

Abstract
Every successful pregnancy requires proper embryo implantation. Low implantation rate is a major problem during infertility treatments using assisted reproductive technologies (ART) 1. Here we report a new molecular influence on implantation through the lysophosphatidic acid (LPA) receptor LPA3 2–4. Targeted deletion of LPA3 in mice resulted in significantly reduced litter size, which could be attributed to delayed implantation and altered embryo spacing. These two events led to delayed embryonic development, hypertrophic placentas shared by multiple embryos, and embryonic death. An enzyme demonstrated to influence implantation, cyclooxygenase-2 (COX-2) 5, was down-regulated in LPA3-deficient uteri during preimplantation. Down regulation of COX-2 led to reduced levels of prostaglandins that are critical for implantation 1. Exogenous administration of the prostaglandins PGE2 and cPGI into LPA3-deficient females rescued delayed implantation but did not rescue defects in embryo spacing. These data identify LPA3 receptor-mediated signalling as a new influence on implantation and further indicate linkage between LPA signalling and prostaglandin biosynthesis.
Multiple factors can adversely affect successful pregnancy. Two of these factors are failed synchronization between embryonic and endometrial development during implantation and occurrence of multiple gestations (especially monochorionic gestation), which can result in fetal demise 1,6–9. These factors are particularly important for the clinical success and efficacy of ART. One molecular factor that has been previously implicated in female reproduction is the small, bioactive phospholipid LPA 10. LPA has a range of influences that are mediated by at least four G protein-coupled receptors, LPA1–4 2. Deletion of LPA1 and LPA2 in mice revealed roles for these receptors in neural development, craniofacial formation, neuropathic pain, and altered cellular signalling, but without obvious effects on female reproduction 11–14. These results suggested that LPA signalling in female reproduction might be mediated by other LPA receptors including LPA3 (formerly known as Edg7) 3,4, LPA4 15, unidentified LPA receptor(s), or possibly non-receptor pathways. Towards identifying LPA-dependent mechanisms affecting reproduction, we targeted LPA3 for deletion, a receptor with distinct signalling properties and a preference for unsaturated LPA species 2–4.
Functional deletion of LPA3 was achieved by replacing a fragment covering the untranslated region and the start codon in exon 2 with a neomycin-resistance gene in reverse orientation in R1 embryonic stem cells (supplementary Fig. S1). The LPA3-deficient mice were born with normal Mendelian frequency without sexual bias (supplementary Table S1), and appeared grossly normal (data not shown). However, LPA3-deficient females produced litter sizes of less than 50% compared to that from wild-type (WT) and LPA3 heterozygote (Het) controls (supplementary Table S2), and showed a statistically significant prolongation of pregnancy (20.9±0.5 days, vs. 19.4±0.7 days in WT/Het controls, P<0.05). These phenotypes were independent of stud genotype, indicating defects in female reproduction.
Towards determining whether LPA3 deletion might directly affect the female reproductive system, expression patterns of LPA3 mRNA were assessed using RT-PCR and in situ hybridization. By RT-PCR, LPA3 mRNA was detected in oviduct, placenta and uterus but not in ovary and eggs (unfertilized eggs and fertilized eggs from one cell to preimplantation blastocyst, data not shown). Within the uterus, LPA3 mRNA expression was up-regulated during postnatal development and varied during the estrous cycle (supplementary Fig. S3a, S3b). Strikingly, LPA3 mRNA levels increased during early pregnancy, peaking around embryonic day 3.5 (E3.5) then returning to basal levels from E4.5 through the end of pregnancy (Fig. 1a). RT-PCR of microdissected E3.5 uterine tissue and in situ hybridization indicated that LPA3 mRNA expression was confined to the luminal endometrial epithelium at E3.5 (Fig. 1b, 1c, 1d, supplementary Fig. S3c). These data suggested that LPA3 loss-of-function resulting in reduced litter sizes could involve direct effects on the female reproductive system.
Figure 1.
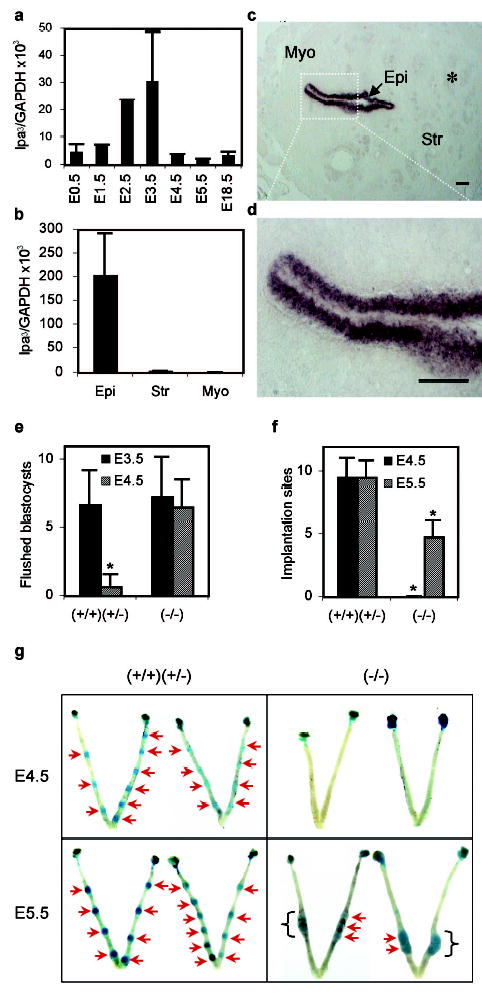
LPA3 mRNA expression in WT uterus and effects of LPA3-deficiency on implantation. a,b. Quantification of uterine LPA3 mRNA during pregnancy and in E3.5 luminal endometrial epithelium (Epi), stroma (Str), and myometrium (Myo). c,d. In situ localization of LPA3 in E3.5 WT uterus. *. Glandular endometrial epithelium. Scale bars=100 μm. e. Flushed blastocysts from E3.5 and E4.5 uteri. f,g. Number and location of implantation sites at E4.5 and E5.5 uteri. Blue bands (arrows): implantation sites; Brackets: clustered implantation sites. *P<0.001. In all figures, error bars are standard deviations, (+/+), (+/−), and (−/−) represent WT, Het, and LPA3-deficient mice, respectively.
To explore this possibility, we examined major events in female reproduction, from ovulation through decidualization. No significant differences were observed in superovulation, fertilization, or decidualization between WT or Het controls and LPA3-deficient female littermates. No significant differences in blastocyst number or developmental stage, isolated from E3.5 uteri, were observed between controls and LPA3-deficient females. These data indicated no obvious defects in ovulation, ovum transportation, and blastocyst development in LPA3-deficient females (Fig. 1e, supplementary Fig. S4).
In notable contrast, embryo implantation studies identified clear phenotypic changes in LPA3-deficient dames: delayed implantation and altered positioning/crowding of embryos. By E4.5, implantation sites identifiable by Evans blue labelling in control animals were absent in uteri of LPA3-deficient dames. Implantation sites became detectable at E5.5 in LPA3-deficient uteri (Fig. 1f, 1g) 16. The number of pre-implantation blastocysts recovered from E4.5 LPA3-deficient uteri was comparable to that from E3.5 control and LPA3-deficient uteri (Fig. 1e, supplementary Fig. S4c), indicating that delayed implantation in LPA3-deficient uteri was ascribable to extra-embryonic influences of LPA signalling. In addition to delayed implantation, LPA3-deficient uteri had a reduced number of implantation sites compared to that in the control uteri in spite of the fact that comparable numbers of blastocysts were available for implantation. The implantation sites in the LPA3-deficient uteri were crowded/clustered in the uterine segments proximal to the cervix (Fig. 1g). This aberrant crowding of embryos in the LPA3-deficient uteri was further demonstrated by findings at later gestational stages. At E10.5, 44% of implantation sites in LPA3-deficient uteri contained 2–4 embryos (averaging 1.65 live embryos per implantation site) (Fig. 2a, 2b). At E18.5, 28% of placentas were shared by 2–3 embryos (Fig. 2c) and associated with placental hypertrophy (Fig. 2d, supplementary Fig. S5). These phenomena were never observed in controls.
Figure 2.

Multiple embryos at individual implantation sites and placental hypertrophy in LPA3-deficient uteri. a. Samples of multiple embryos at individual implantation sites at E10.5. White arrowheads: embryos. Scale bars: 2 mm. b. Cross-sections of E10.5 uteri revealing two less developed embryos sharing one placenta in a LPA3-deficient uterus. Scale bars: 1 mm. c. A placenta shared by three embryos at E18.5 in a LPA3-deficient uterus. Scale bar: 8 mm. b and c, red arrowheads: embryos; yellow arrowheads: placentas. d. Weight of placentas at E18.5.
In addition, embryos isolated from LPA3-deficient uteri (at E10.5 and E18.5) were always smaller than those from WT/Het controls at comparable ages (Fig. 3a, 3b, 3c), although newborns from LPA3-deficient females were, on average, heavier (Fig. 3c), possibly resulting from prolonged pregnancy and/or smaller litter size. The average number of live embryos per animal that could be isolated from LPA3-deficient females decreased following initial implantation (Fig. 3d). Delayed implantation and aberrant embryo spacing were thus associated with both delayed embryonic development and embryonic death, which could account for the reduced litter sizes produced by LPA3 deficiency.
Figure 3.
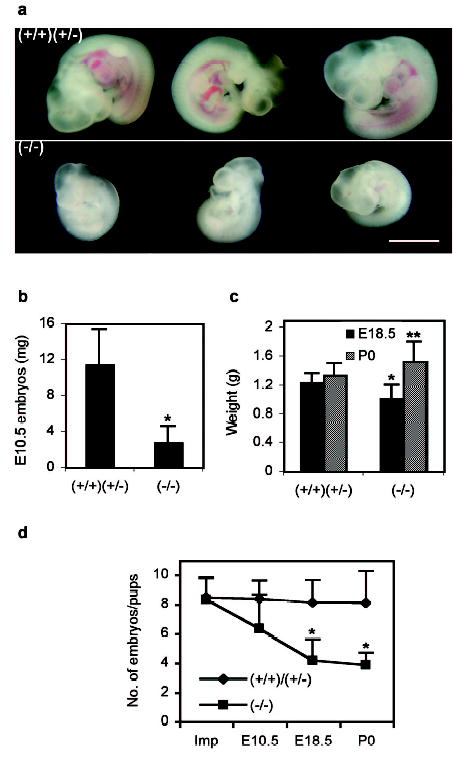
Delayed postimplantational development of embryos and increased embryonic death in LPA3-deficient uteri. a. Representative embryos from E10.5 uteri. Scale bar: 2 mm. b. E10.5 embryo weight. c. Weights of E18.5 embryos and P0 pups. d. The average numbers of embryos implanted (Imp), at E10.5 and E18.5, and P0 pups. The numbers of embryos implanted were calculated as described in Material and Methods. *P<0.001, **P<0.05.
To determine whether LPA3 loss in the embryo itself might contribute to the phenotypes, embryo transfer experiments were pursued. WT blastocysts were transferred to WT or LPA3-deficient pseudopregnant uteri. Transferred WT embryos in WT or LPA3-deficient pseudopregnant uteri were phenotypically indistinguishable for implantation and development compared to embryos produced by natural matings in WT or LPA3-deficient animals. Similarly, when LPA3-deficient blastocysts were transferred to WT pseudopregnant uteri, no implantation or spacing abnormalities were observed (supplementary Fig. S6 and data not shown). WT and LPA3-deficient blastocysts had comparable implantation rates in WT pseudopregnant uteri. Considering the fact that no LPA3 mRNA was detected in WT pre-implantation blastocysts, these results eliminate significant contributions of LPA3 via pre-/post-implantation embryos, and indicate that maternal LPA3 signalling is responsible for the observed phenotypes.
The observed implantation phenotypes of LPA3-deficient mice were strikingly similar to that reported in rats and mice treated with indomethacin 17,18, and mice deficient for cytosolic phospholipase A2α (cPLA2α) 19. Indomethacin is an inhibitor of cyclooxygenases 20, which convert arachidonic acid (AA) to prostaglandin H2 (PGH2) in the biosynthesis of prostaglandins (PGs), while cPLA2α is an important enzyme producing AA. Interestingly, COX-2-deficiency but not COX-1-deficiency in mice results in multiple female reproductive failures, including implantation defects 5,21, although the precise phenotypes can be influenced by genetic background 22,23. Moreover, PGE2 and cPGI (a stable analogue of PGI2) can partially correct implantation defects in both cPLA2α-deficient mice and COX-2-deficient mice 19,24. These data indicate that the cPLA2α – AA – COX – PG pathway is crucial for implantation 1. In view of the phenotypic similarities between LPA3 deficiency and cPLA2α/PG deficiency, we hypothesized that LPA3 might converge on this signalling pathway.
Components of prostaglandin signalling were therefore examined in LPA3-deficient uteri. They include cPLA2α, COX-1 and COX-2, and prostaglandin G protein-coupled receptors EP1–4 and IP 25, along with leukaemia inhibitory factor (LIF) and Hoxa-10, two key regulators in implantation 1. Only COX-2 mRNA levels were significantly reduced in LPA3-deficient uteri (Fig. 4a, supplementary Fig. S7, and data not shown). Since COX-2 is a rate-limiting enzyme for PG biosynthesis, the suppression of COX-2 expression in E3.5 LPA3-deficient uteri resulted in reduced production of PGE2 and PGI2 (Fig. 4b), which may be inadequate for implantation that normally occurs around E4.0 18,26. To rescue this reduction in prostaglandins, we delivered exogenous PGE2 and cPGI to E3.5 LPA3-deficient females, a general approach previously reported 19. Following PG exposure, significantly more LPA3-deficient females with on-time implantation were detected compared to vehicle-controls (Fig. 4c, P=0.003). Notably, this rescue did not affect the uneven embryo spacing nor completely restore the reduction of implantation sites compared to WT controls (Fig. 4d).
Figure 4.
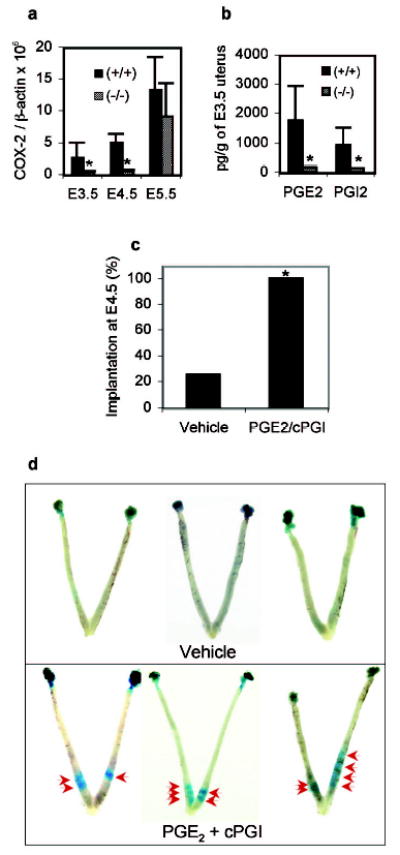
Reduced COX-2 mRNA and prostaglandin levels in LPA3-deficient uteri, and exogenous prostaglandin rescue of delayed implantation. a. Expression of COX-2 during early pregnancy in WT and LPA3-deficient uteri. *P<0.05. b. Reduced PGE2 and PGI2 levels in E3.5 LPA3-deficient uteri. *P<0.05. c. Significantly increased percentage of LPA3-deficient females showing on-time implantation upon PGE2 and cPGI (a stable PGE2 analog) treatment at E3.5. *P=0.003. d. Images of E4.5 LPA3-deficient uteri with or without prostaglandin treatment. Red arrows: implantation sites. Supplementary Figure S1a, S1b, S1c
These findings identify LPA signalling as a new influence on embryo implantation, and are the first to link a lysophospholipid G protein-coupled receptor to prostaglandin biosynthesis, thereby influencing female fertility. As a class, lysophospholipid receptors represent a “drugable” target, as demonstrated by the compound FTY720 that is currently in phase III clinical trials for prevention of transplantation rejection 27. This raises the possibility of creating medicines that influence implantation timing, a critical factor for in vitro fertilization 1,9 and reducing the increased incidence of multi-embryo gestations, especially monochorionic gestations that can result in fetal demise 6. The reduced litter sizes observed in receptor-null mutants for another lysophosopholipid, sphingosine 1-phosphate, suggest that other lysophospholipid receptors may also influence mammalian reproduction through pharmacologically tractable mechanisms 28.
Materials and Methods
Quantitative RT-PCR
Primers used were as described 11,12,29. For amplification of COX-2, the following primers were used: forward, aagcgaggacctgggttca; reverse, aaggcgcagtttatgttgtctgt. Quantitative RT-PCR was performed as described 29. The transcript number of target genes was quantified and normalized against GAPDH or β-actin transcript number.
In Situ Hybridization and Histology
The animals were anesthetized with Halothane inhalation followed by cervical dislocation. In situ hybridization and histology were performed as described 30. Sense and antisense DIG-labelled cRNA probes were generated using appropriate polymerases from a full-length murine LPA3 cDNA.
Mating Study, Embryo Collection, and Localization of Implantation Sites
All the mice used in this study were mixed background (129/SvJ and C57BL/6). Since no difference was observed in all the parameters examined between WT and Het females (Table S2, supplementary Fig. S8a, S8b, and data not shown), females with either one or both WT and Het genotypes were used as controls. Females were naturally mated with WT stud males. The day a plug found was designated as E0.5. Plugged females were anesthetized with Halothane inhalation followed by cervical dislocation. Uteri of pregnant females were dissected at E3.5, E4.5, E5.5, E10.5 and E18.5. Embryos at E10.5 were fixed in 10% Formalin overnight before being weighted. Implantation sites at E4.5 and E5.5 were localized by i.v. injection of Evans blue dye (200 μl, 1% in 1xPBS, Sigma) 16. The numbers of embryos initially implanted in both LPA3-deficient and WT/Het uteri were retrospectively calculated from E10.5 as following: At E10.5, embryos in an average of 1.2 implantation sites (out of 5.0 total) were absorbed in LPA3-deficient uteri, but embryos in only 0.09 implantation sites (out of 8.4 total) were absorbed in WT/Het uteri (P=1.7x10−5). With an average of 1.65 live embryos per implantation site in LPA3-deficient uteri and 1.0 live embryo per implantation site in WT/Het uteri at E10.5, the total number of embryos initially implanted should be 8.3 ((3.8 live + 1.2 absorbed) x 1.65) in LPA3-deficient uteri and 8.4 ((8.31 live + 0.09 absorbed) x 1.0) in WT/Het uteri.
Prostaglandin Measurement
Uteri from E3.5 WT or LPA3-deficient mice were immediately frozen and crushed in liquid nitrogen. Prostaglandins were extracted by ethyl acetate extraction method. The prostaglandin levels of each sample were determined using prostaglandin enzyme-linked immuno assay (EIA) kit (Cayman Chemical). PGI2 was measured as 6-keto-PGF1α.
Prostaglandin Administration
E3.5 LPA3-deficient females were i.p. injected with 100 μl of vehicle (10% ETOH with saline, as control) or 5 μg cPGI and 5 μg PGE2 (Cayman Chemical, in 10% ETOH with saline) at 10:00 am and 6:00 pm. Implantation sites were detected using Evans blue dye at E4.5.
Data Representation
Data were expressed as Mean±SD. Statistical analyses were done using Student’s t test or χ2 test. The significant level was set at P<0.05.
Supplementary Material
Acknowledgments
We thank Drs. F. Liu, S. Kupriyanov, R. Rivera, D. Herr, E. Nilsson, M. Murakami, Y. Kita, and B.C. Paria, Ms. S. Carlson and Q. Chen for technical assistance and insightful suggestions. This work was supported by grants from the National Institute of Mental Health to J.C. and J.J.A.C., the National Institute of Health to M.K.S, Swiss National Science Foundation to B.A., Program for Promotion of Fundamental Studies in Health Sciences of the Pharmaceuticals and Medical Devices Agency (PMDA) and grants-in-aid from the Ministry of Education, Science, Culture and Sports for the 21st Century Center of Excellence Program, Japan to J.A and H.A..
Footnotes
Supplementary Information accompanies the paper on http://www.nature.com/nature.
Competing Interests statement The authors declare that they have no competing financial interests.
References
- 1.Dey SK, et al. Molecular cues to implantation. Endocr Rev. 2004;25:341–73. doi: 10.1210/er.2003-0020. [DOI] [PubMed] [Google Scholar]
- 2.Ishii I, Fukushima N, Ye X, Chun J. LYSOPHOSPHOLIPID RECEPTORS: Signaling and Biology. Annu Rev Biochem. 2004;73:321–354. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 3.Bandoh K, et al. Molecular cloning and characterization of a novel human G-protein-coupled receptor, EDG7, for lysophosphatidic acid. J Biol Chem. 1999;274:27776–85. doi: 10.1074/jbc.274.39.27776. [DOI] [PubMed] [Google Scholar]
- 4.Contos JJ, Chun J. The mouse lp(A3)/Edg7 lysophosphatidic acid receptor gene: genomic structure, chromosomal localization, and expression pattern. Gene. 2001;267:243–53. doi: 10.1016/s0378-1119(01)00410-3. [DOI] [PubMed] [Google Scholar]
- 5.Lim H, et al. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell. 1997;91:197–208. doi: 10.1016/s0092-8674(00)80402-x. [DOI] [PubMed] [Google Scholar]
- 6.Cleary-Goldman J, D’Alton M. Management of single fetal demise in a multiple gestation. Obstet Gynecol Surv. 2004;59:285–98. doi: 10.1097/01.ogx.0000120165.52159.04. [DOI] [PubMed] [Google Scholar]
- 7.Umstad MP, Gronow MJ. Multiple pregnancy: a modern epidemic? Med J Aust. 2003;178:613–5. doi: 10.5694/j.1326-5377.2003.tb05384.x. [DOI] [PubMed] [Google Scholar]
- 8.Daniel Y, et al. Analysis of 104 twin pregnancies conceived with assisted reproductive technologies and 193 spontaneously conceived twin pregnancies. Fertil Steril. 2000;74:683–9. doi: 10.1016/s0015-0282(00)01491-6. [DOI] [PubMed] [Google Scholar]
- 9.Wilcox AJ, Baird DD, Weinberg CR. Time of implantation of the conceptus and loss of pregnancy. N Engl J Med. 1999;340:1796–9. doi: 10.1056/NEJM199906103402304. [DOI] [PubMed] [Google Scholar]
- 10.Tokumura A, Fukuzawa K, Yamada S, Tsukatani H. Stimulatory effect of lysophosphatidic acids on uterine smooth muscles of non-pregant rats. Arch Int Pharmacodyn Ther. 1980;245:74–83. [PubMed] [Google Scholar]
- 11.Contos JJ, Fukushima N, Weiner JA, Kaushal D, Chun J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc Natl Acad Sci U S A. 2000;97:13384–9. doi: 10.1073/pnas.97.24.13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Contos JJ, et al. Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2) Mol Cell Biol. 2002;22:6921–9. doi: 10.1128/MCB.22.19.6921-6929.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kingsbury MA, Rehen SK, Contos JJ, Higgins CM, Chun J. Non-proliferative effects of lysophosphatidic acid enhance cortical growth and folding. Nat Neurosci. 2003;6:1292–9. doi: 10.1038/nn1157. [DOI] [PubMed] [Google Scholar]
- 14.Inoue M, et al. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712–8. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- 15.Noguchi K, Ishii S, Shimizu T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for Lysophosphatidic acid, structurally distant from the Edg family. J Biol Chem. 2003 doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- 16.Paria BC, Huet-Hudson YM, Dey SK. Blastocyst’s state of activity determines the “window” of implantation in the receptive mouse uterus. Proc Natl Acad Sci U S A. 1993;90:10159–62. doi: 10.1073/pnas.90.21.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennedy TG. Evidence for a role for prosaglandins in the initiation of blastocyst implantation in the rat. Biol Reprod. 1977;16:286–91. doi: 10.1095/biolreprod16.3.286. [DOI] [PubMed] [Google Scholar]
- 18.Kinoshita K, et al. Involvement of prostaglandins in implantation in the pregnant mouse. Adv Prostaglandin Thromboxane Leukot Res. 1985;15:605–7. [PubMed] [Google Scholar]
- 19.Song H, et al. Cytosolic phospholipase A2alpha is crucial [correction of A2alpha deficiency is crucial] for ‘on-time’ embryo implantation that directs subsequent development. Development. 2002;129:2879–89. doi: 10.1242/dev.129.12.2879. [DOI] [PubMed] [Google Scholar]
- 20.Frenkian M, Segond N, Pidoux E, Cohen R, Jullienne A. Indomethacin, a COX inhibitor, enhances 15-PGDH and decreases human tumoral C cells proliferation. Prostaglandins. 2001;65:11–20. doi: 10.1016/s0090-6980(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 21.Reese J, Brown N, Paria BC, Morrow J, Dey SK. COX-2 compensation in the uterus of COX-1 deficient mice during the pre-implantation period. Mol Cell Endocrinol. 1999;150:23–31. doi: 10.1016/s0303-7207(99)00033-7. [DOI] [PubMed] [Google Scholar]
- 22.Sibilia M, Wagner EF. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science. 1995;269:234–8. doi: 10.1126/science.7618085. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, et al. Rescue of female infertility from the loss of cyclooxygenase-2 by compensatory up-regulation of cyclooxygenase-1 is a function of genetic makeup. J Biol Chem. 2004;279:10649–58. doi: 10.1074/jbc.M312203200. [DOI] [PubMed] [Google Scholar]
- 24.Lim H, et al. Cyclo-oxygenase-2-derived prostacyclin mediates embryo implantation in the mouse via PPARdelta. Genes Dev. 1999;13:1561–74. doi: 10.1101/gad.13.12.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang ZM, et al. Potential sites of prostaglandin actions in the periimplantation mouse uterus: differential expression and regulation of prostaglandin receptor genes. Biol Reprod. 1997;56:368–79. doi: 10.1095/biolreprod56.2.368. [DOI] [PubMed] [Google Scholar]
- 26.Paria BC, Song H, Dey SK. Implantation: molecular basis of embryo-uterine dialogue. Int J Dev Biol. 2001;45:597–605. [PubMed] [Google Scholar]
- 27.Gabardi S, Cerio J. Future immunosuppressive agents in solid-organ transplantation. Prog Transplant. 2004;14:148–56. doi: 10.1177/152692480401400209. [DOI] [PubMed] [Google Scholar]
- 28.Ishii I, et al. Marked perinatal lethality and cellular signaling deficits in mice null for the two sphingosine 1-phosphate (S1P) receptors, S1P(2)/LP(B2)/EDG-5 and S1P(3)/LP(B3)/EDG-3. J Biol Chem. 2002;277:25152–9. doi: 10.1074/jbc.M200137200. [DOI] [PubMed] [Google Scholar]
- 29.Hama K, et al. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J Biol Chem. 2004;279:17634–9. doi: 10.1074/jbc.M313927200. [DOI] [PubMed] [Google Scholar]
- 30.Ishii I, et al. Selective loss of sphingosine 1-phosphate signaling with no obvious phenotypic abnormality in mice lacking its G protein-coupled receptor, LP(B3)/EDG-3. J Biol Chem. 2001;276:33697–704. doi: 10.1074/jbc.M104441200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.