

Abstract
AZT (3′-azido-3′-deoxythymidine) resistance involves the enhanced excision of AZTMP from the end of the primer strand by HIV-1 reverse transcriptase. This reaction can occur when an AZTMP-terminated primer is bound at the nucleotide-binding site (pre-translocation complex N) but not at the ‘priming’ site (post-translocation complex P). We determined the crystal structures of N and P complexes at 3.0 and 3.1 Å resolution. These structures provide insight into the structural basis of AZTMP excision and the mechanism of translocation. Docking of a dNTP in the P complex structure suggests steric crowding in forming a stable ternary complex that should increase the relative amount of the N complex, which is the substrate for excision. Structural differences between complexes N and P suggest that the conserved YMDD loop is involved in translocation, acting as a springboard that helps to propel the primer terminus from the N to the P site after dNMP incorporation.
Keywords: drug resistance/HIV/nucleotide excision/reverse transcriptase/translocation
Introduction
Human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) is a heterodimer composed of a 560 residue subunit (p66) and a smaller subunit (p51) that contains the N-terminal 440 residues of p66. The C-terminal portion of p66 forms the RNase H domain, the N-terminal portion the polymerase domain. Only p66 has a functional polymerase active site and a DNA-binding cleft formed by the p66 fingers, palm and thumb subdomains. The role of p51 appears to be primarily structural.
Mechanism of DNA polymerization
The mechanism of DNA polymerization by HIV-1 RT (Figure 1) has been studied extensively (Majumdar et al., 1988; Kati et al., 1992; Hsieh et al., 1993; Reardon, 1993). Several crystal structures provide information about the structural changes that occur during polymerization. The structure of unliganded RT has the thumb subdomain in the ‘closed’ position (form ‘E’ in Figure 1) (Rodgers et al., 1995; Hsiou et al., 1996). Binding of template–primer forms the E′/DNA binary complex (step 1) with the thumb moving to accommodate the nucleic acid (Jacobo-Molina et al., 1993; Ding et al., 1998). Subsequent binding of an incoming dNTP forms the E+/DNA/dNTP complex (step 2) that is believed to have the fingers in the open configuration (Figure 1) (Li et al., 1998). There is no crystal structure that corresponds to this complex. The conversion of E+/DNA/dNTP to the activated closed ternary complex (E*/DNA/dNTP) (Kati et al., 1992) involves movement of the p66 fingers subdomain (Huang et al., 1998), a conformational change thought to be the rate-limiting step in polymerization (step 3, Figure 1) (Kati et al., 1992; Hsieh et al., 1993). In all of the complexes, the primer terminus is positioned at what we call the ‘priming site’ (P site) (Figures 1 and 2). This allows the incoming dNTP to bind at the nucleotide-binding site (N site). Nucleophilic attack of the 3′-OH of the primer terminus yields E/DNA–dNMP via phosphodiester bond formation accompanied by pyrophosphate (PPi) release (step 4, Figure 1) and possible opening of the p66 fingers subdomain, events that may occur either concomitantly or in discrete steps. If polymerization is distributive, the enzyme falls off the elongated DNA (Figure 1, step 5′) and polymerization starts from step 1 in Figure 1. However, in processive polymerization, the elongated DNA primer (DNAn + 1) is translocated from the N site to the P site (step 5). The structure of complex N is unknown. Furthermore, because translocation has been kinetically invisible, we know comparatively little about the underlying mechanism. The efficiency of translocation determines whether the enzyme will continue polymerizing (processive synthesis) or will fall off the nucleic acid (distributive synthesis).

Fig. 1. Schematic representation of the mechanism of DNA polymerization indicating the proposed occupancy of sites P (‘priming’ site, in blue) and N (‘nucleotide-binding site’, in red) by the 3′ end of the primer during the course of the reaction. The structures reported here, RT(P)–AZTMP (complex P) and RT(N)–AZTMP (complex N), have AZTMP-terminated primers at the P and N sites, respectively. Step 1: binding of DNA to free enzyme E [RT(apo)] with the 3′ primer end at the P site. Step 2: binding of dNTP to the N site to form an ‘open’ ternary complex [open RT(ter)]. Step 3: formation of a ‘closed’ ternary complex [closed RT(ter)] through conformational change. Step 4: bond formation accompanied by release of pyrophosphate to form the non-translocated complex N [RT(N)–dNMP]. Step 5: in processive synthesis, the primer translocates from the N site to the P site. Step 5′: in non-processive synthesis, DNA dissociates from the enzyme.

Fig. 2. Reaction schemes for cross-linking AZTMP-terminated template–primers at the P (priming) (A) or N (nucleotide-binding) (B) sites of HIV-1 RT. Cross-linking takes place when Cys258 of the αH helix of the p66 thumb of HIV-1 RT is aligned with the cross-linkable nucleotide analog. Complex P [RT(P)–AZTMP complex] is formed after addition of AZTMP (A), whereas complex N [RT(N)– AZTMP complex] is formed with in situ incorporation of dAMP and AZTMP (B). Z is AZTMP.
Excision of AZTMP-based drug resistance and translocation
Nucleoside analog reverse transcriptase inhibitors (NRTIs) inhibit DNA synthesis by blocking DNA elongation because they lack a 3′-OH. Two distinct mechanisms have been reported for RT resistance to NRTIs. In the first mechanism, resistance mutation(s) interfere with the ability of HIV-1 RT to incorporate NRTIs. Resistance to 3TC caused by mutations at M184 is due to steric hindrance that interferes with incorporation of 3TCTP (Sarafianos et al., 1999a; Gao et al., 2000). In the second mechanism, resistance mutations enhance the excision of the analog after it has been incorporated. The basis of 3′-azido-3′-deoxythymidine (AZT) resistance is the enhanced excision of AZTMP. AZT-resistant HIV-1 RT unblocks AZTMP-terminated primers by a mechanism that involves nucleophilic attack by a pyrophosphate donor (Arion et al., 1998), most probably ATP (Meyer et al., 1998, 2002; Boyer et al., 2001). The reaction products are an unblocked primer and a dinucleoside tetraphosphate derived from ATP and the AZTMP at the primer terminus. The excision reaction allows polymerization to continue and causes resistance to AZT (Arion et al., 1998; Meyer et al., 1999; Boyer et al., 2001, 2002a).
Recently, we proposed a model to explain the mechanism of AZT resistance and the role of the resistance mutations. In this model, several of the mutations associated with AZT resistance act primarily to facilitate the binding of ATP. The bound ATP interacts directly with some of the mutated amino acids; its γ-phosphate is near the scissile phosphate (Boyer et al., 2001, 2002a,b). Excision can occur only if the primer terminus is positioned at the N site. If a dideoxy-terminated primer is at the P site it can form a stable, dead-end ternary complex with the incoming dNTP at the N site and the primer terminus at the P site, preventing excision. Conversely, biochemical data show that RT complexed with an AZTMP-terminated primer does not readily form a stable closed ternary complex upon binding of dNTP (Tong et al., 1997; Meyer et al., 1999). In our model, the azido group of an AZTMP-terminated primer interferes either with the ability of AZTMP to occupy the P site, with the ability of the incoming dNTP to bind at the N site or both (Boyer et al., 2001, 2002a). Even in the presence of incoming dNTP, AZTMP-terminated primers have good access to the N site and can be excised more efficiently than primers that do not have a bulky 3′ substituent (e.g. dideoxy or acyclic analogs). To evaluate the model and visualize the relevant structures, we solved the crystal structures of RT in complexes with an AZTMP-terminated template–primer with the 3′ primer end positioned at either the N or the P site.
To permit the isolation and crystallization of complex N, a short-lived intermediate, we covalently trapped the template–primer with the terminal AZTMP at the N site (Figures 1, 2 and 3) to form the ‘open RT(N)–AZTMP complex’ by modifying a cross-linking technique devised by the laboratories of Harrison and Verdine (Huang et al., 1998). [To reduce potential ambiguity, we have abbreviated different states and complexes of HIV-1 RT as RT(P) and RT(N) for HIV-1 RT/DNA binary complexes with their respective primer termini at the P site and N site, RT(apo) for unliganded HIV-1 RT, and RT(ter) for HIV-1 RT/DNA/dNTP ternary complexes. The structures described herein are RT(P)–AZTMP and RT(N)–AZTMP, both terminated with AZTMP. The ternary complex described by Huang et al. (1998) becomes RT(ter)– ddNMP/dTTP.] Central to this procedure are modifications of both the RT (Q258C) and the DNA (Materials and methods). This strategy allowed the generation of RT/DNA complexes that were both stable and homogeneous. Using the same approach, we also prepared a complex of RT with a template–primer bound with its AZTMP terminus at the P site of the enzyme [RT(P)– AZTMP complex].

Fig. 3. Simulated annealing (2Fobs – Fcalc, 1.2σ contours) omit electron density map at the polymerase active site region in complex N (omitting AZTMP and residues within a 5.5 Å radius).
Comparison of the structures of the N and P complexes provides molecular details relevant to the mechanism of ATP-dependent HIV-1 RT resistance to AZT. Because the N and P complexes are trapped at the pre-translocation (complex N) and the post-translocation states (complex P), the structures also provide useful insights into the nature of the transition state and interactions of HIV-1 RT with the product of polymerization prior to translocation.
Results and discussion
Cross-linking of complexes
The methodology used to obtain binary complexes of RT was similar to the protocol developed by the Verdine and Harrison laboratories to prepare the closed RT(ter)– ddNMP/dTTP complex (Huang et al., 1998). In our protocol, the cross-link is with the primer rather than with the template strand, permitting use of the same cross-linkable primer with several different templates. By changing the composition of the reactants, we were able to incorporate an additional nucleotide into the primer strand, generating the N complex.
A modified primer, 5′-ACAGTCCCTGTTCGGG*CGCC-3′, where G* represents a dG residue with a disulfide linker at the exocyclic N2 amino group, was synthesized from a suitably protected, fluorinated oligonucleotide by reaction with bis-(3-aminopropyl) disulfide. We have developed a procedure for preparing this precursor in excellent yield (90%) and high purity (S.G.Sarafianos, A.D.Clark,Jr, S.Tuske, C.Squire, D.Sheng, P.Ilankumaran, A.R.Ramesha, H.Kroth, J.M.Sayer, D.M.Jerina, P.L.Boyer, S.H.Hughes and E.Arnold, in preparation). Previous methodology (Peletskaya et al., 2001) gave lower yields (∼20%).
Screening studies suggested that efficient cross-linking between a modified HIV-1 RT (a cysteine was introduced into the thumb subdomain of p66 at position 258) and nucleic acid takes place when the modified guanosine is at the 6th register upstream from the P site (S.G.Sarafianos et al., in preparation; Figure 2). Optimal cross-linking required the addition of at least one nucleotide, suggesting that the cross-linked complexes are biologically relevant. Thus, for complex P, the template–primer was designed so that the primer contained a modified guanosine on the 5th position upstream from the 3′ end of the primer and the first unpaired base of the template was adenosine, allowing incorporation of an AZTMP followed by translocation and in situ cross-linking. (Figure 2A). For complex N, we used the same modified primer annealed to a template with a thymidine and an adenosine as the first and second nucleotides of the template overhang, which allowed the incorporation of dAMP followed by translocation, incorporation of AZTMP and the covalent trapping by in situ cross-linking prior to a second translocation step (Figure 2B).
Structure determination
The two covalent complexes of RT/DNA with the 3′ ends of the primer strands terminated with AZTMP and trapped at the P [complex P or RT(P)–AZTMP] or at the N [complex N or RT(N)–AZTMP] sites were crystallized as complexes with the monoclonal antibody fragment Fab28 using conditions similar to those used to crystallize the non-cross-linked RT–nucleic acid–Fab complexes (Jacobo-Molina et al., 1993; Ding et al., 1998; Sarafianos et al., 1999a, 2001). The crystals diffracted to comparable resolution, with the same space group symmetry, and had cell dimensions similar to the uncross-linked HIV-1 RT/DNA/Fab complex (Ding et al., 1998). The tethered RT/DNA/Fab crystals, however, yielded sharper diffraction maxima and reduced background scattering, especially when compared after flash cooling. The structures were determined using the molecular replacement method with HIV-1 RT/DNA/Fab as a starting model (Ding et al., 1998). CNS 1.0 (Brünger et al., 1998) and O (Jones et al., 1991) were used for structure refinement and model building. The structures of N and P complexes were refined to 3.0 Å (complex N) and 3.1 Å (complex P) resolution with R-values 0.247 and 0.255, and Rfree values 0.284 and 0.285, respectively.
Description of the structures
Cα superposition of complexes P and N shows that the structures are very similar (the r.m.s.d. between the P and N complexes was 0.5 Å). The differences are concentrated near the polymerase active site. The 3′ end of the AZTMP-terminated primer is at the P site in complex P (Figure 2A) and at the N site in complex N (Figure 2B). The electron density of simulated-annealing omit maps was very clear for the AZTMP at the terminus in both the P and N complexes, except for the azido group (Figure 3). The weak electron density for the azido group suggests it may have multiple conformations.
Structure of complex P
We predicted previously that an incoming dNTP would have unfavorable interactions with the primer terminus of an AZTMP-terminated primer and elements of the polymerase active site, including a divalent metal and the side chain of Asp185 (Boyer et al., 2001). The structure of complex P presented here is consistent with this prediction. Although the azido group may exist in multiple conformations, the conformation that is most consistent with the difference Fourier analysis is similar to that observed in a small molecule crystal structure of AZT. In this conformation, the azido group interacts through the nitrogen closest to the ribose ring (N1′) with Oδ1 of Asp185 (Figure 4). The azido group of AZTMP points in the direction of the p66 fingers subdomain and towards the α-phosphate of the incoming dNTP, which would cause steric conflict with an incoming dNTP in the closed conformation (Figure 5A and C). An incoming dNTP was modeled in two ways: assuming either that the YMDD loop is not displaced when a dNTP reaches the dNTP-binding pocket (alignment of P and ternary complex structures based on residues107–112 and 155–215 of p66; Figure 5C), or that it is displaced together with the AZTMP primer terminus that interacts with the YMDD loop (alignment of complex P and ternary complex structures based on residues 110 and 184–186; data not shown). If the YMDD loop is not displaced (first alignment), the steric conflict is primarily between the side chain of Asp185 and the C5 of the incoming dNTP (Figure 5C). Assuming there is some flexibility of the YMDD loop (second alignment), there would still be steric conflict primarily between the azido group of AZTMP and the α-phosphate of the incoming dNTP (data not shown).

Fig. 4. Ribbon representation of superposed polymerase active sites of complexes P and N (alignment based on p66 residues 107–112 and 155–215). Color scheme: primer strands of complex P (green) and N (magenta), palm subdomains of complexes P (gray) and N (red), and side chains of complexes P (gray) and N (magenta).

Fig. 5. (A) Ribbon representation of superposed polymerase active sites (same basis of superposition as used for Figure 4) of complex N and HIV-1 RT/DNA/dNTP ternary complex [RT(ter)–ddNMP–dTTP complex] (Huang et al., 1998); PDB code 1RTD. Color scheme: side chains of complex N (cyan), primer strand of complex N (magenta), incoming dNTP of the ternary complex (yellow), metals A and B in the ternary complex (yellow). In complex N, the corresponding metals (A′ and B′) are either not seen in the structure and may have been released together with PPi (A′), or are observed (Figure 3) at a position shifted by ∼4.7 Å (metal B′). (B) Superposition of polymerase active sites of the non-terminated [RT(P)–dNMP, green] (Ding et al., 1998; PDB code 2HMI) and AZTMP-terminated P complex [RT(P)–AZTMP, white]. The main structural difference is in the inclination of the terminal nucleotide. (C) Superposition of the polymerase active sites (aligned using p66 residues 107–112 and 155–215) of complex P (in white) on the RT(ter)–ddNMP/dTTP ternary complex (in cyan) (Huang et al., 1998); PDB code 1RTD. The ternary complex YMDD loop is displaced ∼1.0 Å from its position in the P complex. Steric conflicts (in red) are mostly between the C5′ of the incoming dNTP and the side chain of Asp185.
Structure comparison of P complexes that have and have not been cross-linked either to the primer or to the template strand suggests that the protein–DNA cross-link does not perturb the structure significantly (S.Tuske, S.G.Sarafianos, A.D.Clark,Jr, C.Squire, J.P.Ding, K.Das, D.Sheng, P.Ilankumaran, A.R.Ramesha, H.Kroth, J.M.Sayer, D.M.Jerina, P.L.Boyer, S.H.Hughes and E.Arnold, unpublished observations). When the structure of the cross-linked RT(P)–AZTMP complex was compared with that of the uncross-linked complex in which the primer terminus was a normal dNMP [RT(P)–dNMP] (Ding et al., 1998), minor differences were seen in the relative position of the side chain of Asp185 and the AZTMP primer terminus (Figure 5B), suggesting that the polymerase active site can accommodate an AZTMP-terminated template– primer in what is essentially the normal configuration.
Structure of complex N and comparison with the ternary (closed) complex
The AZTMP at the N site interacts with its cognate base on the template strand and with several amino acid residues [Asp113 and Ala114 (main chain), Tyr115 and Q151]. These amino acids are involved in dNTP binding in the RT/DNA/dNTP ternary complex (Huang et al., 1998). The electron density of the azido group in complex N is weak (as it is in the P complex), suggesting that this group may have multiple conformations in the structure. The orientation of the azido group in complex N that is most consistent with the difference Fourier analysis has the azido group projecting almost parallel to the plane of the AZTMP sugar ring, possibly interacting with Asp113 main chain atoms, Ala114 main and side chain atoms, and the AZTMP phosphate.
Comparison of the structures of HIV-1 RT before (Huang et al., 1998) and after (complex N) the catalytic step provides insight into the transition state of DNA polymerization. The N site is occupied by a dNTP and AZTMP in the structures of the ternary complex and complex N, respectively. In the ternary complex, the primer was terminated with a dideoxy nucleotide which prevented incorporation of the bound dNTP (Figure 5A). In complex N, the phosphodiester bond has been formed between the α-phosphate of the incoming AZTTP and the 3′-OH primer terminus. The pyrophosphate produced in the polymerization reaction has been released, resulting in a loss of the contacts of the nucleotide substrate with Lys65 and Arg72, which apparently help to maintain the fingers in a closed conformation in the ternary complex; in the N site complex, the fingers are in the open configuration. It is possible that a conformational change from closed to open form accompanies the release of pyrophosphate (Huang et al., 1998). It is also possible that crystal contacts may contribute to an open-fingers conformation for RT in the RT(N)–AZTMP complex.
There is electron density consistent with the presence of a metal ion at the polymerase active site, at distances of 2.4 Å from Asp110 Oδ1 and 2.3 Å from Asp186 Oδ2. It is likely that this metal corresponds to the metal A in the structure of the ternary complex, displaced (4.7 Å) after the release of the pyrophosphate and the small conformational changes in the chelating aspartates 110 and 186 (Figures 3 and 5A). Hence, it is likely that one of the two divalent metal cations is released with pyrophosphate after incorporation of dNMP, while the other metal remains bound to the active site and shuttles in and out of the coordinating position, accompanied by small torsional movements of residues Asp185 and Asp186. In the structure of the ternary complex, Asp186 did not interact with a metal ion.
The covalent attachment of AZTMP to the primer causes movement of the newly incorporated nucleotide toward the primer strand and the YMDD loop, and away from Tyr115. In addition, the ribose of the AZTMP has undergone a rotation of ∼30°, which permits the newly incorporated phosphate to link the AZTMP phosphate to the 3′ end of the primer (Figure 5). The in-line attack of the 3′-OH of the primer (missing in the structure of the ternary complex) on the α-phosphate of dNTP is accompanied by a stereochemical inversion of the phosphorus substituents, reminiscent of a classical nucleophilic bimolecular substitution (SN2) reaction. These rearrangements result in the loss of an interaction between the side chain of Gln151 and the 3′ group of the incoming nucleotide that was seen in the structure of a ternary complex [RT(ter)–ddNMP/ dTTP]. In the N site complex, there is a hydrogen bond between the side chain of Gln151 and the thymine O2 of the AZTMP base. The hydrogen-bonding ability of the Gln151 side chain may play a role in base selection (Georgiadis et al., 1995).
In the N and ternary complexes, the Oδ2 of Asp185 has close contacts with a non-bridging oxygen of the newly added AZTMP phosphate in the N complex and with the α-phosphate of the incoming dNTP in the ternary complex (∼2.6 Å in both structures). Given the negative charge of these atoms, this interaction should be relatively unfavorable. It is possible, however, that the configuration is stabilized somewhat by an interaction of the Asp185 carboxylate, and two oxygens of the phosphate group (the 3′-O of the penultimate nucleotide of the primer strand and a non-bridging oxygen of the AZTMP phosphate). This interaction is analogous to the frequently observed dicarboxylate interaction where two carboxylates share two protons and have relatively short (∼2.5 Å) oxygen– oxygen contacts (the pH in the crystals was ∼5.6). In the ternary complex, however, the negative charge would be compensated by the presence of a bound metal ion.
Structural changes related to translocation: ‘springboard’ motion of the YMDD motif
Important structural changes occur in the conserved YMDD motif at the polymerase active site of RT during polymerization (Ding et al., 1998). These changes are reminiscent of the motion of a springboard. Superposition of the polymerase active sites of RT (alignment based on p66 residues 107–112 and 155–215) shows that the r.m.s.d. of the Cα positions at the active site for different structures [RT(apo), RT(P)–dNMP, RT(ter)–ddNMP/ dNTP and RT(NNRTI) complexes] is <0.5 Å (Ding et al., 1998). However, portions of the main chain of the YMDD segment are displaced ∼2.8 Å from their position in unliganded HIV-1 RT, which we consider as a reference state of the YMDD motif (Ding et al., 1998) (Figure 6). The largest displacements occur at Asp185, followed by Met184, and result in a local deformation (or partial ‘melting’) of the β6–β10–β9 sheet. The mobility of the YMDD motif combined with the rigidity of the scaffold of the active site suggest a model for translocation: DNA binding is accompanied by a ‘downward’ movement of the YMDD motif by ∼1.7 Å from its position in the unliganded structure (step 1 in Figure 1) (Ding et al., 1998) (distances given for Asp185 Cα). Binding of an incoming dNTP to form the ternary complex (step 2 in Figure 1) and the subsequent reaction to form complex N (step 3 in Figure 1) result in displacements of ∼2.2 and ∼2.7 Å, respectively, relative to unliganded HIV-1 RT (Figure 6C). This motion is analogous to the ‘loading of a springboard’. Conformational strain (including YMDD displacement) may store part of the energy released by phosphodiester bond breakage and dNMP incorporation. Subsequent ‘release’ of the YMDD springboard may supply some of the energy required for translocation: following translocation, the YMDD motif moves upward ∼1.3 Å or approximately halfway between its extreme positions (Figure 6). For the remainder of processive polymerization, the YMDD motif oscillates between the positions of the post-translocation (complex P), pre-incorporation (ternary complex), pre-translocation (complex N) and back to post-translocation position (complex P).

Fig. 6. (A) Conformations of conserved polymerase active site YMDD motif of RT during polymerization. Cα traces of the unliganded [RT(apo), cyan] (Hsiou et al., 1996), complex P [RT(P)–AZTMP, yellow], ternary [RT(ter)–ddNMP/dTTP, white] (Huang et al., 1998) and complex N [RT(N)–AZTMP, magenta] structures. In I, superposition (based on p66 residues 107–112 and 155–215) of unliganded RT(apo) and complex P reveals movement of the YMDD loop in the RT(P)–AZTMP complex relative to RT(apo). In II, superposition of the unliganded RT(apo), complex P [RT(P)–AZTMP] and ternary complex [RT(ter)–ddNMP/dTTP] polymerase active sites shows further displacement of the YMDD loop in the ternary complex. (B) Superposition of RT P and N complexes with AZTMP-terminated nucleic acids shows movement of the YMDD loop following translocation. The Cα of D185 is displaced ∼1.7 Å in complex P, ∼2.2 Å in the ternary complex and ∼2.7 Å in complex N from its reference position in unliganded RT (apo, cyan). (C) Displacement of the Cα carbon of Asp185 of the conserved YMDD motif relative to the unliganded structure of RT(apo) (PDB code: 1DLO). Negatives values in Å correspond to compression. At the lowest point [RT(N)–AZTMP complex], the displacement is ∼2.7 Å. Non-nucleoside reverse transcriptase inhibitor (NNRTI)-bound RT structures have displacements in the opposite direction (YMDD positive), suggesting that conformational changes of YMDD could influence inhibition by NNRTIs (Ding et al., 1998).
During polymerization, RT must accommodate a substantial amount of negative charge proximal to the YMDD motif. The negative charge comes from the side chains of the three catalytic carboxylates (Asp110, Asp185 and Asp186), the three phosphates of the incoming dNTP and, to a lesser extent, from the nucleophilic 3′-OH of the primer terminus and the main chain carbonyl oxygen of Val111. In the RT(ter)–ddNMP/dNTP complex, these negative charges can be accommodated because the two divalent metal ions (Mg2+) help to shield the repulsive forces between the negatively charged species and also help position the crucial groups for efficient catalysis. The chemical step of the reaction results in bond formation between the 3′-O of the primer and the α-phosphate of the dNTP to form the N site complex, concomitant with the release of the β,γ-phosphate as PPi, which may involve residues Lys65 and Arg72 (Sarafianos et al., 1995; Huang et al., 1998; reviewed in Sarafianos et al., 1999b). In the N site complex, there is one divalent metal ion bound at the polymerase active site. Release of one metal ion should reduce the shielding of the local negative charges, generating significant repulsion between the negatively charged and electronegative species that remain close to each other at the active site after release of PPi (the side chain of Asp185, the phosphate of AZTMP and the main chain carbonyl of Val111). We propose that part of this repulsive force may facilitate translocation of the elongated primer. Support for the idea that this repulsive interaction contributes to the translocation mechanism is provided by the close interactions in complex N between Oδ1 and Oδ2 of Asp185, a non-bridging oxygen of the newly incorporated AZTMP phosphate and the bridging 3′-O of the penultimate nucleotide (∼2.6 Å each). In the RT(ter)–ddNMP/dTTP complex, the separation between the Oδ2 of Asp185 and the analogous non-bridging oxygen of the α-phosphate of dNTP (which is not covalently attached in the ternary complex) is 2.7 Å. As the α-phosphorus of dNTP moves closer to the 3′ end of the primer during the approach to the transition state, it will also move closer to the Oδ2 of Asp185 (Figure 5). This structure is stabilized in the ternary complex because the divalent metal shields the repulsion between Oδ2 of Asp185 and the α-phosphate of dNTP. The cross-linking technology made it possible to trap the polymerization product before translocation, although no well-ordered metal remains bound to Asp185 and the phosphate of AZTMP that could neutralize the local concentration of negative charge. The energy required for formation of this relatively unstable intermediate is provided by the breaking of the energy-rich phosphodiester bond linking the α- and β-phosphates in the dNTP, consistent with the hypothesis that cleavage of the dNTP provides the energy to drive translocation (Patel et al., 1995). Other factors potentially involved in translocation include relief of potential steric crowding or conformational strain that may have been introduced during the formation of the N site structure. The equilibrium between the N and P site structures is also influenced by the binding of the incoming dNTP at the N site, which leads to the formation of a complex that prevents the primer end from moving back to the N site (Guajardo and Sousa, 1997; Boyer et al., 2001).
The structure of the pre-translocated RT(N)–AZTMP complex has the fingers in the open conformation and may correspond to a polymerization intermediate after the release of pyrophosphate and fingers opening. It is not clear whether these two events occur concomitantly or if they are discrete steps in the polymerization cycle. It is possible that structural changes involving the fingers may also contribute to translocation. The structure of a catalytically active initiation complex of φ6 RNA polymerase is closed, suggesting the possibility that translocation and insertion of a new NTP (or dNTP in RT) could occur without a major opening of the fingers. If it is possible to obtain a structure of complex N with fingers in the closed conformation [closed RT(N)–AZTMP complex], this could help evaluate the potential contribution of conformational changes in the fingers to the events that occur during processive polymerization.
Similar mobility of active site carboxylate motif in processive polymerases: a general mechanism of translocation?
Structural analysis of complexes of other polymerases with their substrates revealed similar movements in enzymes that contain catalytic carboxylic acids equivalent to the Asp185 and Asp186 of the YMDD motif in RT (Figure 7). The polymerase active sites of the Klenow fragment of Escherichia coli pol I DNA polymerase and T7 RNA polymerase show similar main chain changes in regions that carry the corresponding active site carboxylates. Main chain elements of these enzymes that are equivalent to Met184 and Asp185 show displacements in the presence of nucleic acid (T7 RNA polymerase) or nucleic acid and dNTPs (Klentaq polymerase) similar to what we see for HIV-1 RT (Figure 7A and B). This suggests that the springboard translocation mechanism proposed here for RT may be broadly applicable to other processive polymerases. Pol β, which is evolutionarily distant, has a polymerase active site that is quite different from that of HIV RT, Klenow fragment and T7 RNA polymerase. Pol β is not processive and does not show any analogous conformational changes at the dNTP-binding site (Figure 7C). Further studies will be needed to understand better the role of the movement of the YMDD motif (and related elements of other polymerases) during processive polymerization and translocation.
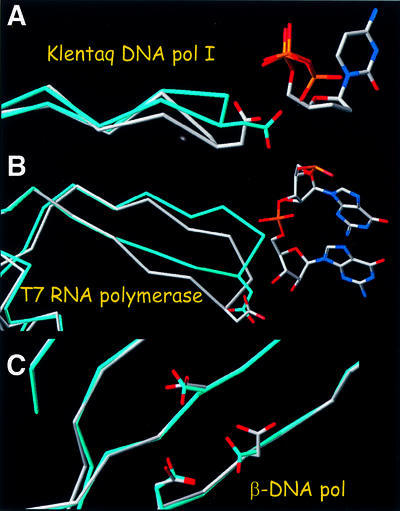
Fig. 7. Structural changes caused by active substrate binding in other processive (Klenow fragment of E.coli pol I and T7 RNA polymerase) and non-processive (human β-pol) polymerases. (A) Structures of the Klenow fragment of Thermus aquaticus DNA polymerase I in the presence (white) and absence (cyan) of nucleic acid substrate (PDB codes 1QTM and 1KTQ). (B) T7 RNA polymerase with nucleic acid present (PDB code 1CEZ) and absent (in cyan; PDB code 1ARO). The 1ARO structure has lysozyme inhibitor bound at a site other than the polymerase site. (C) Structures of human polymerase β in the presence (white) and absence (cyan) of nucleic acid substrate (PDB codes 1BPX and 1BPD).
Structural basis of NRTI resistance by ATP-mediated excision
Excision model. In the model we recently proposed, the pyrophosphate donor in the excision reaction is ATP, and several of the well-characterized AZT resistance mutations enhance the binding of ATP, facilitating excision (Boyer et al., 2001, 2002a,b). ATP is the pyrophosphate donor: the ATP β,γ-pyrophosphate moiety reacts with the AZTMP at the primer terminus. Excision can occur only if the AZTMP-terminated primer is positioned at the N site. The crystal structure of complex N shows the AZTMP-terminated primer bound at the N site and provides the molecular details of this complex [RT(N)–AZTMP complex]. While the crystal structure of complex N does not include ATP, it lends support to our original model in which the β,γ-phosphate moiety of ATP is proximal to the scissile phosphate of the AZTMP-terminated primer, and the adenine moiety of ATP (or other nucleotide bases) makes aromatic π–π interactions with the Tyr215 ring of the AZT-resistant enzyme, stabilized by the Trp210 indole ring when the L210W resistance mutation is present (Boyer et al., 2001; Meyer et al., 2002). Modeling ATP at the same site in the structure of complex P positions the β,γ-phosphate of ATP far from AZTMP.
The original model can be extended to address the factors that affect excision of NRTI-terminated primers. We propose that excision efficiency depends on the extent to which NRTI-terminated template–primer binds in an appropriate configuration for reaction at the N site where excision can occur. Figure 8 summarizes the events that interconnect the structural forms that are relevant for the excision of terminated primers (Figure 8). Steps depicted are: (i) binding of dNTP by inhibitor-terminated complex P yields a closed ternary complex (stability depends on the nature of the template–primer, substrate and enzyme); the 3′ primer terminal NRTI cannot move to the N site and is unavailable for excision; (ii) a fraction of the terminated primer in the P complex translocates from the non-excisable (P) to the excisable (N) position; and (iii) a fraction of the terminated primers at the N site reacts with the pyrophosphate donor, ATP, excising the NRTI to form a dinucleoside tetraphosphate.

Fig. 8. Schematic relationships among events that affect excision-based NRTI resistance (dNTP binding, translocation, excision). Factors that affect any stage will affect the overall equilibrium. X is an NRTI (red), A is ATP (orange) and dNTP (cyan) is the cognate nucleotide triphosphate.
The three states are interconnected; the amount of complex P that is not part of a closed complex determines how much complex N can be formed as a suitable substrate for the excision reaction (Figure 8). Factors that affect any of the above events should affect the excision of an NRTI (or dNMP) from the primer 3′ end.
Factors that may affect ATP-mediated excision. A number of factors may affect the efficiency of ATP-mediated excision. A primary consideration is the ability of the enzyme to bind the substrates of the excision reaction, ATP and NRTI-blocked template–primer. The classic AZT resistance mutations increase the rate of AZTMP excision by improving a pre-existing ATP-binding site; this leads to AZT resistance. However, the issues pertaining to the binding of the nucleic acid are more complex. For excision to occur, the 3′ end of the primer strand must be bound at the N site. Ordinarily, after a dNTP (or the triphosphate form of an NRTI) is incorporated, translocation moves the end of the primer to the P site and there is an equilibrium between binding at the N and P sites. A number of factors can affect the N site/P site equilibrium. These include binding of an incoming dNTP, which can occur when the end of the primer is in the P site. If the last nucleotide at the 3′ end of the primer is a normal deoxynucleotide, the fingers move down onto the incoming dNTP, forming a closed state. The closed state may be transient; after the incoming dNTP is incorporated, pyrophosphate is released and the fingers may open up. However, if the 3′ end of the primer is a dideoxynucleotide, then the closed state [RT–DNAddNMP–dNTP or RT(ter)–ddNMP–dNTP closed complex] is relatively stable (Tong et al., 1997; Meyer et al., 1998; Boyer et al., 2001), shifting the overall equilibrium away from the excision reaction (Figure 8). The opposite is true for AZTMP-terminated primers: because the azido group interferes with the formation of a stable closed com plex [RT–DNAAZTMP–dNTP or RT(ter)–AZTMP–dNTP closed complex] the dNTP binding equilibrium shifts to the right, favoring formation of the binary complex and AZT resistance. Hence, AZTMP is excised much more efficiently than are the corresponding dideoxy chain terminators. Similarly, a major effect of mutations of the fingers insertions is to destabilize the closed configuration of HIV-1 RT. As a consequence, the fingers insertions show enhanced excision of a variety of NRTIs (Mas et al., 2000; Boyer et al., 2002b). Interactions of the primer terminus at the N or P site may also affect the translocation equilibrium; it is possible that the interactions of the azido group of an AZTMP-terminated primer seen in the structure of the N complex contributes to an increase in the relative amount of complex N. So far, it has not been possible to monitor biochemically translocation from the N to the P site; however, the interactions of RT with an AZTMP-terminated primer may affect translocation.
Template–primer positioning. Even when the end of the primer is in the N site, the precise positioning of the reactive components helps to define the rate of excision. Both the nature of nucleic acid sequence/structure and the particular NRTI can influence the precise position of the 3′ end of the primer relative to the polymerase active site. In addition, mutations in HIV-1 RT (e.g. M184V) can affect the positioning of the template–primer (Sarafianos et al., 1999a; Boyer et al., 2001, 2002a). All of these affect the precise position of the nucleic acid relative to the polymerase active site. Although there is no direct evidence, it is likely that some of the mutations that contribute to enhanced ATP binding also influence the precise mode of ATP binding and the excision efficiency.
Excision versus polymerization. In thinking about the ways in which all these factors help to determine the excision efficiency, it is also important to keep in mind that the excision reaction is closely related to polymerization. The excision reaction is closely related to pyrophosphorolysis except that the pyrophosphate donor is ATP instead of PPi. A number of factors that influence the rate of excision can also affect the rate of polymerization. Most of the well-studied mutations that are known to enhance excision are selective: they preferentially enhance the rate of excision, but do not similarly enhance the rate of polymerization. This is essential for drug resistance; a comparable increase in the rates of incorporation and excision of an NRTI will not necessarily cause resistance. As such, it is important, when considering the mechanisms that underlie resistance, to focus on mechanisms that will enhance excision selectively without causing a similar increase in the rate of polymerization. The enhanced binding of ATP caused by the classical AZT resistance mutations is selective for excision because ATP, which is a substrate for the excision reaction, is not a product of the polymerization reaction. This will be an important thread in elucidating the other factors that contribute to the enhanced excision of NRTIs: the mechanisms must somehow affect the excision reaction preferentially.
Finally, the model predicts that the efficiency of the excision reaction and the resulting resistance to NRTIs would be influenced by changes in the concentrations of dNTP, ATP, blocked or unblocked template–primer and dinucleoside tetraphosphate product. Factors that change the concentrations of these compounds (cell cycle, ATPases, enzymes that catabolize tetraphosphates, etc.) will also influence the rate of excision.
In conclusion, the crystal structures of RT with AZTMP-terminated template–primers bound in excisable (N site) and non-excisable (P site) modes are consistent with our model for ATP-mediated excision (Boyer et al., 2001, 2002a). The structures provide useful information on the interactions that are involved and on the specific contacts and roles of various residues at the polymerase active site of RT. The excision model explains the available biochemical data and makes predictions about factors that affect the excision-based NRTI resistance. Comparison of the pre-translocation and post-translocation complexes offers structural insights for the translocation mechanism of DNA polymerization.
Materials and methods
Template–primer design
To immobilize RT covalently on template–primers, we modified the cross-linking technique developed by the laboratories of Harrison and Verdine (Huang et al., 1998). In this procedure, Gln258 of the p66 subunit is mutated to cysteine (Gln258Cys), and the N2 amino group of a guanine is modified to contain a three-methylene tether with a disulfide linker group attached (Figure 2B). In the original protocol (Huang et al., 1998), the nucleic acid had the modified guanine on the template strand. Inspection of the distances and interactions of residue 258 at the minor groove of the bound nucleic acid (Ding et al., 1998) suggested that cross-linking of Cys258 might also be possible if the disulfide group is attached on the primer strand. The sequences of the nucleic acid substrates and the strategy for their design are shown in Figure 3. Synthesis of the specifically modified oligonucleotide will be described elsewhere (S.G.Sarafianos et al., in preparation).
RT–DNA cross-linking
RT containing the C280S mutation in both p66 and p51 subunits and the Q258C mutation only in the p66 subunit was prepared using an expression system described previously (Boyer et al., 1994) with the p66 and p51 subunits expressed from two separate coding regions. The last HIV-1 RT-encoded amino acid at the p51 C-terminus is 428, followed by two glycines and seven histidines, allowing purification using immobilized metal affinity chromatography. For cross-linking reactions, ∼1 mg of purified RT was mixed with the appropriate template–primer at a 1:1.5 RT:template–primer molar ratio. Cross-linking mixtures also contained 1 mM dATP and 1 mM AZTTP to prepare the N site complex and 1 mM AZTTP to prepare the P site complex in a final volume of ∼4 ml. The in situ addition and cross-linking reactions were initiated by the addition of 5 mM MgCl2 and incubated for 2 h at room temperature. The mixture contained the desired product (HIV-1 RT with the p66 subunit cross-linked to the template–primer), together with uncross-linked RT, uncross-linked template–primer, unreacted dATP, AZTTP and MgCl2. The mixture was loaded onto an immobilized Ni2+ affinity column charged with 100 mM nickel sulfate, allowing separation of the uncross-linked template–primer, free nucleotides and other reaction components. Elution with 100 mM imidazole onto a tandemly connected heparin column trapped the uncross-linked RT p66/p51, while the DNA cross-linked RT p66-DNA/p51 species passed through the column (see Figure 4). The peak fractions containing the DNA cross-linked RT, p66–DNA–p51, were pooled, concentrated to 20 mg/ml and mixed with Fab28 at 1:0.8 mass ratio (Jacobo-Molina et al., 1993). Hanging drops were prepared by mixing equal volumes of the complex and crystallization solutions (100 mM cacodylate pH 5.6, 31–34% saturated ammonium sulfate) at 4°C. Before being flash cooled in liquid nitrogen, crystals were soaked in solutions containing 37% saturated ammonium sulfate, 18% glucose (w/v) and 18% glycerol (v/v), reaching the final concentration in two steps of 20 min each. Diffraction data sets were collected at the Advanced Photon Source (APS) BioCars BM-C13 beam line (Table I).
Table I. Summary of data collection and refinement satistics.
| RT(N)–AZTMP | RT(P)–AZTMP | |
|---|---|---|
| Temperature (°C) | –165 | –165 |
| Resolution range (Å) | 40–3.0 | 40–3.1 |
| Space group | P3212 | P3212 |
| Unit cell (a, b, c; Å) | 166.35, 166.35, 220.96 | 166.70, 166.70, 221.12 |
| Rsym | 0.10 | 0.11 |
| Reflections (total/unique) | 496 353/64 902 | 930 560/61 682 |
| Completeness % (total/ highest resolution shell) | 92.6/64.0 | 96.7/82.7 |
| Radiation source | APS (BioCars 13) | APS (BioCars 13) |
| Refinement statistics | ||
| Resolution range (Å) | 20.0–3.0 | 20.0–3.1 |
| No. of atoms | 12 112 | 12 108 |
| Rcryst/Rfree | 0.247/0.284 | 0.255/0.285 |
| R.m.s.d. bonds (Å) | 0.010 | 0.014 |
| R.m.s.d. angles (°) | 1.7 | 1.8 |
| Luzzati coordinate error (Å) | 0.46 | 0.45 |
Structure solution
The structures of the N and P site complexes [RT(N)–AZTMP and RT(P)–AZTMP] were solved by molecular replacement with HIV-1 RT–DNA–Fab as a starting model (Ding et al., 1998). To reduce model bias, we calculated simulated annealing maps with the DNA and the polymerase active site omitted from the model. The DNA position was clearly indicated in difference Fourier maps (Fobs – Fcalc). The structures were refined using the torsion-restrained slow-cooling protocol in CNS (Brünger et al., 1998), followed by positional and individual isotropic thermal parameter refinement. Later stages of refinement incorporated resolution-dependent normalization and bulk solvent correction of structure factors. Statistics of X-ray data collection and refinement are given in Table I. Coordinates and structure factors are available from the Protein Data Bank. The PDB codes for the structures of complex N and complex P are 1N6Q and 1N5Y, respectively.
Acknowledgments
Acknowledgements
We thank other Arnold laboratory members and Keith Brister and staff at the Argonne Photon Source for help in synchrotron data collection, and Chris Squire, Dequan Sheng, Jianping Ding, Ananda Bhattacharya and Pat Clark for assistance and helpful discussions. E.A.’s laboratory has been supported by NIH grants AI 27690 (MERIT award) and GM 56690. S.H.H.’s laboratory has been supported by NIGMS and the NCI.
References
- Arion D., Kaushik,N., McCormick,S., Borkow,G. and Parniak,M.A. (1998) Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry, 37, 15908–15917. [DOI] [PubMed] [Google Scholar]
- Boyer P.L., Ding,J., Arnold,E. and Hughes,S.H. (1994) Drug resistance of HIV-1 reverse transcriptase: subunit specificity of mutations that confer resistance to nonnucleoside inhibitors. Antimicrob. Agents Chemother., 38, 1909–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer P.L., Sarafianos,S.G., Arnold,E. and Hughes,S.H. (2001) Selective excision of AZTMP by drug-resistant HIV reverse transcriptase. J. Virol., 75, 4832–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer P.L., Sarafianos,S.G., Arnold,E. and Hughes,S.H. (2002a) The M184V mutation reduces the selective excision of zidovudine 5′-monophosphate (AZTMP) by the reverse transcriptase of HIV-1. J. Virol., 76, 3248–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer P.L., Sarafianos,S.G., Arnold,E. and Hughes,S.H. (2002b) Nucleoside analog resistance caused by insertions in the fingers of HIV-1 reverse transcriptase involves ATP-mediated excision. J. Virol., 76, 9143–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Ding J., Das,K., Hsiou,Y., Sarafianos,S.G., Clark,J.,A.D., Jacobo-Molina,A., Tantillo,C., Hughes,S.H. and Arnold,E. (1998) Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with double-stranded DNA and an antibody Fab fragment at 2.8 Å resolution. J. Mol. Biol., 284, 1095–1111. [DOI] [PubMed] [Google Scholar]
- Gao H.Q., Boyer,P.L., Sarafianos,S.G., Arnold,E. and Hughes,S.H. (2000) The role of steric hindrance in 3TC resistance of HIV-1 reverse transcriptase. J. Mol. Biol., 300, 403–418. [DOI] [PubMed] [Google Scholar]
- Georgiadis M.M., Jessen,S.M., Ogata,C.M., Telesnitsky,A., Goff,S.P. and Hendrickson,W.A. (1995) Mechanistic implications from the structure of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase. Structure, 3, 879–892. [DOI] [PubMed] [Google Scholar]
- Guajardo R. and Sousa,R. (1997) A model for the mechanism of polymerase translocation. J. Mol. Biol., 265, 8–19. [DOI] [PubMed] [Google Scholar]
- Hsieh J., Zinnen,S. and Modrich,P. (1993) Kinetic mechanism of the DNA-dependent DNA polymerase activity of HIV reverse transcriptase. J. Biol. Chem., 268, 24607–24613. [PubMed] [Google Scholar]
- Hsiou Y., Ding,J., Das,K., Clark,A.D.,Jr, Hughes,S.H. and Arnold,E. (1996) Structure of unliganded HIV-1 reverse transcriptase at 2.7 Å resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure, 4, 853–860. [DOI] [PubMed] [Google Scholar]
- Huang H., Chopra,R., Verdine,G.L. and Harrison,S.C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science, 282, 1669–1675. [DOI] [PubMed] [Google Scholar]
- Jacobo-Molina A. et al. (1993) Crystal structure of HIV-1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc. Natl Acad. Sci. USA, 90, 6320–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kati W.M., Johnson,K.A., Jerva,L.F. and Anderson,K.S. (1992) Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem., 267, 25988–25997. [PubMed] [Google Scholar]
- Li Y., Korolev,S. and Waksman,G. (1998) Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase: structural basis for nucleotide incorporation. EMBO J., 17, 7514–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar C., Abbotts,J., Broder,S. and Wilson,S.H. (1988) Studies on the mechanism of HIV reverse transcriptase: steady-state kinetics, processivity and polynucleotide inhibition. J. Biol. Chem., 263, 15657–15665. [PubMed] [Google Scholar]
- Mas A., Parera,M., Briones,C., Soriano,V., Martinez,M.A., Domingo,E. and Menendez-Arias,L. (2000) Role of a dipeptide insertion between codons 69 and 70 of HIV-1 reverse transcriptase in the mechanism of AZT resistance. EMBO J., 19, 5752–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer P.R., Matsuura,S.E., So,A.G. and Scott,W.A. (1998) Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl Acad. Sci. USA, 95, 13471–13476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer P.R., Matsuura,S.E., Mian,A.M., So,A.G. and Scott,W.A. (1999) A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell, 4, 35–43. [DOI] [PubMed] [Google Scholar]
- Meyer P.R., Matsuura,S.E., Tolun,A.A., Pfeifer,I., So,A.G., Mellors,J.W. and Scott,W.A. (2002) Effects of specific zidovudine resistance mutations and substrate structure on nucleotide-dependent primer unblocking by HIV-1 reverse transcriptase. Antimicrob. Agents Chemother., 46, 1540–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P.H., Jacobo-Molina,A., Ding,J., Tantillo,C., Clark,A.D.,Jr, Raag,R., Nanni,R.G., Hughes,S.H. and Arnold,E. (1995) Insights into DNA polymerization mechanisms from structure and function analysis of HIV-1 reverse transcriptase. Biochemistry, 34, 5351–5363. [DOI] [PubMed] [Google Scholar]
- Peletskaya E.N., Boyer,P.L., Kogon,A.A., Clark,P., Kroth,H., Sayer,J.M., Jerina,D.M. and Hughes,S.H. (2001) Cross-linking of the fingers subdomain of HIV-1 reverse transcriptase to template–primer. J. Virol., 75, 9435–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon J.E. (1993) HIV reverse transcriptase. J. Biol. Chem., 268, 8743–8751. [PubMed] [Google Scholar]
- Rodgers D.W., Gamblin,S.J., Harris,B.A., Ray,S., Culp,J.S., Hellmig,B., Woolf,D.J., Debouck,C. and Harrison,S.C. (1995) The structure of unliganded reverse transcriptase from the HIV-1. Proc. Natl Acad. Sci. USA, 92, 1222–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos S.G., Pandey,V.N., Kaushik,N. and Modak,M.J. (1995) Site-directed mutagenesis of Arg72 of HIV-1 reverse transcriptase. J. Biol. Chem., 270, 19729–19735. [DOI] [PubMed] [Google Scholar]
- Sarafianos S.G., Das,K., Clark,A.D.,Jr, Ding,J., Boyer,P.L., Hughes,S.H. and Arnold,E. (1999a) Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with β-branched amino acids. Proc. Natl Acad. Sci. USA, 96, 10027–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos S.G., Das,K., Ding,J., Boyer,P.L., Hughes,S.H. and Arnold,E. (1999b) Touching the heart of HIV-1 drug resistance: the fingers close down on the dNTP at the polymerase active site. Chem. Biol., 6, R137–R146. [DOI] [PubMed] [Google Scholar]
- Sarafianos S.G., Das,K., Tantillo,C., Clark,A.D.,Jr, Ding,J., Whitcomb,J.M., Boyer,P.L., Hughes,S.H. and Arnold,E. (2001) Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J., 20, 1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong W., Lu,C.D., Sharma,S.K., Matsuura,S., So,A.G. and Scott,W.A. (1997) Nucleotide-induced stable complex formation by HIV-1 reverse transcriptase. Biochemistry, 36, 5749–5757. [DOI] [PubMed] [Google Scholar]