor at 26 000 r.p.m. (∼120 000 g) for 2 h. Virus was resuspended in 3 ml of phosphate-buffered saline (PBS) containing 1% Triton X-100 and mixed with an equal volume of 7 M urea, 2% SDS. Genomic RNA was extracted three times with acid phenol and once with diethyl ether, and was recovered by ethanol precipitation. Total cytoplasmic RNA was isolated from BEV-infected cells according to Spaan et al. (1981).
Immunoaffinity purification of capped RNA
mAb H20, though raised against the rare nucleoside 2,2,7-trimethylguanosine, cross-reacts with 7-methylguanosine (m7G) cap structures, allowing its application in the characterization and IAP of m7G-capped transcripts (Bochnig et al., 1987). mAb H20 was a kind gift from Dr Reinhard Lührmann (Department of Cellular Biochemistry; Max Planck Institute for Biophysical Chemistry). mAb R78, which served as an isotype-matched negative control, was kindly provided by Peter van Kooten (Immunology Unit, Faculty of Veterinary Medicine, Utrecht University).
IAP of capped RNAs was performed as described previously (Kabrane Lazizi et al., 1999). Protein G–Sepharose beads (Gammabind G-Sepharose; Pharmacia, Uppsala, Sweden; 500 µl of a 50% suspension) were washed twice in 5 M guanidine–HCl for 30 min at room temperature to remove RNases, followed by three additional washes in RNA binding buffer (10 mM NaPi pH 7, 150 mM NaCl, 10 mM EDTA). The beads were collected by low speed centrifugation and taken up in 500 µl of RNA binding buffer containing 150 µg of either mAb H20 or R78. Incubation was for 16 h at 4°C. The antibody-coupled beads were washed three times in RNA binding buffer and resuspended as a 50% solution. SDS–PAGE analysis of aliquots of the beads confirmed that equal amounts of mAbs H20 and R78 had been coupled.
Total cytoplasmic RNA extracted from the equivalent of 4 × 106 BEV-infected cells was added to a 150 µl reaction volume of RNA binding buffer, containing 25% mAb-coupled beads and 2000 U/ml RNAguard (Pharmacia). Incubation was for 1 h at 4°C under constant rotation. Subsequently, beads were washed four times with RNA binding buffer supplemented with 0.5% NP-40 (Fluka, Buchs, Germany). RNA bound to the mAb-coupled beads was eluted with 1% SDS, 3.5 M urea in TES for 5 min at room temperature, extracted twice with acid phenol and once with diethyl ether, and finally recovered by ethanol precipitation. The RNA pellet was dissolved in 10 µl of water and analyzed by hybridization with BEV-specific 32P-labeled oligonucleotide probes.
To confirm the specificity of the procedure, total RNAs purified from cells infected with the coronavirus mouse hepatitis virus (MHV) strain A59 were subjected to IAP. The capped MHV mRNAs exclusively bound to mAb H20-coupled beads. In addition, synthetic RNAs were produced by in vitro transcription of plasmid DNA with T7 RNA polymerase in either the presence or absence of the cap analog M7G(5′)ppp(5′)G (Amersham). Again, these transcripts only bound to mAb H20-coupled beads and only when provided with a 5′ cap structure (see Supplementary data available at The EMBO Journal Online).
Xrn1p assay
Xrn1p digestion was performed essentially as described by Boeck et al. (1998). Total intracellular RNA, extracted from the equivalent of 2.5 × 106 BEV-infected cells, was incubated with 2 µg of purified Xrn1p (generous gift of N.Cozzarelli, University of California, Berkely) in a final volume of 10 µl of 33 mM Tris–HCl pH 8.0, 50 mM NaCl, 2.5 mM MgCl2, 0.2 mM dithiothreitol in either the presence or absence of 5 mM EDTA. Incubation was for 30 min at 37°C.
RNA hybridization analysis
RNA was separated in 1% agarose gels containing 2.2 M formaldehyde in MOPS buffer (10 mM MOPS pH 7, 5 mM sodium acetate, 1 mM EDTA) as described by Sawicki and Sawicki (1990). Gels were dried under a vacuum at 58°C and hybridized with 5′-end-labeled oligonucleotide probes for 16 h at 5°C below the Tm (Meinkoth and Wahl, 1984), unless stated otherwise.
RLM-RACE RT–PCR
The true 5′ termini of capped mRNAs can be determined by advanced RLM-RACE PCR (Maruyama and Sugano, 1994). This technique involves the attachment of synthetic adaptor oligonucleotide to the 5′ end of the mRNA. The adaptor is transcribed and included into the first-strand cDNA copy only when reverse transcription proceeds across the 5′ terminus of the mRNA. To ensure that the adaptor is ligated exclusively to mRNAs with an intact 5′ end, the RNA sample is pre-treated with calf intestinal phosphatase. Thus, contaminating rRNA, tRNA, degraded mRNA fragments and DNA are dephosphorylated and excluded from ligation reactions. mRNAs with an intact, capped 5′ end are then decapped by treatment with tobacco acid pyrophosphatase, leaving a monophosphorylated 5′ terminus and allowing subsequent ligation of the adaptor using T4 RNA ligase. RLM-RACE was performed with the FirstChoice™ RLM-RACE kit (Ambion, Austin, TX) according to the manufacturer’s instructions. The amounts of intracellular and genomic RNA used per reaction corresponded to that extracted from 2 × 104 BEV-infected Ederm cells and 3 × 107 TCID50 units, respectively. RT–PCR with gene- and adaptor-specific primers was either performed with M-MuLV reverse transcriptase (Superscript II) and Taq polymerase (Gibco-BRL, Gaithersburg, MD) or with Tth DNA polymerase (Roche, Mannheim, Germany) according to the manufacturer’s instructions. PCR DNAs were excised from agarose–0.5× TAE gels, purified using the Qiagen gel extraction kit (Qiagen, Hilden, The Netherlands), ligated in vector pGEM-T easy (Promega, Madison, WI) and cloned in Escherichia coli strain pc2495. Plasmid DNA, extracted from individual clones, was subjected to sequence analysis using a T7 RNA polymerase sequencing kit (Pharmacia). For each construct, 6–12 clones were characterized.
Primer extension analysis
Ten nanograms of 5′-end-labeled oligonucleotide was annealed to intracellular RNA, extracted from 2 × 106 BEV-infected cells. Annealing and first-strand cDNA synthesis were carried out with M-MuLV reverse transcriptase (Superscript II; Gibco-BRL) according to Fichot and Girard (1990), except that dideoxynucleotides were omitted from the reaction mixture. Extension products were separated in 6% acrylamide–7.5 M urea gels.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We are grateful to Jolanda Mijnes, Robbert van der Most and Willem Luytjes for helpful discussions and for critical reading of the manuscript, Nancy Crisona and Nicholas Cozzarelli for generously providing purified Xrn1p, Reinhard Lührmann and Peter van Kooten for their kind gift of purified mAbs H20 and R78, respectively, and Yamina Kabrane for helpful suggestions concerning immunoaffinity purification of capped RNAs.
References
- Baric R.S. and Yount,B. (2000) Subgenomic negative-strand RNA function during mouse hepatitis virus infection. J. Virol., 74, 4039–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Stohlman,S.A. and Lai,M.M. (1983) Characterization of replicative intermediate RNA of mouse hepatitis virus: presence of leader RNA sequences on nascent chains. J. Virol., 48, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Stohlman,S.A., Razavi,M.K. and Lai,M.M. (1985) Characterization of leader-related small RNAs in coronavirus-infected cells: further evidence for leader-primed mechanism of transcription. Virus Res., 3, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Shieh,C.K., Stohlman,S.A. and Lai,M.M. (1987) Analysis of intracellular small RNAs of mouse hepatitis virus: evidence for discontinuous transcription. Virology, 156, 342–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochnig P., Reuter,R., Bringmann,P. and Luhrmann,R. (1987) A monoclonal antibody against 2,2,7-trimethylguanosine that reacts with intact, class U, small nuclear ribonucleoproteins as well as with 7-methylguanosine-capped RNAs. Eur. J. Biochem., 168, 461–467. [DOI] [PubMed] [Google Scholar]
- Boeck R., Lapeyre,B., Brown,C.E. and Sachs,A.B. (1998) Capped mRNA degradation intermediates accumulate in the yeast spb8-2 mutant. Mol. Cell. Biol., 18, 5062–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brian D.A. (2001) Nidovirus genome replication and subgenomic mRNA synthesis. Pathways followed and cis-acting elements required. In Lavi,E., Weiss,S.R. and Hingley,S.T. (eds), The Nidoviruses, Vol. 494. Kluwer Academic/Plenum, New York, NY, pp. 415–428. [DOI] [PubMed]
- Brian D.A. and Spaan,W.J.M. (1997) Recombination and coronavirus defective interfering RNAs. Semin. Virol., 8, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. et al. (1993) The Coronaviridae now comprises two genera, coronavirus and torovirus: report of the Coronaviridae Study Group. Adv. Exp. Med. Biol., 342, 255–257. [DOI] [PubMed] [Google Scholar]
- Chang R.Y., Krishnan,R. and Brian,D.A. (1996) The UCUAAAC promoter motif is not required for high-frequency leader recombination in bovine coronavirus defective interfering RNA. J. Virol., 70, 2720–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I.R., Ostrovsky,M., Zhang,G. and White,K.A. (2001) Regulatory activity of distal and core RNA elements in tombusvirus subgenomic mRNA2 transcription. J. Biol. Chem., 276, 41761–41768. [DOI] [PubMed] [Google Scholar]
- Cowley J.A., Dimmock,C.M., Spann,K.M. and Walker,P.J. (2000) Gill-associated virus of Penaeus monodon prawns: an invertebrate virus with ORF1a and ORF1b genes related to arteri- and coronaviruses. J. Gen. Virol., 81, 1473–1484. [DOI] [PubMed] [Google Scholar]
- Cowley J.A., Dimmock,C.M. and Walker,P.J. (2002) Gill-associated nidovirus of Penaeus monodon prawns transcribes 3′-coterminal subgenomic mRNAs that do not possess 5′-leader sequences. J. Gen. Virol., 83, 927–935. [DOI] [PubMed] [Google Scholar]
- de Vries A.A.F., Horzinek,M.C., Rottier,P.J.M. and de Groot,R.J. (1997) The genome organization of the Nidovirales: similarities and differences between arteri-, toro- and coronaviruses. Semin. Virol., 8, 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boon J.A., Snijder,E.J., Chirnside,E.D., de Vries,A.A., Horzinek, M.C. and Spaan,W.J. (1991) Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. J. Virol., 65, 2910–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boon J.A., Kleijnen,M.F., Spaan,W.J. and Snijder,E.J. (1996) Equine arteritis virus subgenomic mRNA synthesis: analysis of leader–body junctions and replicative-form RNAs. J. Virol., 70, 4291–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichot O. and Girard,M. (1990) An improved method for sequencing of RNA templates. Nucleic Acids Res., 18, 6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi Y. and Shatkin,A.J. (2000) Viral and cellular mRNA capping: past and prospects. Adv. Virus Res., 55, 135–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser A.L., de Vries,A.A.F., Raamsman,M., Horzinek,M.C. and Rottier,P.J.M. (1999) An infectious cDNA clone of equine arteritis virus: a tool for future fundamental studies and vaccine development. In Plowright,W., Rossdale,P.D. and Wade,J.F. (eds), Proceedings of the 8th International Conference on Equine Infectious Diseases, Dubai 1998. R&W Publications, Newmarket, UK, pp. 166–176.
- Gowda S., Satyanarayana,T., Ayllon,M.A., Albiach-Marti,M.R., Mawassi,M., Rabindran,S., Garnsey,S.M. and Dawson,W.O. (2001) Characterization of the cis-acting elements controlling subgenomic mRNAs of citrus tristeza virus: production of positive- and negative-stranded 3′-terminal and positive-stranded 5′-terminal RNAs. Virology, 286, 134–151. [DOI] [PubMed] [Google Scholar]
- Kabrane Lazizi Y., Meng,X.J., Purcell,R.H. and Emerson,S.U. (1999) Evidence that the genomic RNA of hepatitis E virus is capped. J. Virol., 73, 8848–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan R., Chang,R.Y. and Brian,D.A. (1996) Tandem placement of a coronavirus promoter results in enhanced mRNA synthesis from the downstream-most initiation site. Virology, 218, 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroneman A., Cornelissen,L.A., Horzinek,M.C., de Groot,R.J. and Egberink,H.F. (1998) Identification and characterization of a porcine torovirus. J. Virol., 72, 3507–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulpa D., Topping,R. and Telesnitsky,A. (1997) Determination of the site of first strand transfer during Moloney murine leukemia virus reverse transcription and identification of strand transfer-associated reverse transcriptase errors. EMBO J., 16, 856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C. and Cavanagh,D. (1997) The molecular biology of coronaviruses. Adv. Virus Res., 48, 1–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M. and Stohlman,S.A. (1981) Comparative analysis of RNA genomes of mouse hepatitis viruses. J. Virol., 38, 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M., Patton,C.D. and Stohlman,S.A. (1982a) Further characterization of mRNAs of mouse hepatitis virus: presence of common 5′-end nucleotides. J. Virol., 41, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M., Patton,C.D. and Stohlman,S.A. (1982b) Replication of mouse hepatitis virus: negative-stranded RNA and replicative form RNA are of genome length. J. Virol., 44, 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S., Joo,M. and Makino,J.K. (1991) A system for study of coronavirus messenger RNA synthesis: a regulated, expressed subgenomic defective interfering RNA results from intergenic site insertion. J. Virol., 65, 6031–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama K. and Sugano,S. (1994) Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene, 138, 171–174. [DOI] [PubMed] [Google Scholar]
- Meinkoth J. and Wahl,G. (1984) Hybridization of nucleic acids immobilized on solid supports. Anal. Biochem., 138, 267–284. [DOI] [PubMed] [Google Scholar]
- Miller W.A. and Koev,G. (2000) Synthesis of subgenomic RNAs by positive-strand RNA viruses. Virology, 273, 1–8. [DOI] [PubMed] [Google Scholar]
- Miller W.A., Dreher,T.W. and Hall,T.C. (1985) Synthesis of brome mosaic virus subgenomic RNA in vitro by internal initiation on (–)sense genomic RNA. Nature, 313, 68–70. [DOI] [PubMed] [Google Scholar]
- Molenkamp R., Greve,S., Spaan,W.J. and Snijder,E.J. (2000) Efficient homologous RNA recombination and requirement for an open reading frame during replication of equine arteritis virus defective interfering RNAs. J. Virol., 74, 9062–9070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.O., Gultyaev,A.P., Spaan,W.J. and Snijder,E.J. (2000) Genetic manipulation of arterivirus alternative mRNA leader–body junction sites reveals tight regulation of structural protein expression. J. Virol., 74, 11642–11653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.O., van den Born,E., Spaan,W.J. and Snijder,E.J. (2001) Sequence requirements for RNA strand transfer during nidovirus discontinuous subgenomic RNA synthesis. EMBO J., 20, 7220–7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P.H. and Preston,B.D. (1994) Marked infidelity of human immunodeficiency virus type 1 reverse transcriptase at RNA and DNA template ends. Proc. Natl Acad. Sci. USA, 91, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagripanti J.L., Zandomeni,R.O. and Weinmann,R. (1986) The cap structure of simian hemorrhagic fever virion RNA. Virology, 151, 146–150. [DOI] [PubMed] [Google Scholar]
- Sawicki S.G. and Sawicki,D.L. (1990) Coronavirus transcription: subgenomic mouse hepatitis virus replicative intermediates function in RNA synthesis. J. Virol., 64, 1050–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki S.G. and Sawicki,D.L. (1995) Coronaviruses use discontinuous extension for synthesis of subgenome-length negative strands. Adv. Exp. Med. Biol., 380, 499–506. [DOI] [PubMed] [Google Scholar]
- Sawicki S.G. and Sawicki,D.L. (1998) A new model for coronavirus transcription. Adv. Exp. Med. Biol., 440, 215–219. [DOI] [PubMed] [Google Scholar]
- Sawicki D., Wang,T. and Sawicki,S. (2001) The RNA structures engaged in replication and transcription of the A59 strain of mouse hepatitis virus. J. Gen. Virol., 82, 385–396. [DOI] [PubMed] [Google Scholar]
- Schaad M.C. and Baric,R.S. (1994) Genetics of mouse hepatitis virus transcription: evidence that subgenomic negative strands are functional templates. J. Virol., 68, 8169–8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethna P.B., Hung,S.L. and Brian,D.A. (1989) Coronavirus subgenomic minus-strand RNAs and the potential for mRNA replicons. Proc. Natl Acad. Sci. USA, 86, 5626–5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethna P.B., Hofmann,M.A. and Brian,D.A. (1991) Minus-strand copies of replicating coronavirus mRNAs contain antileaders. J. Virol., 65, 320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit T.L., Vaewhongs,A.A. and Lommel,S.A. (1998) RNA-mediated trans-activation of transcription from a viral RNA. Science, 281, 829–832. [DOI] [PubMed] [Google Scholar]
- Snijder E.J. and Horzinek,M.C. (1993) Toroviruses: replication, evolution and comparison with other members of the coronavirus-like superfamily. J. Gen. Virol., 74, 2305–2316. [DOI] [PubMed] [Google Scholar]
- Snijder E.J. and Meulenberg,J.J. (1998) The molecular biology of arteriviruses. J. Gen. Virol., 79, 961–979. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., den Boon,J.A., Bredenbeek,P.J., Horzinek,M.C., Rijnbrand,R. and Spaan,W.J. (1990a) The carboxyl-terminal part of the putative Berne virus polymerase is expressed by ribosomal frameshifting and contains sequence motifs which indicate that toro- and coronaviruses are evolutionarily related. Nucleic Acids Res., 18, 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Den Boon,J.A., Spaan,W.J., Weiss,M. and Horzinek,M.C. (1990b) Primary structure and post-translational processing of the Berne virus peplomer protein. Virology, 178, 355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Horzinek,M.C. and Spaan,W.J. (1990c) A 3′-coterminal nested set of independently transcribed mRNAs is generated during Berne virus replication. J. Virol., 64, 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., den Boon,J.A., Horzinek,M.C. and Spaan,W.J. (1991) Characterization of defective interfering RNAs of Berne virus. J. Gen. Virol., 72, 1635–1643. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Horzinek,M.C. and Spaan,W.J. (1993) The coronaviruslike superfamily. Adv. Exp. Med. Biol., 342, 235–244. [DOI] [PubMed] [Google Scholar]
- Spaan W.J., Rottier,P.J., Horzinek,M.C. and van der Zeijst,B.A. (1981) Isolation and identification of virus-specific mRNAs in cells infected with mouse hepatitis virus (MHV-A59). Virology, 108, 424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Most R.G. and Spaan,W.J.M. (1995) Coronavirus replication, transcription and RNA recombination. In Siddell,S.G. (ed.), The Coronaviridae. Plenum Press, New York, NY, pp. 11–31.
- van der Most R.G., de Groot,R.J. and Spaan,W.J. (1994) Subgenomic RNA synthesis directed by a synthetic defective interfering RNA of mouse hepatitis virus: a study of coronavirus transcription initiation. J. Virol., 68, 3656–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle G., Dobbe,J.C., Gultyaev,A.P., Luytjes,W., Spaan,W.J. and Snijder,E.J. (1999) Arterivirus discontinuous mRNA transcription is guided by base pairing between sense and antisense transcription-regulating sequences. Proc. Natl Acad. Sci. USA, 96, 12056–12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K.A. and Morris,T.J. (1995) RNA determinants of junction site selection in RNA virus recombinants and defective interfering RNAs. RNA, 1, 1029–1040. [PMC free article] [PubMed] [Google Scholar]