

Abstract
Prior observations of phage-host systems in vitro have led to the conclusion that susceptible host cell populations must reach a critical density before phage replication can occur. Such a replication threshold density would have broad implications for the therapeutic use of phage. In this report, we demonstrate experimentally that no such replication threshold exists and explain the previous data used to support the existence of the threshold in terms of a classical model of the kinetics of colloidal particle interactions in solution. This result leads us to conclude that the frequently used measure of multiplicity of infection (MOI), computed as the ratio of the number of phage to the number of cells, is generally inappropriate for situations in which cell concentrations are less than 107/ml. In its place, we propose an alternative measure, MOIactual, that takes into account the cell concentration and adsorption time. Properties of this function are elucidated that explain the demonstrated usefulness of MOI at high cell densities, as well as some unexpected consequences at low concentrations. In addition, the concept of MOIactual allows us to write simple formulas for computing practical quantities, such as the number of phage sufficient to infect 99.99% of host cells at arbitrary concentrations.
It has long been observed that when bacteriophage are mixed with susceptible host bacteria, the number of phage in the culture supernatant does not increase until after an eclipse period of, generally, 30 to 40 min at 37°C has passed. This period of time is explained as the time the phage requires to inject its genome into the host, express its genes, and assemble progeny phage and release them into the environment. Additionally, when host cell densities are very low, it has been observed that there is a longer delay before phage numbers increase over the numbers of input phage. This period has been explained as the time needed for the host cells to reach a “replication threshold” (16) or “proliferation threshold” (7, 8) density. This density has been reported to be approximately 104 cells per ml for the multiple phage-host combinations tested (16) and has been said to have broad implications for the propagation of phages in natural environments and in terms of their use as antimicrobial therapies (7, 8). The mechanism of this delay in phage replication has not been widely investigated or discussed.
One explanation for the apparent threshold density would be a requirement on the part of the phage for the host cell to be in a particular metabolic state and that this state is only reached when the cell density is 104 CFU/ml or more. Small molecules called autoinducers or quorum factors are known to be secreted into the environment by bacteria and, by their accumulation as the number of cells increases, to allow the bacteria to monitor their local population density (3). These soluble signaling molecules alter the expression of dozens of genes and thereby regulate the metabolic state when the sensing bacteria are exposed to them at a sufficient concentration. Quorum factors could therefore explain the dependence of phage replication on cell density if, for example, molecules that serve as phage receptors are expressed in response to quorum factors. However, the data to be presented below demonstrate no detectable quorum factor effect on the ability of phage to infect bacteria.
Here we propose an alternative explanation for the phenomenon that has been interpreted as a replication threshold density that can be extracted from the mathematical model of Schlesinger (12) and Stent (13). This model makes the assumption that phage rely entirely on chance encounters with their hosts, and so, in liquid culture at least, their ability to infect and reproduce can be entirely predicted by the equations that describe the movements and coagulation (irreversible binding) of inert colloidal particles under the influence of Brownian motion (12). In this model, one finds that
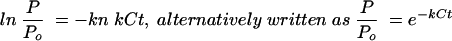 |
(1) |
where P/Po is the fraction of phage that remains unbound at time t (in minutes); C is the concentration of host cells per cubic centimeter, which remains constant over time t; and k is an adsorption rate constant (in cubic centimeters per minute) that can be determined experimentally for a given phage-host combination. Variations between phage-host systems in the number of phage binding sites per cell, the diffusion rate constant of the virus, and the efficiency with which collisions between cells and phage result in infection are accounted for by empirical determination of the adsoption rate constant, k, for each system (see references 12 and 13 for a description of the method). For example, T-even phages, which can utilize up to 300 binding sites per host cell, have an adsorption rate constant of 2.4 × 10−9 cm3/min (13) while the adsorption rate constant for filamentous phage M13, which has only 2 or 3 binding sites per cell, is 3 × 10−11 cm3/min (2, 14).
It follows from equation 1 that, if the concentration of host cells, C, is lower, all else being equal, a larger fraction of the phage will remain unbound at time t. This has important consequences for the utility of the common term multiplicity of infection (MOI), which will be discussed in detail. To our knowledge, verification of Schlesinger's model of phage-host interactions over a range of host cell concentrations has never been carried out, no doubt in part because of the difficulty in quantitating the number of host cells infected by a lytic phage. We report here a series of experiments designed to rigorously test the model by using transducing phages M13K07 and P1 in which phage injection of reporter phagemids served as a surrogate marker for phage infection. As a result of these experiments, it will be demonstrated that, as predicted by the model, the rate of phage infection is dependent solely on host cell density and follows very closely the theoretical expectations given for interactions of two types of colloidal particles. In addition, it will be shown that at phage densities sufficient to ensure infection, low host cell density does not affect phage replication rates. In light of the significant impact host cell density has on infection rates, we propose that the term MOI with regard to bacteriophage be further refined so that MOIinput indicates MOI in its traditional sense, i.e., the simple ratio of input phage to input cells. The designation MOIactual would indicate the number of phage calculated to be bound per host cell at the end of the adsorption period according to Schlesinger's model and therefore the effective MOI in a given experiment. Finally, a simple method for calculating MOIactual is given and the implications for phage therapy applications are discussed.
MATERIALS AND METHODS
M13 phagemid system.
The pBluescript II KS+ phagemid was purchased from Stratagene. The pBlue-GFPuv phagemid was constructed by ligating the smaller EcoRI/KpnI fragment of plasmid pGFPuv (Clontech) into pBluescript II KS+, which was also digested with EcoRI/KpnI. The pBlue-GFPuv phagemid was transformed into and maintained in NovaBlue (Novagen) host cells. Escherichia coli ER2738 and M13K07 helper phage were purchased from New England Biolabs (Beverly, Mass.). ER2738 cells were grown in Luria broth (LB) with 20 μg of tetracycline/ml (Sigma) to maintain the F′ episome. Both M13 phagemids were maintained and selected with 80 μg of carbenicillin (Novagen) per ml. M13K07 phage lysates were made by infecting cells carrying either pBluescript or pBlue-GFPuv with M13K07 in accordance with standard methods (11) at an approximate MOIactual of 0.1, except without kanamycin in the medium. Lysates were cleared of cell debris by centrifugation and cleared of any remaining bacteria by filtration with a 0.2-μm-pore-size filter. Infectious phage titers were determined by plaque formation on ER2738 cells by the agar overlay technique. Transducing phage titers were determined by incubation of diluted lysates with log phase ER2738 cells that had been concentrated by centrifugation to approximately 109 CFU/ml. After 30 min of incubation at 37°C, the entire mixture was plated on LB agar containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), isopropyl-β-d-thiogalactopyranoside (IPTG), and carbenicillin (80 μg/ml) and incubated at 37°C until blue colonies could be quantitated (16 to 20 h). Each phagemid delivery event produces a carbenicillin-resistant blue colony or a blue and green fluorescent protein-positive colony on this medium. M13 phage do not kill their host, and therefore no immunity in the target cells is necessary (10). Since the helper phage M13K07 packaged the phagemid pBluescript 500 times more efficiently than its own genome, the M13 lysates contained 99.8% transducing phage. X-Gal and IPTG were obtained from Gold Biotechnology and used at 1.7 and 330 μM, respectively. Serial dilutions of cells and phage were carried out with LB and aerosol barrier tips (Fisher Scientific).
P1 phagemid system.
A P1 phagemid was constructed with the pBBR122 vector (MoBiTec, Düsseldorf, Germany), which carries a kanamycin resistance gene for transfer detection, a broad-host-range origin of replication, and essential elements for P1 packaging. E. coli C600 host cells (Stratagene) susceptible to P1 infection were transformed with this phagemid and then infected with the wild-type phage P1 kc to produce a P1 phage lysate containing approximately 90% infectious phage and 10% transducing phage carrying the phagemid. E. coli C600 cells lysogenized with the P1 mutant P1CmC1.100 (9) are referred to as P1C600 cells and are immune to infectious P1 phage but are ready acceptors of P1-delivered phagemid (C. Westwater, unpublished results). P1C600 cells were grown in LB containing 17.5 μg of chloramphenicol/ml to maintain the lysogen. Phagemid delivery into P1C600 cells results in a kanamycin- and chloramphenicol-resistant colony when the bacteria are plated on 50 μg of kanamycin/ml and 17.5 μg of chloramphenicol/ml (Sigma). Dilutions of cells and phage lysate were performed with LB containing 10 mM MgSO4 and 5 mM CaCl2 with aerosol barrier tips.
RESULTS
Host cell density, not quorum factors, determines the phage infection rate.
It seems reasonable that the metabolic state of cells at low population densities may affect their susceptibility to phage infection. We addressed this hypothesis by comparing pBluescript phagemid delivery by M13K07 transducing phage to cells diluted from actively growing cultures into either fresh medium, conditioned medium from actively growing cultures, or conditioned medium from saturated cultures. Two final cell densities were also tested in each medium by placing the same number of cells and phage in either a 0.1- or a 1-ml final volume. Contrary to expectations if quorum factors were necessary for phage binding and infection, there was a small decrease in phagemid delivery in the presence of either conditioned medium (Fig. 1). In contrast, concentrating identical mixtures of cells and virus into a smaller volume of the same respective media improved efficiency three- to fivefold (Fig. 1). Similar results (not shown) were obtained with P1 phage. This result indicated that cell density itself, rather than factors secreted into the medium of densely populated cells, determines the phage-host interaction.
FIG. 1.

Effect of conditioned medium on transduction efficiency. Actively growing E. coli (ER2738) cells in LB containing 20 μg of tetracycline/ml, in order to maintain the F′ plasmid, were briefly chilled on ice before being diluted 10,000-fold in either fresh LB containing tetracycline or filter-sterilized conditioned medium isolated from logarithmic growth or saturated cultures of the same cells. Transducing M13K07 phage carrying plasmid pBlue-GFPuv were added to the corresponding cell suspensions. Duplicate transduction mixtures were set up in parallel that contained the same number of cells and phage but in 1/10 of the volume. All mixtures were incubated at 37°C for 30 min, and then aliquots were plated on LB agar containing IPTG in triplicate. After overnight incubation, all colonies were counted and the number of transduced colonies was detected by expression of green fluorescent protein. The mean percentage of colonies transduced under each condition is shown.
Definition of MOIactual and 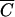 .
.
Traditionally, MOI is defined as the average number of virus particles infecting each cell, calculated as the ratio of the number of infectious virus particles divided by the number of host cells. When infection of all of the cells in a culture is desired, an MOI of 10 is often used. This is based on the fact that the virus particles are not distributed exactly evenly among the cells, and it can be calculated that an MOI of 10 gives each cell a better than 99.99% chance of being bound and infected by at least one virus particle (see appendix A). However, a crucial assumption in the derivation of this rule is that all of the virus particles will find and infect a cell within the adsorption period allowed. It is important to ask under what conditions this assumption is valid. The colloidal particle model of phage-cell interactions (equation 1) predicts that the number of phage that remain unbound even after an average adsorption period of 30 min can be quite high, depending primarily on the concentration of susceptible cells. The traditional MOI in a particular experiment, the MOIinput, can be more accurately thought of then as the maximum possible MOI that will be experienced by the cells. If the actual average number of phage expected to bind per cell under the experimental conditions is written as MOIactual, then the relationship between MOIinput and MOIactual is given by the following equation:
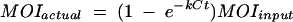 |
(2) |
since, from Schlesinger's model, the fraction of phage bound at time t equals 1 − P/Po = 1 − e−kCt and it is therefore also possible to calculate the number of phage bound at time t as follows:
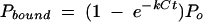 |
(3) |
Note that, as with traditional calculations of MOI, the calculations are based on starting conditions and cell proliferation is not accounted for. Therefore, the model is only accurate when the adsorption period, t, is sufficiently shorter than the doubling time of the cells. (Our experiments all used 30 min for E. coli, and good agreement between expected and observed results was obtained.)
It follows from equation 2 that as C or t become larger, MOIactual approaches MOIinput. This is logical since when the cell density is higher or more time is allowed for adsorption, more phage would be expected to bind. It is because MOIinput and MOIactual are essentially the same at high cell densities that the need for MOIactual may not have been previously apparent. The breakpoint at which MOIinput and MOIactual diverge can be specifically defined by introducing the special concentration as follows:
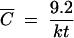 |
Simply, 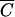 is the lowest cell density at which MOIinput is equal to MOIactual for a given k and t. (For the derivation of
is the lowest cell density at which MOIinput is equal to MOIactual for a given k and t. (For the derivation of 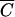 , see appendix B.) In other words, if the host cell density is at this concentration or a higher concentration (C ≥
, see appendix B.) In other words, if the host cell density is at this concentration or a higher concentration (C ≥ 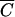 ), then MOIinput is equal to MOIactual for practical purposes and it is reasonable to assume that every phage added will bind to a cell within the adsorption period, t. Conversely, if C is less than
), then MOIinput is equal to MOIactual for practical purposes and it is reasonable to assume that every phage added will bind to a cell within the adsorption period, t. Conversely, if C is less than 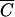 , then MOIactual is noticeably less than MOIinput and MOIactual should be used. Note that
, then MOIactual is noticeably less than MOIinput and MOIactual should be used. Note that 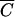 is dependent on t and decreases as t increases for a given phage-host system.
is dependent on t and decreases as t increases for a given phage-host system.
Predictive value of MOIactual.
We claim that using the traditional MOI, MOIinput, will result in incorrect expectations of infection rates in any case in which C is less than 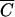 and that using MOIactual in its place will provide an accurate means of estimating the infection rate at any cell density. These claims were tested by using phage transduction of reporter phagemids as a surrogate for phage infection.
and that using MOIactual in its place will provide an accurate means of estimating the infection rate at any cell density. These claims were tested by using phage transduction of reporter phagemids as a surrogate for phage infection.
In the first experiment, host cells and transducing P1 phage lysates were combined over a range of cell densities by mixing a fixed number of phage with an equal volume of one of several 10-fold dilutions of cells. After 30 min of incubation, half of each mixture was plated on agar selective for cells that received the phagemid and half was plated on nonselective agar to control for cell death and growth. To test the model against the data collected in this experiment, the following formula was used to convert the ratio of phage to cells into the number of transduced cells: Expected fraction of cells infected = 1 − e−MOI (see appendix A). To obtain the expected results based on the traditional MOI, the MOIinput for a given incubation was used in the formula and the fraction obtained was multiplied by the number of cells in that incubation mixture. To obtain the expected results predicted by the model presented in this report, the same computations were performed using MOIactual in place of the MOIinput. Figure 2A shows the number of infected cells found experimentally and the number predicted by assuming that the ratio of bound phage to cells is given by the MOIactual or the MOIinput, respectively. Note that predictions of the MOIactual fit the data better both qualitatively and quantitatively. Also, as predicted, the graphs are essentially the same at cell densities higher than 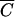 . At low cell densities, the expected results for the MOIinput decrease because cell numbers are limiting.
. At low cell densities, the expected results for the MOIinput decrease because cell numbers are limiting.
FIG. 2.

A fixed number of phage with serial dilutions of host cells. (A) Approximately 400 PFU of P1 transducing phage were incubated with serial dilutions of P1C600 host cells at the cell densities indicated for 30 min and then plated on kanamycin to select for cells that had been transduced with the reporter phagemid. (B) Approximately 400 PFU of transducing phage M13K07 were incubated with serial dilutions of ER2738 host cells at the cell densities indicated for 30 min and then plated on carbenicillin and X-Gal to select for cells that had been transduced with reporter phagemid pBlue-GFPuv. The expected number of transductants for each cell density was calculated as either N(1 − e−MOIactual) (○) or N(1 − e−MOIinput) (□). ▴, observed numbers of transductants.
For the phage-host system used in this experiment, the value 2.3 × 10−9 cm3/min for k was determined experimentally and then used to compute the expected values predicted by the model. Note that there is no logical problem with assuming the validity of the model in order to determine the value of k since the model simply claims that there exists some constant for which equation 2 is valid. If the model were not valid, one would find that the value of k must depend on the concentration of cells. However, Fig. 2A demonstrates the validity of the model with this fixed value of k for concentrations ranging from 102 to 109 cells/ml.
Figure 2B shows the same type of experiment performed with phage M13K07. Again, predictions based on the MOIactual modeled actual results much more accurately than those based on the MOIinput. Because of the very small k (3 × 10−11 cm3/min) and, consequently, the very large 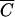 for this phage, it was not practical to achieve cell densities at which the MOIinput and MOIactual are essentially the same.
for this phage, it was not practical to achieve cell densities at which the MOIinput and MOIactual are essentially the same.
Universal infection at low cell densities.
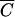 for M13 phage, based on a k of 3 × 10−11 (14) and a time t of 30 min, is 1010 cells/ml.
for M13 phage, based on a k of 3 × 10−11 (14) and a time t of 30 min, is 1010 cells/ml. 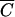 for P1 phage, based on a k of 2.3 × 10−9 and a time t of 30 min, is 1.3 × 108 cells/ml. Therefore, the model predicts and the previous experiment confirmed that at any cell density lower than these concentrations, an MOIinput of 10 will be inadequate to achieve infection of 99.99% of the cells. However, this does not necessarily imply that it is impossible to achieve universal infection at cell densities significantly lower than
for P1 phage, based on a k of 2.3 × 10−9 and a time t of 30 min, is 1.3 × 108 cells/ml. Therefore, the model predicts and the previous experiment confirmed that at any cell density lower than these concentrations, an MOIinput of 10 will be inadequate to achieve infection of 99.99% of the cells. However, this does not necessarily imply that it is impossible to achieve universal infection at cell densities significantly lower than 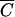 . In fact, it is possible to explicitly determine the MOIinput required to achieve an MOIactual of 10 for arbitrary cell densities. By using equation 2, the necessary numbers of phage were calculated for phages P1 and M13 over a range of cell densities (Table 1). Theoretically, using these values should result in the infection of nearly every cell in a given sample, regardless of cell density.
. In fact, it is possible to explicitly determine the MOIinput required to achieve an MOIactual of 10 for arbitrary cell densities. By using equation 2, the necessary numbers of phage were calculated for phages P1 and M13 over a range of cell densities (Table 1). Theoretically, using these values should result in the infection of nearly every cell in a given sample, regardless of cell density.
TABLE 1.
Predicted number of input phage sufficient for infection of 99.99% of cells according to cell densitya
| C | M13
|
P1
|
||
|---|---|---|---|---|
| Theoretical MOIinput for MOIactual of 10 | Theoretical Po needed for MOIactual of 10 | Theoretical MOIinput for MOIactual of 10 | Theoretical Po needed for MOIactual of 10 | |
| 1010 | 10.0 | 100,000,000,000 | 10.0 | 100,000,000,000 |
| 109 | 10.0 | 10,001,234,250 | 10.0 | 10,000,000,000 |
| 108 | 16.9 | 1,685,117,750 | 10.0 | 1,001,008,802 |
| 107 | 116.2 | 1,161,861,009 | 20.1 | 200,632,421 |
| 106 | 1,116.1 | 1,116,118,611 | 150.1 | 149,985,032 |
| 105 | 11,116.1 | 1,111,611,186 | 1,454.3 | 145,428,111 |
| 104 | 111,116.1 | 1,111,161,112 | 14,497.8 | 144,977,542 |
| 103 | 1,111,116.1 | 1,111,116,111 | 144,932.5 | 144,932,536 |
| 102 | 11,111,116.1 | 1,111,111,611 | 1,449,280.4 | 144,928,036 |
| 10 | 111,111,116.1 | 1,111,111,161 | 14,492,758.6 | 144,927,586 |
| 1 | 1,111,111,116.1 | 1,111,111,116 | 144,927,541.2 | 144,927,541 |
| Limiting value | 1,111,111,111 | 144,927,536 | ||
The theoretical MOIinput needed to realize an MOIactual of 10 for phages M13 and P1 was calculated for cell densities of 1 to 1010 cells per cm3 and an adsorption time of 30 min by using equation 2. The theoretical minimum number of phage necessary to attain an MOIactual of 10 in 30 min at each cell density in a volume of 1 cm3 was calculated by using equation 4 for Pmin. Notice that the theoretical minimums slowly approach but never reach the limiting value, or lower bound, of Pmin (from equation 5). In addition, note that Pmin changes very little for any cell concentration at or below 106/cm3 for M13 or 105/cm3 for P1.
In order to test this hypothesis, an experiment was carried out in which the number of cells per cubic centimeter was held constant and the number of phage added was increased in 10-fold increments. After an incubation time of 30 min, each reaction mixture was plated on an indicator plate for determination of β-galactosidase activity and the percentages of infected (blue) colonies and uninfected (white) colonies were determined by direct counting. The results are plotted along with the expected values in Fig. 3. Again, there was very close agreement between the expected and observed results. Furthermore, note that 100% infection (100% transformation and expression of the gene for β-galactosidase [blue colonies]) was achieved in the samples with the highest phage densities without loss of cell viability. This demonstrates that it is possible for phage to deliver a plasmid to all of the cells in a sample, even at low cell densities, provided that the number of phage used is sufficient. The MOIinput in the case of the highest phage concentration is 106, which previously might have been considered an unacceptable dose because of the likelihood of cell death by nonspecific lysis from without. However, no cell mortality was observed, and this is not surprising since we know from equation 2 that the MOIactual experienced by the cells was less than 100.
FIG. 3.

A fixed number of cells with serial dilutions of phage. Nine dilutions of transducing phage M13K07 lysate carrying phagemid pBlue-GFPuv were used to infect nine aliquots of 200 CFU of ER2738 host cells at a cell density of 1,000 CFU/cm3. After 30 min of incubation at 37°C, each reaction mixture was plated on nonselective LB agar containing IPTG and X-Gal. The percentages of blue colonies and green fluorescent protein-positive colonies were determined by direct counting and are plotted as the observed values (closed symbols). Expected values (open symbols) were calculated by finding the MOIactual for each set of reaction conditions in the experiment and multiplying the total number of colonies observed in each sample by its respective value for 1 − e−MOIactual. The results of two independent dilution series are shown. (tu, transducing units.)
Phage replication at low host cell densities.
As mentioned previously, a replication threshold density has been reported to exist in all of the phage-host systems tested, such that progeny phage are not produced when the host cell density is lower than 104/ml. However, the data presented in the preceding section suggest that phage infection, and therefore replication, would be expected to happen at any host cell density, provided that there are sufficient phage present to ensure that the cells present come into contact with and productively bind at least one phage. Proving this at low host cell densities, however, presents a practical problem for detection since unbound input phage will outnumber progeny phage to such an extent that the progeny phage will not be detectable as an increase in titer. This is because of both the high input phage numbers required and the low number of host cells producing progeny phage.
This problem of detection is overcome if the progeny phage are different from the input phage. The strategy utilized here was to make use of the M13K07 helper phage's 500- to 1,000-fold preference for packaging phagemids over its own defective genome (4). If the host cells carry the phagemid pBluescript, then the majority of the progeny phage will produce blue, ampicillin-resistant colonies when the titer is determined on susceptible cells, whereas the input phage will produce only white, kanamycin-resistant colonies, easily allowing differentiation of the progeny phage from the input phage.
This experiment was set up such that actively growing pBluescript-carrying host cells were diluted in 10-fold increments and each dilution was mixed with 1010 M13K07 phage, which is an amount adequate to ensure infection of most of the cells even at 10 CFU/cm3 or a lower concentration (Table 1). Blue colonies representing the output titer at each cell density after 60 min are plotted in Fig. 4. The experiment was performed twice with different host cell lines and with slightly different phagemids with very similar results. Most importantly, the straight lines evident on this log-log plot indicate that, on average, the same number of progeny phage are being produced per host cell regardless of the cell density. This observation is inconsistent with the existence of a replication threshold density.
FIG. 4.

Phage replication at low host cell densities. Actively growing NovaBlue E. coli bacteria carrying the pBlue-GFPuv phagemid ( ) or ER2738 E. coli bacteria carrying the pBluescript phagemid (
) or ER2738 E. coli bacteria carrying the pBluescript phagemid ( ) were serially diluted in fresh medium and then infected with 1010 PFU of helper phage M13K07. Aliquots were removed at 30 and 60 min postinfection, the host cells were removed by centrifugation and filtered with a 0.2-μm-pore-size filter, and in the resulting supernatants, the titers of the progeny phage that transduced ampicillin resistance and β-galactosidase expression were determined. No progeny phage were detectable at 30 min. Titers at 60 min are plotted as a function of the initial cell concentration in each culture. The slopes of the lines were plotted by linear regression and were 1.103 and 0.904, respectively. (tu, transducing units.)
) were serially diluted in fresh medium and then infected with 1010 PFU of helper phage M13K07. Aliquots were removed at 30 and 60 min postinfection, the host cells were removed by centrifugation and filtered with a 0.2-μm-pore-size filter, and in the resulting supernatants, the titers of the progeny phage that transduced ampicillin resistance and β-galactosidase expression were determined. No progeny phage were detectable at 30 min. Titers at 60 min are plotted as a function of the initial cell concentration in each culture. The slopes of the lines were plotted by linear regression and were 1.103 and 0.904, respectively. (tu, transducing units.)
The Pmin function: the minimum number of phage needed to achieve a given infection rate for any cell density, C.
In the preceding section, it was shown that it is possible to predict how many input phage are actually required to infect the input cells in a given adsorption time if the concentration of those cells is known, as well as a suitable adsorption constant, k. However, a more practical calculation is the following: given k, C, and the total volume of a reaction, V (in cubic centimeters), find the number of phage needed (Po) to achieve a chosen MOIactual. To answer this question, we define the function Pmin (M, N) as the minimum total number of phage that needs to be added (minimum Po) to result in an MOIactual of M for N bacteria by time t. Substitution into equation 2 of M for the desired MOIactual, N/V for C, and Po/N for the MOIinput and solving for Po yields the following equation:
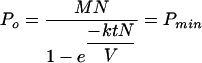 |
(4) |
When Pmin was plotted over a range of N’s (Fig. 5) for phages M13 and P1 with M = 10, t = 30 min, and V = 1 cm3, several interesting features of this function were observed. First, when C is greater than 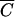 , the graph of the function is essentially linear and indistinguishable from the function MN. However, at cell concentrations lower than C̄, Pmin behaves very differently. As shown in Fig. 5, it has a lower bound and is almost horizontal for most N's giving concentrations less than
, the graph of the function is essentially linear and indistinguishable from the function MN. However, at cell concentrations lower than C̄, Pmin behaves very differently. As shown in Fig. 5, it has a lower bound and is almost horizontal for most N's giving concentrations less than 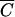 . This has the unexpected consequence that almost the same number of phage are needed to infect 99.99% of 106 cells as to infect 10 cells. By using L'Hospital's rule for computing limits of indeterminate forms, it can be deduced that no matter how small the number of cells (N) is, M cannot be achieved with less than
. This has the unexpected consequence that almost the same number of phage are needed to infect 99.99% of 106 cells as to infect 10 cells. By using L'Hospital's rule for computing limits of indeterminate forms, it can be deduced that no matter how small the number of cells (N) is, M cannot be achieved with less than
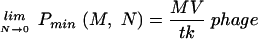 |
(5) |
FIG. 5.

The Pmin (M, N) function as related to cell density. The Pmin function (equation 4) for phages M13 (•) and P1 (▪) is plotted as a function of host cell density for an adsorption time of 30 min, a volume of 1 cm3, and an MOIactual of 10. Note that for all cell concentrations less than 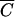 , Pmin is essentially the same.
, Pmin is essentially the same.
In other words, Pmin will not be less than MV/tk for any cell concentration less than 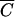 . This correlation is illustrated in Table 1, where it is evident that the number of phage needed to achieve an MOIactual of 10 decreases very little with large decreases in cell density, approaching but not reaching MV/tk for each phage. Moreover, although Pmin does increase for larger N’s approaching the amount needed to get a concentration of
. This correlation is illustrated in Table 1, where it is evident that the number of phage needed to achieve an MOIactual of 10 decreases very little with large decreases in cell density, approaching but not reaching MV/tk for each phage. Moreover, although Pmin does increase for larger N’s approaching the amount needed to get a concentration of 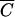 , it does not increase by much. Pmin at
, it does not increase by much. Pmin at 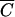 is only 5.31 times higher than the observed lower bound.
is only 5.31 times higher than the observed lower bound.
DISCUSSION
By using phagemid delivery and transduction as a surrogate marker for bacteriophage infection, we have been able to conclusively demonstrate for filamentous phage M13 and tailed phage P1 that phage-host interactions in liquid culture follow the kinetics of colloidal particles subject to Brownian motion, as proposed by Schlesinger (12). Since phages P1 and M13 have very different morphologies and binding site specificities, it seems reasonable to expect that the model applies to any phage-host system until proven otherwise. Since Schlesinger's model is based, in turn, on a mathematical model for the collisions of inert particles, this has several implications regarding bacteriophage biology. First, although it is not generally in question, it demonstrates beyond doubt that phage act as inert particles dependent on random Brownian motion in their interactions with bacteria and not as predators capable of active pursuit. Second, as the equations predict irreversible binding events and the predictions very closely match the observed number of infected host bacteria, nearly every such binding event must result in a successful transfer of phagemid or phage DNA into the host. This finding of the remarkable efficiency of phage-mediated delivery of DNA was arrived at previously by Stent by theoretical means (13). Third, the rate at which a phage infects a host cell is dependent solely on the density of the host cell population. Therefore, the concept of MOI, which, as currently defined, incorporates no indication of reaction volume or cell density, is inadequate in bacteriophage biology where a range of cell densities of greater than 10 orders of magnitude is possible and probable due to the small sizes of the particles involved. MOI is also often inadequate in the case of eukaryotic viruses, which is a problem that has been addressed by Valentine and Allison (15) and more recently by Andreadis et al. (1) and Morgan et al. (5). Because of the larger relative size of the cells and infection of attached cell monolayers rather than cell suspensions, our method for calculating the MOIactual does not appear to be directly applicable to eukaryotic systems.
Our finding that there is no detectable replication threshold for the M13 bacteriophage in E. coli is at odds with the findings of Wiggins and Alexander (16), who consistently saw a lag in phage production when host cell densities were lower than 104/ml. A partial explanation, as mentioned above, may lie in the difficulty of detecting progeny phage against a background of input phage. In addition, since the three phages Wiggins and Alexander used to demonstrate a replication threshold density were all somatic phages, adsorption constants would be expected to be similar in magnitude to that for phage P1, and so it is possible to reanalyze their data with our present mathematical model. Based on their graphs of phage and cell concentrations over time, starting phage concentrations in their experiments ranged from approximately 200 to 1,100 PFU/ml in a volume of 50 ml for a total input phage population of 10,000 to 55,000 PFU. It can be easily deduced from equation 3 that at cell densities lower than 104/ml, these initial conditions would result in an average of 0.7 to 4 phage binding cells within 30 min, respectively (the fraction of phage bound multiplied by the number of input phage equals 0.00007207 × 10,000 or 55,000). This would result in insufficient progeny phage to be detected against the input phage population. At 104 cells/ml, 7 to 40 phage will bind cells within 30 min, which would produce a 7 to 9% increase in the phage population, respectively, after the latency period, assuming a burst size of 100. Since phage removal from solution by adsorption to cells is negligible at this cell density, this increase would be detectable against the input phage population, resulting in the apparent existence of a replication threshold density of 104 cells/ml. It should be clear from our results that an increase in the number of input phage would have resulted in a decrease in the apparent replication threshold density. Likewise, a phage with a lower adsorption rate constant, such as M13, would have resulted in a higher apparent replication threshold density of approximately 106 cells per ml under the same conditions.
The confirmation of this mathematical model also has implications for the use of phage as antimicrobial therapeutic agents. Payne et al. (7, 8) developed a mathematical model of lytic phage therapy treatment of bacterial infections in vivo in which they argue that administration of therapeutic phage must be carefully timed to coincide with a sufficiently dense bacterial population to support phage proliferation in order to be effective, similar to the replication threshold. Our results confirm that host cell density is essential to estimating what percentage of administered phage will find target cells within a given period of time and that progeny phage will be detectable sooner at higher initial cell densities. However, our data demonstrate that there is no replication threshold density of host cells independent of the phage population and adsorption constant. In vivo bacterial infections are also complicated by nonuniform dispersal of the target bacteria in the body and the absence of any technique by which to rapidly enumerate and map the distribution of such populations. In addition, the larger the target bacterial population at the time that it is treated and killed, the larger the release of endotoxin and the chance that lethal septic shock will follow. Therefore, given that we have demonstrated that it is possible to infect every target cell in a given volume even when the cells are at very low densities and that it takes no more phage to do so than to infect all of the cells in a much denser population, it seems that the safest course of action in vivo is always to use as many phage as necessary to infect all of the target cells with the first dose. Then treatment can begin at the earliest possible time and replication of the phage is not necessary.
It has also been observed that many phages that proliferate on a given host in vitro fail to do so in vivo (7). This fact, as well as concerns that lytic phage may facilitate the transfer of toxic genes from pathogenic bacteria to commensal flora, has encouraged the development of nonreplicating phage therapeutics. In the lethal agent delivery system (6), nonreplicating phage function as molecular syringes that inject phagemids expressing bacterial suicide genes resulting in cell death. Due to the inability of lethal agent delivery systems to proliferate in vivo, there is no question of the necessity of being able to deliver such a lethal phagemid to every bacterial cell in a given culture, or infected individual, for a therapeutic antimicrobial result. Our results showing transduction of every cell in aliquots of bacteria in vitro suggest that delivery of bactericidal agents by nonreplicating phage may indeed be a viable antimicrobial strategy.
In conclusion, the rate at which phage bind to cells is very accurately predicted by Schlesinger's equation (equation 1). Four factors determine how many bacteria will be infected or transduced by phage in a given situation: the density of host cells, the adsorption constant of the phage, the number of phage, and the length of time for which they interact. The simple ratio of virus particles to cells given by the MOI is meaningless in the absence of information about these other parameters. Furthermore, the virologist's rule of thumb that an MOI of 10 be used in order to ensure that 99.99% of the host cells are infected by at least one virus particle is dependent on the assumption that all of the virus added will be bound to cells by the end of the adsorption period. We have shown that this is only a valid assumption for bacteriophage when host cells are present at densities equal to or greater than concentration 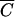 , which is, in turn, dependent on the adsorption constant of the phage and the length of the adsorption period. We have also shown, however, that it is possible to accurately estimate the fraction of phage bound at any cell concentration and arbitrary time. More generally, adoption of the value MOIactual in place of the value MOI results in a rule of thumb in which the fraction of input phage bound in the time allowed is taken into account and reproducibility between experiments will be greatly enhanced.
, which is, in turn, dependent on the adsorption constant of the phage and the length of the adsorption period. We have also shown, however, that it is possible to accurately estimate the fraction of phage bound at any cell concentration and arbitrary time. More generally, adoption of the value MOIactual in place of the value MOI results in a rule of thumb in which the fraction of input phage bound in the time allowed is taken into account and reproducibility between experiments will be greatly enhanced.
Acknowledgments
This research was supported by Hexal Gentech Forschungs GmbH (Munich, Germany), except for A. Kasman, who was supported solely by the College of Charleston.
We thank M. B. Yarminlinsky for providing bacteriophage P1CmC1.100 and Phillip A. Werner for preparing the P1 phagemid-containing lysate.
APPENDIX A
Elementary probability states that, given N cells and M bound phage per cell, the probability that a given cell will be infected is [1 − (1/N)]MN. The old rule of thumb that M = 10 is sufficient regardless of N follows from the remarkable but well-established fact that
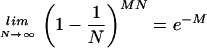 |
and so, for a large N, one needs MOIactual = −ln(0.0001) = 9.21 to get an expected infection rate of 99.99% (which has presumably been rounded up to M = 10 for convenience). However, for lower cell numbers, an MOIactual of 10 is still more than sufficient. The observant reader may notice that for extremely low cell numbers, one can get nearly 100% infection with an MOIactual noticeably smaller than 10. However, since the limit above converges quickly, for N > 10, one can say that e−MOIactual is a good approximation of the expected percentage of uninfected cells. Consequently, the rule that an MOI of 10 is sufficient to ensure that nearly all of the cells are infected should remain true assuming that one now means an MOIactual, and not an MOIinput, of at least 10.
APPENDIX B
In general, it is possible that the MOIactual is significantly less than the MOIinput. If this is the case, then the ratio of MOIactual to MOIinput would be significantly smaller than 1. In contrast, we will say that the two are essentially equal if this ratio has a value of greater than 0.9999. It is easily determined that this will be the case for any concentration C that is greater than C̄ with the equation C̄ = [ln(10,000)/kt] ≈ 9.2/kt. In fact, since −kC̄t = [ln(10,000)kt]/kt = −ln(10,000) = ln(0.0001), it follows that for any C that is greater than C̄ as defined above, MOIactual/MOIinput = (1 − e−kCt) > (1 − e−kC̄t) = [1 − eln(0.0001)] = 1 − 0.0001 = 0.9999.
REFERENCES
- 1.Andreadis, S., T. Lavery, H. E. Davis, J. M. Le Doux, M. L. Yarmush, and J. R. Morgan. 2000. Toward a more accurate quantitation of the activity of recombinant retroviruses: alternatives to titer and multiplicity of infection. J. Virol. 74:1258-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caro, L. G., and M. Schnos. 1966. The attachment of the male-specific bacteriophage F1 to sensitive strains of Escherichia coli. Proc. Natl. Acad. Sci. USA 56:126-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Kievit, T. R., and B. H. Iglewski. 2000. Bacterial quorum sensing in pathogenic relationships. Infect. Immun. 68:4839-4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Messing, J., B. Gronenborn, B. Muller-Hill, and P. Hans Hopschneider. 1977. Filamentous coliphage M13 as a cloning vehicle: insertion of a HindIII fragment of the lac regulatory region in M13 replicative form in vitro. Proc. Natl. Acad. Sci. USA 74:3642-3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan, J. R., J. M. LeDoux, R. G. Snow, R. G. Tompkins, and M. L. Yarmush. 1995. Retrovirus infection: effect of time and target cell number. J. Virol. 69:6994-7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norris, J. S., C. Westwater, and D. Schofield. 2000. Prokaryotic gene therapy to combat multidrug resistant bacterial infection. Gene Ther. 7:723-725. [DOI] [PubMed] [Google Scholar]
- 7.Payne, R. J. H., D. Phil, and V. A. A. Jansen. 2000. Phage therapy: the peculiar kinetics of self-replicating pharmaceuticals. Clin. Pharm. Ther. 68:225-230. [DOI] [PubMed] [Google Scholar]
- 8.Payne, R. J. H., and V. A. A. Jansen. 2001. Understanding phage therapy as a density dependent kinetic process. J. Theor. Biol. 208:37-48. [DOI] [PubMed] [Google Scholar]
- 9.Rosner, J. L. 1972. Formation, induction, and curing of bacteriophage P1 lysogens. Virology 48:679-680. [DOI] [PubMed] [Google Scholar]
- 10.Salivar, W. O., H. Tzagoloff, and D. Pratt. 1964. Some physical-chemical and biological properties of the rod-shaped coliphage M13. Virology 24:359-371. [DOI] [PubMed] [Google Scholar]
- 11.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 12.Schlesinger, M. 1932. Adsorption of phages to homologous bacteria. II. Quantitative investigations of adsorption velocity and saturation. Estimation of the particle size of the bacteriophage. Z. Hyg. Immunitaetsforsch. 114:149-160. Translated from German and reprinted in G. S. Stent (ed.), Papers on bacterial viruses, 2nd ed., p. 26-36. Little Brown and Company, Boston, Mass., 1965. [Google Scholar]
- 13.Stent, G. S. 1963. Molecular biology of bacterial viruses, p. 88-96. W. H. Freeman and Company, San Francisco, Calif.
- 14.Tzagoloff, H., and D. Pratt. 1964. The initial steps in infection with coliphage M13. Virology 24:372-380. [DOI] [PubMed] [Google Scholar]
- 15.Valentine, R. C., and A. C. Allison. 1959. Virus particle adsorption. I. Theory of adsorption and experiments on the attachment of particles to nonbiological surfaces. Biochim. Biophys. Acta 34:10-23. [DOI] [PubMed] [Google Scholar]
- 16.Wiggins, B. A., and M. Alexander. 1985. Minimum bacterial density for bacteriophage replication: implications for significance of bacteriophages in natural ecosystems. Appl. Environ. Microbiol. 49:19-23. [DOI] [PMC free article] [PubMed] [Google Scholar]