

Abstract
In different eukaryotic model systems, chromatin and gene expression are modulated by post-translational modification of histone tails. In this in vivo study, histone methylation and acetylation are investigated along the imprinted mouse genes Snrpn, Igf2r and U2af1-rs1. These imprinted genes all have a CpG-rich regulatory element at which methylation is present on the maternal allele, and originates from the female germ line. At these ‘differentially methylated regions’ (DMRs), histone H3 on the paternal allele has lysine-4 methylation and is acetylated. On the maternally inherited allele, in contrast, chromatin is marked by hypermethylation on lysine-9 of H3. Allele-specific patterns of lysine-4 and lysine-9 methylation are also detected at other regions of the imprinted loci. For the DMR at the U2af1-rs1 gene, we establish that the methyl-CpG-binding-domain (MBD) proteins MeCP2, MBD1 and MBD3 are associated with the maternal allele. These data support the hypothesis that MBD protein-associated histone deacetylase/chromatin-remodelling complexes are recruited to the parental allele that has methylated DNA and H3-K9 methylation, and are prevented from binding to the opposite allele by H3 lysine-4 methylation.
Keywords: histone methylation/Igf2r/imprinting/Snrpn/U2af1-rs1
Introduction
The expression of imprinted genes in mammals depends on whether the allele is inherited from the mother or the father (Surani et al., 1998). DNA methylation is essential for imprinted gene expression, and at most imprinted loci there are key regulatory sequences that are methylated on one of the parental alleles only (Reik and Walter, 2001). At several of these ‘differentially methylated regions’ (DMRs), the allelic methylation originates from the female or male germ line and is maintained throughout development. DNA methylation cannot, however, be the only determinant of imprints. Allelic methylation at imprinted loci must somehow be protected from the demethylation that occurs following fertilization and during pre-implantation development and, at later stages, from the acquisition of de novo methylation. In addition, some DMRs lose their methylation during pre-implantation development and regain allelic methylation at later stages (Reik and Walter, 2001). Histone deacetylase (HDAC) activities can be linked to DNA methylation (Bird and Wolffe, 1999), but converse mechanisms, involved in protecting the unmethylated allele against the action of DNA methyltransferases and HDACs, remain largely unknown (Bird, 2002).
At several imprinted mouse loci, differences in the histone acetylation status of the maternal and paternal alleles have been detected in cell lines. At the 5′-portion of the Snrpn gene, a DMR that corresponds to the imprinting-control centre involved in the neurogenetic syndrome Prader–Willi Syndrome (PWS; Nicholls et al., 1998), histones H3 and H4 are more highly acetylated on the unmethylated paternal than on the methylated maternal allele (Gregory et al., 2001). At the imprinted Igf2/H19 domain, it is the DMR upstream of H19 that has underacetylation of H4 on the methylated allele (Grandjean et al., 2001). Histone H4, associated with the intronic DMR of the IGF2-receptor (Igf2r) gene is underacetylated on the methylated maternal allele (Hu et al., 2000). Also the DMR of the splice-factor-encoding U2af1-rs1 gene displays pronounced allelic acetylation differences (Gregory et al., 2001).
Methylation, phosphorylation and ubiquitylation of histones are thought to regulate chromatin organization and gene expression as well (Strahl and Allis, 2000; Jenuwein and Allis, 2001). Several recent studies have focused on methylation of histone H3 at lysines 4 (K4) and 9 (K9) (Bannister et al., 2002). We set out to investigate H3-K4 and H3-K9 methylation along imprinted mouse loci, and we focused our in vivo chromatin immunoprecipitation (ChIP) approach on the Snrpn, U2af1-rs1 and Igf2r genes. All three comprise a regulatory DMR with maternal DNA methylation, which originates from the female germ line and is maintained in all somatic lineages (Stöger et al., 1993; Shemer et al., 1997; Shibata et al., 1997). At these germ line DMRs, H3-K4 methylation was detected exclusively on the allele that lacks DNA methylation. This allelic K4 methylation was associated with H3 acetylation and was present in all tissues analysed. On the opposite parental allele, which comprises methylated DNA, a strong enrichment of H3-lysine-9 methylation was observed with an antiserum to a branched peptide comprising methylated K9. Together with in vivo evidence for selective association of methyl-CpG-binding (MBD) proteins, these data reveal a complex regulation of active and repressed chromatin states at imprinted loci, involving histone methylation, histone acetylation, DNA methylation and MBD proteins.
Results
Differential histone methylation at the Snrpn imprinting-control centre
The Snrpn gene is expressed from the paternal allele, encodes an RNA splicing factor and is part of the bicistronic Snurf–Snrpn locus on chromosome 7. It comprises a DMR with maternal allele-specific DNA methylation at the 5′-portion of the Snrpn gene. This maternal methylation originates from the egg and is maintained throughout development (Shemer et al., 1997). This DMR (DMR1; Figure 1A) regulates the paternal expression of Snrpn and flanking genes and cor responds to the imprinting-control centre involved in PWS in humans (Bielinska et al., 2000; Shemer et al., 2000).

Fig. 1. Differential histone methylation and acetylation at Snrpn. (A) Schematic presentation of Snrpn showing exons (vertical bars) and DMRs (horizontal lines). Regions analysed by ChIP are indicated underneath (small horizontal bars). A summary of the maternal (M) and/or paternal (P) allele specificity of the precipitated chromatin is presented underneath for ChIP assays on H3 acetylation on lysines 9 and 14 (H3-AcK9+14), H4 acetylated at lysines 5, 8, 12 and 16 (H4-Ac), H3 methylated at K4 (H3-MeK4) and H3 methylated at K9 (H3-MeK9). (B) ChIP at the DMR1 in brain. Chromatin was extracted from brain tissue dissected from mice that were (C57BL/6 × M.spretus) F1 for chromosome 7, and was precipitated with antisera to H3-AcK9+14, H4-Ac, H3-MeK4 and H3-MeK9. DNAs extracted from antibody-bound (B) and unbound (U) fractions were used for amplification at the DMR1. PCR products were denatured and run through an SSCP gel. Maternal (M) and paternal (P) bands are indicated. To the left, PCR products from control C57BL/6 (D) and M.spretus (S) genomic DNAs and from the input chromatin (I) used for ChIP. (C) The DMR2 in brain. The same bound and unbound fractions as in panel B were used for PCR amplification from the DMR2. (D) The DMR1 in liver. Liver chromatin was precipitated with different antisera, and extracted DNA samples were amplified with the primer pair from the DMR1. (E) The DMR2 in liver. The same extracted DNA samples as in panel D were used as template for amplification with primers from the DMR2.
We analysed chromatin from tissue material dissected from adult mice that were (C57BL/6 × Mus spretus) F1 for chromosome 7. ChIP was carried out on native chromatin fragments of, on average, 2–5 nucleosomes in length. DNA samples were extracted from antibody-bound (B) and unbound (U) fractions, and the relative abundance of maternal and paternal alleles was analysed by radioactive PCR followed by electrophoretic detection of single-strand conformation polymorphisms (SSCP) in the amplification products. For each of the precipitated samples we determined the ratio of the paternal versus maternal (P:M) band intensities. These ratios were corrected for the P:M ratios in the chromatin that was used for precipitation (input chromatin, I). It was essential to perform this correction, since at some regions, the maternal and paternal alleles were not equally represented in the input chromatin. Possibly, at these regions, one of the parental alleles is more sensitive to nucleases, leading to differential chromatin fractionation and purification. Representative results from different experiments are shown in Figures 1–4 and a summary of the corrected P:M ratios is presented in Table I.

Fig. 4. Paternal H3 acetylation and K4 methylation at U2af1-rs1. (A) Schematic presentation of the intronless U2af1-rs1 gene (filled box) with the DMR indicated above. Regions analysed by ChIP are indicated underneath (small horizontal bars). (B) The 5′-UTR. Chromatin extracted from liver was precipitated with different antisera, and after extraction of DNA from the bound and unbound fractions, amplification was with primers from the 5′-UTR. (C) The 3′-UTR. The same precipitation samples and control DNAs as in (B) were used for amplification with a primer pair from the 3′-UTR. (D) Chromatin upstream of U2af1-rs1. The same DNA samples as in (B) were used for amplification with a primer pair from a region at 2 kb upstream of the gene.
Table I. Summary of ChIP data presented as the ratio of paternal over maternal (P:M) band intensities in antibody-bound fractions, corrected for the P:M ratio in the input chromatin.
| Snrpn | DMR1 | DMR2 | ||
|---|---|---|---|---|
| Liver | ||||
| H3: | AcK9+K14 | >10 | 4.1 | |
| H4 | AcK5,8,12,16 | 3.6 | >10 | |
| H3: | MeK4 | >10 | 0.6 | |
| H3: | MeK9 (α-linear) | 1.2 | 1.4 | |
| H3: | MeK9 (α-branched) | 0.1 | 0.1 | |
| Brain | ||||
| H3: | AcK9+K14 | >10 | 5.2 | |
| H4 | AcK5,8,12,16 | >10 | 4.4 | |
| H3: | MeK4 | >10 | 4.7 | |
| H3: | MeK9 (α-linear) | 0.5 | 0.9 | |
| H3: |
MeK9 (α-branched) |
0.5 |
0.3 |
|
|
Igf2r |
|
DMR1 |
DMR2 |
exon-48 |
| Liver | ||||
| H3: | AcK9+K14 | 0.2 | 2.6 | 0.5 |
| H4 | AcK5,8,12,16 | 0.4 | 1.9 | 0.3 |
| H3: | MeK4 | <0.1 | >10 | 1.3 |
| H3: | MeK9 (α-linear) | 1.2 | 0.4 | 3.0 |
| H3: | MeK9 (α-branched) | 2.9 | 0.3 | 3.0 |
| Brain | ||||
| H3: | AcK9+K14 | 1.0 | 5.2 | 1.2 |
| H4 | AcK5,8,12,16 | 1.0 | 4.4 | 1.0 |
| H3: | MeK4 | 1.2 | >10 | 1.1 |
| H3: | MeK9 (α-linear) | 1.2 | 0.6 | 1.1 |
| H3: |
MeK9 (α-branched) |
0.8 |
0.3 |
1.0 |
|
U2af1-rs1 |
|
upstream |
5′-UTR |
3′-UTR |
| Liver | ||||
| H3: | AcK9+K14 | 3.1 | >10 | 4.1 |
| H4 | AcK5,8,12,16 | 2.1 | 4.2 | 1.6 |
| H3: | MeK4 | 3.2 | >10 | >10 |
| H3: | MeK9 (α-linear) | 2.1 | 0.4 | 0.9 |
| H3: | MeK9 (α-branched) | 0.2 | <0.1 | <0.1 |
| Brain | ||||
| H3: | AcK9+K14 | 2.2 | 4.1 | 1.3 |
| H4 | AcK5,8,12,16 | 2.1 | 1.7 | 0.3 |
| H3: | MeK4 | 3.5 | 5.2 | 5.1 |
| H3: | MeK9 (α-linear) | 0.8 | 0.4 | 0.2 |
| H3: | MeK9 (α-branched) | – | 0.2 | 0.3 |
In brain, a tissue with high (paternal) expression of Snrpn, there is methylation at lysine-4 of histone H3 (H3-MeK4) and acetylation on lysines 9 and 14 (H3-AcK9+14) exclusively on the paternal chromosome at the DMR1 (Figure 1B). The same was found in a precipitation with an antiserum that recognizes histone H4 acetylated on lysines 5, 8, 12 and 16 (H4-Ac). These modifications were present in a high proportion of cells since a depletion of the paternal allele was apparent in the corresponding unbound fractions. To analyse H3 lysine-9 methylation (H3-MeK9), we used an antiserum raised against a linear K9-dimethylated peptide. Chromatin precipitated with this ‘α-linear’ antiserum showed a minor enrichment of the maternal allele at the DMR1. At a second DMR, comprising exon-7 of Snrpn, DNA is methylated on the paternal chromosome in brain (Shemer et al., 1997). Also this DMR, DMR2, has paternal H3-MeK4 and H3-AcK9+14 (Figure 1C), and this finding implies that these modifications are present on the allele at which the DNA is methylated. The same was observed for hyperacetylation of histone H4 (H4-Ac), which was detected on the expressed paternal allele only. With the α-linear antiserum to H3-MeK9, no allelic differences were apparent.
In liver, a tissue with little or no expression of the imprinted Snrpn gene, chromatin at the DMR1 is comparable with that in brain in that H3-MeK4, H3-AcK9+14 and H4-Ac are present on the paternal allele exclusively (Figure 1D). Pronounced allelic differences were not detected at the DMR1 with the α-linear antiserum to K9 methylation. In liver, the intragenic DMR2 has comparable levels of DNA methylation on maternal and paternal chromosomes (Gregory et al., 2001). As in brain, however, H3-Ac9+14 and H4-Ac were detected on the paternal allele at this region, but no allelic differences were apparent in K4 methylation, or in the K9 methylation precipitated by the α-linear antibody (Figure 1E).
Since no pronounced allelic differences had been detected with the α-linear antiserum to H3-MeK9 methyl ation, we performed a systematic comparison with another antiserum against a branched peptide with four fingers of the K9-dimethylated TARKST sequence in the N-terminus of histone H3 (Lachner et al., 2001). Apart from the peptides used for their derivation (linear versus branched), a difference between the two antisera is that the α-linear antiserum recognizes H3-K9 dimethylation (Nakayama et al., 2001; Maison et al., 2002) whereas the α-branched antiserum detects both K9 di- and tri-methylation (data not shown, provided by T.Jenuwein). By western blotting, we confirmed that both the antisera react with histone H3 only (data not shown). ChIP was performed on native liver chromatin, and we included the antiserum to H3-K4 methylation as a control. Input-corrected P:M ratios were determined, and we applied real-time PCR to quantify the precipitated chromatin (Figure 2). At the DMR1 of Snrpn, the α-branched antiserum precipitated a similar fraction of chromatin (3%, versus ∼0.2% in a no-antibody control precipitation) as the α-linear antiserum (1.4% precipitation). In contrast to the α-linear antiserum, however, the α-branched antiserum gave strong enrichment of the maternal allele in liver (Figure 2A) and in brain (Table I). Also at the intragenic DMR2, the α-branched antibody gave clear enrichment of the maternal allele, again, both in liver and brain (Table I).
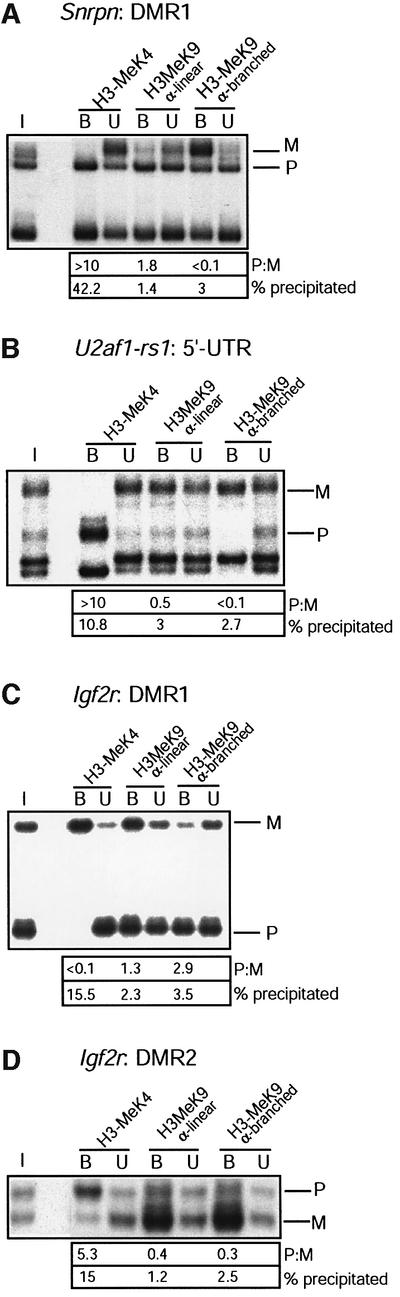
Fig. 2. H3-K9 hypermethylation on chromatin associated with the DNA-methylated allele of DMRs. Liver chromatin was precipitated with antisera to H3-K4 dimethylation (H3-MeK4), to a linear peptide with K9 dimethylation (α-linear) and to a branched peptide with K9 dimethylation (α-branched). (A) Maternal K9 methylation at the Snrpn imprinting-control centre. DNAs extracted from bound and unbound fractions were amplified with primers from the Snrpn DMR1; to the left, amplification from the input chromatin (I). P:M ratios (corrected for P:M in the input chromatin) are indicated. Real-time PCR was applied on the bound fractions to determine the percentage of chromatin precipitated. (B) Maternal K9 methylation at U2af1-rs1. PCR was with the primers from the 5′-UTR. (C) Paternal K9 methylation at the Igf2r DMR1 region. PCR was with the primers from the DMR1. (D) Maternal K9 methylation at the DMR2 of Igf2r. Amplification was with the DMR2 primers.
Analysis of livers from genetically reciprocal mice that were (M.spretus × C57BL/6) F1 for chromosome 7, established that the allelic histone modifications at Snrpn are parent-of-origin dependent and not mouse strain-dependent (data not shown).
Differential histone methylation at Igf2r
In most tissues, the imprinted IGF2/cation-independent mannose-6-phosphate-receptor gene (Igf2r) is expressed from the maternal allele only. This imprinted expression is regulated by a DMR in the second intron (Wutz et al., 1997). The maternal DNA methylation at this DMR (DMR2) originates from the egg and is maintained in all somatic lineages (Stöger et al., 1993). On the unmethyl ated paternal allele, the DMR2 expresses an anti-sense transcript, Air, which is essential for the repression of the paternal Igf2r (sense) promoter (Sleutels et al., 2002). The promoter of Igf2r constitutes a DMR (DMR1), which acquires its paternal DNA methylation during early post-implantation development, as a consequence of the Air-induced repression of the paternal allele. Chromatin was analysed at three regions along Igf2r: the DMR1, the DMR2 and an intragenic region at exon 48 (Figure 3A).

Fig. 3. Allele-specific histone methylation at the Igf2r gene. (A) Schematic presentation of the Igf2r gene, showing the DMR1 (paternal DNA methyl ation), DMR2 (maternal DNA methylation), the transcription initiation sites for sense and antisense transcription (arrows), and the position of exons (filled boxes). Regions analysed by ChIP are indicated underneath (small horizontal bars). (B) The DMR1 in liver. Presentation of results is as in Figure 1. (C) The DMR2 in liver. The same samples were analysed by PCR amplification with the primers from the DMR2. (D) 3′-end of the gene. The same samples as in Figure 2B were analysed by PCR amplification with primers at exon 48. (E) The DMR1 in brain. Chromatin extracted from brain was immunoprecipitated and analysed with the primers from the DMR1. (F) The DMR2 in brain. The precipitated samples as in Figure 2C were analysed with the primers at the DMR2. (G) 3′-end of the gene. The same samples as in Figure 2B were analysed by PCR amplification with primers at exon 48.
Tissues were dissected from mice that were (C57BL/6 × M.spretus) F1 for chromosome 17 (where Igf2r resides). In liver, which has maternal expression of Igf2r, chromatin at the DMR1 has maternal H3-AcK9+14, H4-Ac and H3-K4 methylation (Figure 3B). The α-linear antibody to K9 methylation gave precipitation from both the parental chromosomes. In liver, there were also strong allelic differences at the DMR2 (Figure 3C). At this intronic DMR, acetylation and K4 methylation of histone H3 were present on the paternal allele. Pronounced allelic differences were not apparent with the α-linear antiserum to K9 methylation.
At exon-48, a different pattern of histone modification was detected (Figure 3D). In liver, H3-AcK9+14 is moderately enriched on the expressed maternal allele, and H3-K9 methylation detected by the α-linear antibody is enriched on the repressed paternal allele, but no differential H3-MeK4 was apparent at this intragenic region lacking differential DNA methylation.
In brain, in which the expression of Igf2r occurs from both the parental alleles, the same pronounced H3-MeK4 and H3-Ac differences were detected at the DMR2 as in liver. Also in brain, chromatin at the imprinting-control centre has paternal K4 methylation and acetylation on H3 (Figure 3F). At the Igf2r promoter (DMR1), active on both the parental alleles in brain (Hu et al., 1999), no allelic differences in histone methylation or acetylation were apparent (Figure 3E).
In the comparative ChIP experiments with the K9-methylation antisera (Figure 2), major allelic differences were detected at Igf2r. At the promoter/DMR1, it was the paternal allele, comprising methylated DNA, that was enriched by the α-branched antiserum, whereas the α-linear antiserum gave precipitation of both the parental chromosomes (Figure 2B). At the imprinting-regulating DMR2, the α-branched and the α-linear antisera to H3-K9 methylation gave both enrichment of the maternal chromosome (Figure 2C). On the exon-48 region, K9 methylation was enriched on the repressed paternal allele by both the antisera, in liver but not in brain (Table I).
H3-K9 hypermethylation marks the maternal, and K4 methylation the paternal allele, of U2af1-rs1
The splice factor-encoding U2af1-rs1 gene on mouse chromosome 11 is expressed from the paternal allele. It has maternal DNA methylation, which is established at the 5′-portion of the gene in the female germ line, and spreads across a larger region during early development (Shibata et al., 1997). It is unclear at present whether this germ line DMR is an imprinting-control centre. Analysis of the U2af1-rs1 gene was performed on tissues from congenic mice (line SP11) that were (C57BL/6 × M.spretus) F1 for proximal chromosome 11. Chromatin was analysed in three regions. Two of these, at the 5′-untranslated region (UTR) and the 3′-UTR, are within the region of maternal DNA methylation (Figure 4A). The third region is at 2 kb upstream of the gene, where both the parental chromosomes are methylated (Feil et al., 1997).
We first analysed adult liver, a tissue in which there is no expression of U2af1-rs1 (Gregory et al., 2002). At the 5′-UTR, H3-AcK9+14, H4-Ac and H3-MeK4 are detected on the paternal allele exclusively (Figure 4B). With the α-linear antiserum to H3-MeK9, a slight enrichment of the maternal allele was achieved at the 5′-UTR (Table I). At the 3′-UTR, the paternal allele has K4 methylation and acetylation on H3, and these modifications are absent on the maternal allele (Figure 4C). Levels of H3-K9 methylation detected by the α-linear antiserum at the 3′-UTR appeared similar on maternal and parental alleles. In contrast to the H3 acetylation, the paternal hyperacetyl ation of H4 did not extend into the 3′-UTR, where levels of H4 acetylation were similar on maternal and paternal chromosomes. Chromatin at the region upstream of the gene also displayed paternal K4 methylation and acetyl ation on H3. Unlike at the 5′-UTR, however, no clear enrichment of the maternal allele was obtained with the α-linear K9-methylation antiserum (Figure 4D).
In brain, a tissue with high (paternal) U2af1-rs1 expression, we detected paternal H3-K4 methylation along the domain of differential DNA methylation. Like in liver, this paternal H3-MeK4 correlated with H3-Ac9+14, but not with paternal H4-Ac, which was detected at the 5′-UTR but not the 3′-UTR. As in liver, the paternal K4 methylation and acetylation on H3 seem to spread outside the domain of differential histone methylation, since they are also detected at the region upstream of the U2af1-rs1 gene (Table I).
Comparative studies on K9 methylation (Figure 2C) established that at the 5′-UTR of U2af1-rs1, the α-linear antiserum precipitated a similar fraction of chromatin (3%, compared with 0.2% in a negative control precipitation) as the α-branched antiserum (∼2.6% precipitation). However, whereas the first precipitated both the parental alleles with an ∼2-fold enrichment of the maternal allele, the α-branched antiserum precipitated the maternal allele exclusively. The same was observed at the region upstream of the gene (Table I). At U2af1-rs1, the difference between the K9-methylation antisera may depend on the chromatin preparation protocol used, since it was not apparent when we used formaldehyde crosslinked liver chromatin, on which both the antisera gave an ∼2-fold enrichment of the maternal allele (Figure 5A).

Fig. 5. MBD proteins associate with the maternal allele of U2af1-rs1. (A) Analysis of H3-K9 methylation on crosslinked chromatin. Formaldehyde crosslinked chromatin was precipitated with the α-linear and α-branched antisera to H3-MeK9. DNA from the bound and unbound fractions was amplified with primers from the 5′-UTR. To the left, amplification from input chromatin (I). P:M ratios, corrected for P:M in the input chromatin, are indicated, as well as the percentage of chromatin precipitated at U2af1-rs1. (B) The newly derived antisera αMeCP2 N-t, αMBD1 C-t, αMBD2 N-t and αMBD3 C-t were tested on a western blot with total nuclear extract from a murine cell line. The density markers indicate a pre-stained standard (Kaleidoscope, Bio-Rad). Comparable results were obtained with a human cell line (data not shown). (C) Crosslinked chromatin was precipitated with antisera to MeCP2, MBD1 and MBD3. Corrected P:M ratios are indicated for the precipitated fractions.
MBD proteins associate with the maternal allele of U2af1-rs1
Association of MBD proteins to chromatin may be linked not only to histone deacetylation (Bird and Wolffe, 1999), but possibly also to H3-K9 methylation (Bannister et al., 2002). To explore such possible links in vivo, we performed ChIP on crosslinked chromatin. The U2af1-rs1 DMR was selected for this study because we had found earlier that its constitutive maternal DNA methylation is linked to deacetylation of histone H3, but not H4 (Gregory et al., 2001). Novel antisera were derived against MBD1, MBD2, MBD3 and MeCP2 (see Materials and methods). These displayed good specificity by western blotting and produced bands with mobilities comparable with the expected sizes (Figure 5B; Ballestar and Wolffe, 2001). The MeCP2 antiserum gave a signal of ∼85 kDa, matching exactly with that obtained with another, commercial, antibody that we used (data not shown). The MBD1 antiserum produced a band at ∼70 kDa, as expected (Ng et al., 2000). The N-terminus-specific MBD2 antiserum recognized the long form of MBD2 (MBD2a). The MBD3 antiserum, derived using a peptide from the C-terminus of the protein, gave a doublet signal at ∼36 kDa, compatible with the reported sizes of the two variants in mouse, human and Xenopus (Wade et al., 1999; Zhang et al., 1999).
ChIP was performed on crosslinked chromatin from liver, a tissue in which all four MBD proteins are expressed (Hendrich and Bird, 1998). MeCP2, which recruits SIN3A and associated HDAC1/2 activities to chromatin in vitro (Jones et al., 1998; Nan et al., 1998), was found to be associated with the methylated maternal allele (Figure 5C). This allelic association was apparent with the novel antiserum to the N-terminus of the protein (αMeCP2 N-t), and with a commercial antiserum directed to the C-terminus (αMeCP2 Ct). MBD1, a methylation-dependent transcriptional repressor, which acts by recruiting HDAC activities other than HDAC1 (Ng et al., 2000), was also preferentially associated with the maternal allele. No significant levels of precipitation were obtained at U2af1-rs1 with two different antisera to MBD2, one against the N-terminus specific to MBD2a (αMBD2 N-t) and another against the C-terminus (Figure 5C, data not shown). MBD protein MBD3 does not bind to methylated DNA (Hendrich and Bird, 1998), but is part of the nucleosome remodelling and deacetylation complex NuRD/MeCP1, and can be recruited onto chromatin by MBD2 and other nuclear factors (Ng et al., 1999; Zhang et al., 1999; Feng and Zhang, 2001; Hendrich et al., 2001). We found MBD3 to be associated with the maternal U2af1-rs1 allele.
Discussion
By using a combination of ChIP, PCR and discrimination of parental alleles by SSCP, patterns of histone methyl ation and acetylation were investigated along the imprinted Igf2r, Snrpn and U2af1-rs1 genes in the mouse. To prevent epigenetic alterations as a consequence of in vitro culture of cells, and to approach best the in vivo situation, all the studies were performed on freshly dissected tissue material. Snrpn, Igf2r and U2af1-rs1 are comparable in that they all have a DMR with maternal DNA methylation, present in all somatic lineages. Chromatin is similarly organized at these germ line DMRs. On the methylated maternal allele, histone H3 has lysine-9 hypermethylation and histones H3 and H4 are underacetylated. On the opposite parental allele, the DNA is unmethylated and the associated chromatin has K4 methylation and hyperacetylation on H3. These data present one of the first examples of differential histone methylation in mammalian gene regulation. In addition, the pronounced histone differences at these key DMRs, and elsewhere at the imprinted loci analysed, suggest that histone methylation and acetylation are involved in at least the somatic maintenance of imprinting. Previous examples from other eukaryotic model systems, of the involvement of K4 and K9 methylation on H3 in heritable gene repression/activity include the mating-type loci in fission yeast (Noma et al., 2001), the β-globin locus in chicken (Litt et al., 2001), mammalian pericentric heterochromatin (Peters et al., 2001a) and the inactive X-chromosome in female mammals (Boggs et al., 2001; Heard et al., 2001; Peters et al., 2001b).
DMRs at Igf2r, Snrpn and U2af1-rs1 have allelic K4 and K9 methylation on histone H3. At the imprinting-control centre of Snrpn, a DMR with maternal DNA methylation, chromatin has high levels of paternal K4 methylation and acetylation on H3, in a tissue with high expression (brain) and in a tissue with little or no expression (liver). Histone H3 associated with the maternal allele, in comparison, has no detectable histone acetylation or K4 methylation, but displays a strong enrichment of K9 methylation with the α-branched antiserum. This agrees with a recent human study on PWS and Angelman Syndrome cell lines, in which maternal K9 methylation was detected at the imprinting-control centre of SNRPN (Xin et al., 2001). Although similar amounts of chromatin were precipitated with the α-linear antiserum at the DMR1, only a minor enrichment in K9 methylation was observed on the maternal allele. Possibly, K9 methylation is present on both the parental chromosome, but only on the maternal allele is there a level/configuration of K9 methylation that is recognized by the α-branched antiserum. The finding that the α-linear antiserum precipitates K9 dimethylation on both the parental chromosomes at the Snrpn DMRs (and at DMRs of Igf2r and U2af1-rs1), implies that at the parental allele lacking DNA methylation, K9 dimethylation may occur on chromatin fragments that also have K9+K14 acetylation. This was investigated in more detail by performing ChIP with an antiserum directed specifically to K9 acetylation. At all the DMRs, this antiserum gave the same allele-specific precipitation as the antiserum to K9+14 acetylation (data not shown). Similar evidence for the co-existence of methylation and acetylation at K9 on chromosomal fragments was presented in a recent study on pericentric heterochromatin (Maison et al., 2002).
An alternative explanation for the differential behaviour of the two antisera is that the α-linear antiserum recognizes K9 di-methylation only, whereas the α-branched antibody recognizes both dimethylation and tri-methylation of K9 (T.Jenuwein, personal communication). The allele-specific precipitation with the α-branched antiserum could thus indicate that at the germ line DMRs analysed, tri-methylation of K9 is present on the maternal allele only. The allelic occurrence of tri- rather than di-methylation of K9 needs testing once appropriate antisera are available, and evokes the possibility that this K9 ‘hypermethylation’ constitutes a maternal allele-specific imprint which originates from the female germ line. Importantly, in all ChIP experiments with the α-branched antiserum we obtained precipitation of the parental allele opposite to that precipitated by the antiserum to H3-K4 dimethylation. This suggests that the K9 hypermethylation is mutually exclusive with K4 dimethylation on H3.
At the intragenic DMR2 of Snrpn, allelic patterns of H3-K4 methylation do not correlate with DNA methylation. In brain, where the DMR2 has DNA methylation on the expressed paternal allele, there is paternal K4 methylation on H3. In liver, where the gene is not expressed and the DMR2 has equal DNA methylation on the parental chromosomes, there is no differential K4 methylation. These results also establish that on regions other than the germ line DMRs, K4 methylation may occur on chromatin that comprises methylated DNA sequences. Interestingly, maternal hypermethylation of H3-K9 was detected at the DMR2 in both liver and brain. It seems, therefore, that this maternal mark is present on the gene independent of the level of gene expression.
Also at the imprinting-regulating DMR2 of Igf2r there is paternal K4 methylation and acetylation on histone H3. In contrast, on the maternal allele there is an enrichment in H3-K9 hypermethylation and no acetylation is apparent. A curious implication of this finding is that transcription on the maternal allele can proceed through a chromatin domain with underacetylation of histones H3 and H4 and H3-K9 hypermethylation. The DMR1/promoter region displayed pronounced differences between liver and brain, and histone modifications in this region seem to reflect the maternal versus biallelic Igf2r expression in these respective tissues. Hence, in liver, where only the maternal promoter is active, there is maternal K4 methylation and H3 and H4 acetylation, whereas the paternal allele has H3-K9 hypermethylation. In brain, where both the maternal and the paternal promoter are active (Hu et al., 1999), no allelic differences in histone methylation or acetylation were detected at the DMR1. At the intragenic exon-48 region, no pronounced allelic differences in K4 methyl ation or acetylation on H3 were apparent. Acetylation of histone H4, however, was higher on the expressed maternal than on the repressed paternal allele in liver, suggesting that modifications at the 3′-portion of the gene reflect the expression status rather than the parental origin of the gene.
Also at a third germ line DMR with maternal DNA methylation, the CpG island of the U2af1-rs1 gene, there is K4 methylation and acetylation on H3 on the paternal allele, and H3-K9 hypermethylation on the maternal allele. In the different tissues analysed, these modifications seem spread across a larger domain, since they include the region upstream of the gene. This finding is interesting relative to non-mammalian model systems, in which domains with specific H3 methylation and acetylation patterns have been described (Noma et al., 2001; Litt et al., 2001).
Allelic recruitment of HDAC/chromatin remodelling complexes by MBD proteins
It has been reported that the nucleosome-remodelling and deacetylase complex NuRD can bind to the N-terminus of histone H3, and that this association is prevented by K4 methylation (Nishioka et al., 2002; Zegerman et al., 2002). NuRD is a multi-protein complex comprising HDAC1 and HDAC2, as well as the MBD protein MBD3 (Zhang et al., 1999). By performing ChIP on crosslinked chromatin, we found MBD3 to be associated with the maternal U2af1-rs1 allele. Since MBD3 does not bind to methylated DNA sequences, but is recruited indirectly to the chromatin by other nuclear proteins (Hendrich and Bird, 1998; Saito and Ishikawa, 2002), this finding is unlikely to be an artefact of the ChIP procedure. In addition, there has been no evidence from other studies of artefactual association of MBD proteins in formaldehyde crosslinked chromatin (Lorincz et al., 2001; Drewell et al., 2002). Rather, this finding suggests that NuRD is bound to the allele that comprises methylated DNA, and is excluded from the opposite parental allele by H3-K4 methylation. NuRD can be recruited to chromatin by MBD2 (Zhang et al., 1999). However, using antisera to different epitopes of MBD2, we did not obtain evidence for allele-specific association of this MBD protein to U2af1-rs1, although we did observe allele-specific binding to another imprinted mouse gene (A.M.Hever, K.Delaval, C.Fournier and R.Feil, unpublished data). Possibly, NuRD is recruited to U2af1-rs1 by MBD2-independent mechanism(s) (Hendrich et al., 2001). Alternatively, NuRD binding may have obscured MBD2 in the crosslinked chromatin.
MeCP2 interacts with SIN3A/B and associated HDAC1 and HDAC2 activities in vitro (Jones et al., 1998; Nan et al., 1998). The observed binding of MeCP2 to the maternal U2af1-rs1 allele thus constitutes a second mechanism by which HDAC activities can be recruited, and extends an earlier report on the allelic association of MeCP2 to the imprinted H19 gene (Drewell et al., 2002). Additionally, we found MBD1 to be associated with the maternal U2af1-rs1 allele. This MBD protein represses genes by histone deacetylation, but here the mechanism seems to involve a histone deacetylase other than HDAC1 (Ng et al., 2000). Collectively, our data support the hypothesis that several MBD proteins are associated with chromatin on the maternal U2af1-rs1 allele. These would recruit different HDACs to the chromatin. How the maternal CpG methylation at U2af1-rs1 is linked to deacetylation of histone H3, but not of H4 (Gregory et al., 2001), remains unclear. Possibly, HDAC activities recruited to the locus act on H3 preferentially. Alternatively, specific histone modifications recruit HDAC activities to H3, rather than H4. The observed H3-K9 hypermethyl ation, for instance, might be involved in the recruitment of NuRD and other HDAC complexes.
Maintenance of opposite epigenetic states at imprinted genes
Our combined data lead us to propose a model for the maintenance of the parental allele-specific chromatin states at DMRs which is based on the interdependence and mutual exclusion of different epigenetic modifications (Figure 6).

Fig. 6. A working model for the maintenance of opposite epigenetic states at DMRs. Methylation (M) of DNA is present on one of the parental alleles only (at many DMRs, this is the maternal allele). This methylation is recognized by MBD proteins, which recruit HDAC/chromatin-remodelling complexes to the chromatin. HDACs (filled circles) ensure deacetylation of histones and may be associated with DNMTs, which maintain the DNA methylated. MBD protein binding may also be mechanistically linked to the H3-K9 hypermethylation on this parental allele, and this could constitute a second element in the maintenance of its DNA methylation. The opposite parental allele has no DNA methylation and is not accessible to HDAC complexes and associated DNMT activities, possibly in part because of the H3-K4 methylation. H3-K4 methylation could thus prevent DNA methylation from becoming established. In contrast, HATs and K4 histone methyltransferase (HMT) have access to the chromatin and this maintains the acetylation and K4 methylation on this allele.
On the methylated parental allele, CpG methylation is recognized by MBD proteins. This effects the recruitment of HDAC- and chromatin-remodelling activities, which maintain the histones in an underacetylated and compacted state. HDACs may be associated with DNA methyltransferases in vivo (Fuks et al., 2000) and these are important for maintaining the CpG methylation. In addition, there is H3-K9 hypermethylation and this could be another factor in the maintenance of DNA methylation. In the fungus Neurospora crassa, for instance, ablation of an H3–K9 methyltransferase leads to loss of DNA methylation, and the same phenotype was obtained when lysine-9 in H3 was replaced with another amino acid (Tamaru and Selker, 2001). In Arabidopsis, loss of H3-K9 methylation leads to a substantial decrease in DNA methylation as well (Gendrel et al., 2002). Given the putative role of H3-K9 hypermethylation in the maintenance of DNA methylation at DMRs, the question arises whether this histone modification is part of the imprint which originates from the germ line. It has been reported that the chromosomes in female germ cells have K9 methylation on histone H3. This persists after fertilization, whereas the male genome acquires comparable levels of K9 methylated only at around the two-cell stage (Arney et al., 2002; Cowell et al., 2002). Another role of H3-K9 hypermethylation might be to prevent other modifications, including acetylation, from becoming established. In a previous study, the U2af1-rs1 and Snrpn DMRs were found to be extremely resistant to trichostatin A, an inhibitor of HDACs. Particularly, on the maternal allele, the allele that has H3-K9 hypermethylation, there was no apparent acquisition of histone acetylation upon inhibition of HDACs (Gregory et al., 2002).
On the opposite parental chromosome, chromatin-mediated mechanisms prevent the DNA from becoming methylated. One putative candidate here is the K4 methylation on H3. This histone modification might exert its action by having a negative effect directly on DNA methyltransferases (DNMTs), such that these cannot methylate the associated DNA sequences, or indirectly, via inhibition of histone deacetylation and chromatin-remodelling activities. Support for the latter comes from the observation that K4 methylation prevents association of the HDAC1/2 containing NuRD complex to the chromatin (Zegerman et al., 2002). Additionally, K4 methylation might be required for the establishment of lysine acetylation on H3 (Bernstein et al., 2002). Acetylation together with the K4 methylation may preclude K9 on H3 from becoming methylated (Nishioka et al., 2002), and could thereby prevent establishment or maintenance of CpG methylation. Indeed, at the DMRs analysed, K4 methylation and the K9 hypermethylation were never detected on the same parental chromosome. Other, non-exclusive, mechanisms that have been proposed to prevent the DNA at CpG islands from becoming methylated (on one of the parental alleles at imprinted loci) include non-histone protein binding and transcriptional activity, possibly in particular during early development (Feil and Khosla, 1999; Bird, 2002).
Future work should unravel the precise inter-relationships between histone methylation and other modifications at imprinted mammalian loci, and when during development the allelic patterns of H3-K9 and H3-K4 methylation become established. Another avenue concerns the enzymes that are involved in the regulation of the differential K9 and K4 methylation. Recent studies have shown, for instance, that the SET domain protein SUV39H is essential for K9 methylation at mammalian pericentric heterochromatin (Peters et al., 2001a), but other SET domain methyltransferases appear to be involved in its regulation at facultative heterochromatin (Peters et al., 2001b) and at euchromatic regions (Tachibana et al., 2002). SET domain activities may also relate to the issue as to whether the allelic K9 hypermethylation on imprinted loci is recognized by the heterochromatin protein-1 or other proteins, and may thus effect chromatin compaction, like at pericentric heterochromatin.
Materials and methods
Animals
Mice, interspecific hybrids for specific chromosomal regions, were obtained by mating C57BL/6 females with M.spretus males, and by back-crossing against C57BL/6 males for several generations. Congenic line SP11 has an M.spretus proximal chromosome 11 on a C57BL/6 background (Gregory et al., 2001).
Antisera, chromatin preparation and ChIP
Polyclonal rabbit antisera were made against linear peptides corresponding to aa 6–25 of human MeCP2 (antiserum αMeCP2 N-t), 2–20 of human MBD2 (αMBD2 N-t), 254–270 of human MBD3 (αMBD3 C-t) and the KQEPPDPEEDKEENKDDSAS peptide common to all the MBD1 isoforms (αMBD1 C-t). Commercial antisera were also used for ChIP: an H3 antibody to a branched peptide with four fingers of the K9-dimethylated TARKST sequence (Lachner et al., 2001); an antiserum to aa 6–13 of H3 with a dimethylated K9 (Upstate Biotechnolgy 07–212); an antiserum against aa 1–8 of H3 dimethylated at K4 (Upstate Biotechnology; Litt et al., 2001); an antiserum against a peptide comprising aa 1–20 of H3 acetylated at K9 and K14 (Upstate Biotechnology); an antiserum against a peptide with aa 1–12 of H3 acetylated at K9 (Upstate Biotechnology); an antiserum to aa 2–19 of H4 acetylated at lysines 5, 8, 12 and 18 (Upstate Biotechnology); an antiserum to aa 465–478 of MeCP2 (Upstate Biotechnology) and an antiserum to aa 152–262 of MBD2 (Upstate Biotechnology). Purification of nuclei, fractionation of chromatin with MNase to obtain chromatin fragments of 1–5 nucleosomes in length, and ChIP, were performed as described previously (Gregory et al., 2001). Approximately 40 µg of native chromatin were used for each ChIP, using ∼4 µg of affinity-purified antibody. For preparation of crosslinked chromatin, tissue was fractionated in a glass homogenizer, passed through cheesecloth, and incubated for 5 min at 25°C in phosphate-buffered saline (PBS) with 1% formaldehyde, 1 mM phenylmethyl sulfonyl fluoride (PMSF), 5 mM Na-butyrate. Crosslinking was stopped by addition of glycine to 125 mM and cells were washed in PBS, 1 mM PMSF, 5 mM Na-butyrate. All subsequent procedures were performed on ice, with buffers containing 1 mM PMSF, 5 mM Na-butyrate and a protease inhibitor mixture (Roche Diagnostics). Cells were lysed in 5 mM PIPES pH 8, 85 mM KCl, 0.5% NP40 and nuclei were harvested by centrifugation, followed by lysis in 50 mM Tris–HCl pH 8, 10 mM EDTA, 1% SDS. Ultrasound sonication was performed to generate fragments of up to ∼1 kb in size. ChIP was performed with 125–250 µg of (Sepharose-A precleared) chromatin in 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl pH 8, 167 mM NaCl, with ∼5 µg of affinity-purified antiserum. Antibody-bound chromatin was brought down with protein A–Sepharose beads, followed by extensive washing and elution with 1% SDS, 100 mM NaHCO3.
PCR–SSCP and quantitative PCR
For PCR–SSCP, genomic DNA was extracted from antibody-bound and unbound fractions, and 50 ng were used to PCR amplify in the presence of [32P]dCTP (1% of total dCTP). Amplification was from three regions in U2af1-rs1: a 192 bp upstream region, a 293 bp region at the 5′-UTR and a 163 bp 3′-UTR region. Amplification at Snrpn was from a 228 bp region at the DMR1 and a 499 bp region in DMR2 (Gregory et al., 2001). Igf2r primers amplified from three regions (T-annealing: 68°C): a 162 bp fragment at the DMR1 (forward, GGYGCTGGACGGGGAAACTGA GGT; reverse, CCAGTCCCGGGTCACATGAGCATCG), a 169 bp fragment at the DMR2 (forward, CGTGTAGTTCAGAACACTGGTG AGC, reverse, TACGCGAGGTGAGGGTTCCACTGAT), and a 225 bp fragment at exon-48 (forward, GTCAGCAAGGAGGAGGAG ACAGATG; reverse, CTCCGCTCCTCGGCCTGAGTGAACT). After denaturation, PCR products were resolved by non-denaturing acrylamide gel electrophoresis. Bands on exposed X-ray films were quantified using the Aida-2 imaging software. Antibody-bound fractions were also analysed by quantitative PCR (LightCycler, Roche Diagnostic), using SYBR green dye (Qiagen). The primers used for this were the same as those described for PCR–SSCP (see above).
Acknowledgments
Acknowledgements
We are grateful to Thomas Jenuwein for antiserum to H3-K9 methylation and for providing details about its specificity, to Jamal Tazi, Marie-Luce Vignais and Thierry Forné for reading the manuscript, to Sara Salinas for help with western blotting, and to the IGMM animal unit for expert mouse breeding. R.F. acknowledges grant support from the CNRS, the Human Frontier Science Program and the Fondation pour la Recherche Médicale.
References
- Arney K.L, Bao,S., Bannister,A.J., Kouzarides,T. and Surani,M.A. (2002) Histone methylation defines epigenetic asymmetry in the mouse zygote. Int. J. Dev. Biol., 46, 317–320. [PubMed] [Google Scholar]
- Ballestar E. and Wolffe,A.P. (2001) Methyl-CpG-binding proteins. Targeting specific gene repression. Eur. J. Biochem., 268, 1–6. [DOI] [PubMed] [Google Scholar]
- Bannister A.J., Schneider,R. and Kouzarides,T. (2002) Histone methylation. Dynamic or static? Cell, 109, 801–806. [DOI] [PubMed] [Google Scholar]
- Bernstein B.E., Humphrey,E.L., Erlich,R.L., Schneider,R., Bouman,P., Liu,J.S., Kouzarides,T. and Schreiber,S.L. (2002) Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl Acad. Sci. USA, 99, 8695–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielinska B, Blaydes,S.M., Buiting,K., Yang,T., Krajewska-Walasek,M., Horsthemke,B. and Brannan,C.I. (2000) De novo deletions of SNRPN exon 1 in early human and mouse embryos result in a paternal to maternal imprint switch. Nat. Genet., 25, 74–78. [DOI] [PubMed] [Google Scholar]
- Bird A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev., 16, 6–21. [DOI] [PubMed] [Google Scholar]
- Bird A.P. and Wolffe,A.P. (1999) Methylation-induced repression—belts, braces and chromatin. Cell, 99, 451–454. [DOI] [PubMed] [Google Scholar]
- Boggs B.A., Chueng,P., Heard,E., Spector,D.L., Chinault,A.C. and Allis,C.D. (2001) Differentially methylated forms of histone H3 show unique association patterns with inactive human X chromosome. Nat. Genet., 30, 73–80. [DOI] [PubMed] [Google Scholar]
- Cowell I.G. et al. (2002) Heterochromatin, HP1 and methylation at lysine 9 of histone H3 in animals. Chromosoma, 111, 22–36. [DOI] [PubMed] [Google Scholar]
- Drewell R.A., Goddard,C.J., Thomas,J.O. and Surani,M.A. (2002) Methylation-dependent silencing at the H19 imprinting control centre by MeCP2. Nucleic Acids Res., 30, 1139–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R. and Khosla,S. (1999) Genomic imprinting in mammals, an interplay between chromatin and DNA methylation? Trends Genet., 15, 431–435. [DOI] [PubMed] [Google Scholar]
- Feil R., Boyano,M.D., Allen,N.D. and Kelsey,G. (1997) Parental chromosome-specific chromatin conformation in the imprinted U2af1-rs1 gene in the mouse. J. Biol. Chem., 272, 20893–20900. [DOI] [PubMed] [Google Scholar]
- Feng Q. and Zhang,Y. (2001) The MeCP1 complex represses transcription through preferential binding, remodeling and deacetylating methylated nucleosomes. Genes Dev., 15, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F., Burgers,W.A., Brehm,A., Hughes-Davies,L. and Kouzarides,T. (2000) DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet., 24, 88–91. [DOI] [PubMed] [Google Scholar]
- Gendrel A.V., Lippman,Z., Yordan,C., Colot,V. and Martienssens,R. (2002) Dependence of heterochromatic histone H3 methylation patterns on the Arabidopsis gene DDM1. Science, 20, 20. [DOI] [PubMed] [Google Scholar]
- Grandjean V., O’Neill,L., Sado,T., Turner,B. and Ferguson-Smith,A. (2001) Relationship between DNA methylation, histone H4 acetylation and gene expression in the mouse imprinted Igf2-H19 domain. FEBS Lett., 488, 165–169. [DOI] [PubMed] [Google Scholar]
- Gregory R.I., Randall,T.E., Johnson,C.A., Khosla,S., Hatada,I., O’Neill, L.P., Turner.B.M. and Feil,R. (2001) DNA methylation is linked to deacetylation of histone H3, but not H4, on the imprinted genes Snrpn and U2af1-rs1. Mol. Cell. Biol., 21, 5426–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory R.I., O’Neill,L.P., Randall,T.E., Fournier,C., Khosla,S., Turner, B.M. and Feil,R. (2002) Inhibition of histone deacetylases alters chromatin conformation at the imprinted mouse U2af1-rs1 locus in mouse embryonic stem cells. J. Biol. Chem., 277, 11728–11734. [DOI] [PubMed] [Google Scholar]
- Heard E., Rougeulle,C., Arnaud,D., Avner,P., Allis,C.D. and Spector,D.L. (2001) Methylation of histone H3 at lys-9 is an early mark on the X chromosome during X inactivation. Cell, 107, 727–738. [DOI] [PubMed] [Google Scholar]
- Hendrich B. and Bird,A.P. (1998) Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol., 18, 6538–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B., Guy,J., Ramsahoye,B., Wilson,V.A. and Bird,A. (2001) Closely related proteins MBD2 and MBD3 play distinctive but interactive roles in mouse development. Genes Dev., 15, 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.F., Balaguru,K.A., Ivaturu,R.D., Oruganti,H., Li,T., Nguyen,B.T., Vu,T.H. and Hoffman,A.R. (1999) Lack of reciprocal genomic imprinting of sense and antisense RNA of mouse insulin-like growth factor II receptor in the central nervous system. Biochem. Biophys. Res. Commun., 257, 604–608. [DOI] [PubMed] [Google Scholar]
- Hu J.-F., Pham,J., Dey,I., Li,T., Vu,T.H. and Hoffman,A.R. (2000) Allele-specific histone acetylation accompanies genomic imprinting of the insulin-like growth factor II receptor gene. Endocrinology, 141, 4428–4435. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. and Allis,C.D. (2001) Translating the histone code. Science, 293, 1074–1079. [DOI] [PubMed] [Google Scholar]
- Jones P.L., Veenstra,G.J., Wade,P.A., Vermaak,D., Kass,S.U., Landsberger, N., Strouboulis,J. and Wolffe,A.P. (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet., 19, 187–191. [DOI] [PubMed] [Google Scholar]
- Lachner M., O’Carroll,D., Rea,S., Mechtler,K. and Jenuwein,T. (2001). Methylation of histone H3 lysine-9 creates a binding site for HP1 protein. Nature, 410, 116–120. [DOI] [PubMed] [Google Scholar]
- Litt M.D., Simpson,M., Gaszner,M., Allis,C.D. and Felsenfeld,G. (2001) Correlation between histone lysine methylation and developmental changes at the chicken β-globin locus. Science, 293, 2453–2455. [DOI] [PubMed] [Google Scholar]
- Lorincz M.C., Schübeler.D. and Groudine,M. (2001) Methylation-mediated proviral silencing is associated with MeCP2 recruitment and localized histone H3 deacetylation. Mol. Cell. Biol., 21, 7913–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison C., Bailey,D., Peters,A.H., Quivy,J.P., Roche,D., Taddei,A., Lachner,M., Jenuwein,T. and Almouzni,G. (2002) Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat. Genet., 19, 329–334. [DOI] [PubMed] [Google Scholar]
- Nakayama J., Rice,J.C., Strahl,B.D., Allis,C.D. and Grewal,S.I. (2001). Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science, 292, 110–113. [DOI] [PubMed] [Google Scholar]
- Nan X., Ng,H.-H., Johnson,C.A., Laherty,C.D., Turner,B.M., Eisenman, R.N. and Bird,A. (1998) Transcriptional repression by the methyl CpG binding protein MeCP2 involves a histone deacetylase complex. Nature, 393, 386–389. [DOI] [PubMed] [Google Scholar]
- Ng H.-H., Zhang,Y., Hendrich,B., Johnson,C.A., Turner,B.M., Erdjument-Bromage,H., Tempst,P., Reinberg,D. and Bird,A. (1999) MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet., 23, 58–61. [DOI] [PubMed] [Google Scholar]
- Ng H.-H., Jeppesen,P. and Bird,A. (2000) Active repression of methylated genes by the chromosomal protein MBD1. Mol. Cell. Biol., 20, 1394–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls R.D., Saitoh,S. and Horsthemke,B. (1998) Imprinting in Prader–Willi and Angelman syndromes. Trends Genet., 14, 194–200. [DOI] [PubMed] [Google Scholar]
- Nishioka K., Chuikov,S., Sarma,K., Erdjument-Bromage,H., Allis,D., Tempst,P. and Reinberg,D. (2002) Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev., 16, 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K-I., Allis,C.D. and Grewal,S.I.S. (2001) Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science, 293, 1150–1155. [DOI] [PubMed] [Google Scholar]
- Peters A.H. et al. (2001a) Loss of Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell, 107, 323–337. [DOI] [PubMed] [Google Scholar]
- Peters A.H., Mermoud,J.E., O’Carroll,D., Pagani,M., Schweizer,D., Brockdorff,N. and Jenuwein,T. (2001b) Histone H3 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet., 30, 77–80. [DOI] [PubMed] [Google Scholar]
- Reik W. and Walter,J. (2001) Genomic imprinting: parental influence on the genome. Nat. Rev. Genet., 2, 21–32. [DOI] [PubMed] [Google Scholar]
- Saito M. and Ishikawa,F. (2002) The mCpG-binding domain of human MBD3 does not bind to mCpG but interacts with NuRD/Mi2 components HDAC1 and MTA2. J. Biol. Chem., 277, 35434–35439. [DOI] [PubMed] [Google Scholar]
- Shemer R., Birger,Y., Riggs,A.D. and Razin,A. (1997) Structure of the imprinted mouse Snrpn gene and establishment of its parental-specific methylation pattern. Proc. Natl Acad. Sci. USA, 94, 10267–10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemer R., Hersko,A.Y., Perk,J., Mostoslavsky,R., Tsuberi,B.Z., Cedar,H., Buiting,K. and Razin,A. (2000) The imprinting box of the Prader–Willi/Angelman syndrome domain. Nat. Genet., 26, 440–443. [DOI] [PubMed] [Google Scholar]
- Shibata H. et al. (1997) An oocyte-specific methylation imprint center in the mouse U2afbp-rs/U2af1-rs1 gene marks the establishment of allele-specific methylation during preimplantation development. Genomics, 44, 171–178. [DOI] [PubMed] [Google Scholar]
- Sleutels F., Zwart,R. and Barlow,D.P. (2002) The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature, 415, 810–813. [DOI] [PubMed] [Google Scholar]
- Stöger R., Kibicka,P., Liu,C.G., Kafri,T., Razin,A., Cedar,H. and Barlow,D. (1993) Maternal-specific methylation of the imprinted mouse Igf2r locus identifies the expressed allele as carrying the imprinting signal. Cell, 73, 61–71. [DOI] [PubMed] [Google Scholar]
- Strahl B.D. and Allis,C.D. (2000) The language of covalent histone modifications. Nature, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Surani M.A. (1998) Imprinting and the initiation of gene silencing in the germ line. Cell, 93, 309–312. [DOI] [PubMed] [Google Scholar]
- Tachibana M. et al. (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev., 16, 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaru H. and Selker,E.U. (2001) A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature, 414, 277–283. [DOI] [PubMed] [Google Scholar]
- Wade P.A., Gegonne,A., Jones,P.L., Ballestar,E., Aubry,F. and Wolffe,A.P. (1999) Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat. Genet., 23, 62–66. [DOI] [PubMed] [Google Scholar]
- Wutz A., Smrka,O.W., Schweifer,N., Schellander,K., Wagner,E.F. and Barlow,D.P. (1997) Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature, 389, 745–749. [DOI] [PubMed] [Google Scholar]
- Xin Z., Allis,C.D. and Wagstaff,J. (2001) Parent-specific complementary patterns of histone H3 lysine 9 and H3 lysine 4 methylation at the Prader–Willi syndrome imprinting center. Am. J. Hum. Genet., 69, 1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegerman P., Canas,B., Pappin,D. and Kouzarides,T. (2002) Histone H3 lysine 4 methylation disrupts the binding of the nucleosome remodelling and deacetylase (NuRD) repressor complex. J. Biol. Chem., 277, 11621–11624. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Ng,H.-H., Erdjument-Bromage,H., Tempst,P., Bird,A. and Reinberg,D. (1999) Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev., 13, 1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]