

Abstract
Phosphorylation of the eukaryotic initiation factor eIF4E in response to mitogenic stimuli and cytokines is implicated in the regulation of the initiation step of translation. It still remains unclear how the phosphorylation of eIF4E regulates the translation. To address this problem, we applied a unique technique in protein engineering, intein-mediated protein ligation, to synthesize eIF4E, which is selectively phosphorylated at Ser 209. Using selectively chosen synthetic cap analogs, we compared quantitatively the cap affinity for phosphorylated and unphosphorylated eIF4E by a fluorometric time-synchronized titration method. A 1.5- to 4.5-fold reduction of the cap affinity for phosphorylated eIF4E was observed, depending on the negative charge of the 5′-to-5′ phosphate chains as well as the presence of a longer tetraribonucleotide strand. Possible implications for understanding the regulation of eIF4E functioning, cap complex formation, and stability, are discussed.
Keywords: expressed protein ligation, fluorescence, intein, mRNA 5′ cap, translation initiation
INTRODUCTION
Eukaryotic mRNA translation initiation is a complicated, highly regulated process involving assembly of a large protein–mRNA complex that directs the ribosome to the initiation codon. In most cases, translation begins with the eukaryotic initiation factor 4E recognizing the m7(5′)Gppp (5′)N (where N is any nucleotide) cap structure at the mRNA 5′ terminus (Mathews et al. 2000). eIF4E forms a complex (termed eIF4F) with two other proteins: eIF4A, an RNA helicase, and eIF4G, a scaffolding protein that binds other factors to recruit the ribosome. The N terminus of eIF4G, apart from interacting with eIF4E, binds the poly(A)-binding protein, whereas its C terminus contains binding sites for eIF4A, eIF3, and Mnk1 or Mnk2 kinase (Lamphear et al. 1995; Imataka et al. 1998; Pyronnet et al. 1999). The interaction of these factors and the 40S small ribosomal subunit is required for the assembly of the 48S ribosomal preinitiation complex.
The eIF4E amount is limited as compared with other initiation factors (Duncan et al. 1987), which renders eIF4E an important target for the translation regulation (Sonenberg 1996). The accessibility of eIF4E is controlled through reversible interaction with small eIF4E-binding proteins, 4E-BPs, which block the association between eIF4E and eIF4G, thereby preventing the formation of the active eIF4F complex and inhibiting protein synthesis. The binding of the 4E-BPs to eIF4E is regulated by their specific phosphorylation state. The hyperphosphorylation of 4E-BPs in response to cell stimulation by hormones and growth factors abrogates their interaction with eIF4E (Gingras et al. 2001). Mammalian eIF4E is phosphorylated at a single residue, serine 209 (Joshi et al. 1995), by Mnk1 and Mnk2 kinases, which are bound to eIF4G (Flynn & Proud 1995, Waskiewicz et al. 1999, Scheper et al. 2001). Phosphorylation of Drosophila eIF4E is important for growth (Lachance et al. 2002). However, the mechanism by which the eIF4E phosphorylation might affect translation is not understood. It was reported that phosphorylated eIF4E, purified from rabbit reticulocytes, exhibits a three- to fourfold higher binding affinity for cap analogs (Minich et al. 1994). Contrary to these findings, a recent report by Scheper et al. (2002) showed reduction of the phosphorylated eIF4E affinity for capped mRNA. Unphosphorylated recombinant eIF4E can initiate translation in vitro (Svitkin et al. 1996). This suggests that the phosphorylation is not absolutely required for eIF4E to function. Most of the eIF4E (∼80–85%) in the 48S preinitiation complex is in the phosphorylated form, whereas the unbound protein is phosphorylated only in ∼50% (Lamphear & Panniers 1990). Numerous studies report a correlation between eIF4E phosphorylation and the rate of protein synthesis, implying that phosphorylation has a positive effect on cap-dependent translation (Joshi-Barve et al. 1990; Lamphear and Panniers 1990; Kleijn et al. 1998). Based on the crystal structure of the complex of unphosphorylated murine eIF4E with m7GDP (Marcotrigiano et al. 1997), it has been postulated that a phosphate group at Ser 209 forms a salt bridge with the ɛ-amino group of Lys159, which “clamps” the mRNA chain to eIF4E. This interaction was postulated to enhance the eIF4E–cap interaction.
To elucidate the modulation of the eIF4E cap-binding activity by phosphorylation, we have measured spectroscopically the affinity of several cap analogs for phosphorylated eIF4E versus its unphosphorylated counterpart by means of the fluorescence quenching method (Niedzwiecka et al. 2002). The series of the analogs used in the study included those with increasing negative charge as a result of phosphate chain elongation, and one with increasing number of the nucleotides in the RNA chain (Fig. 1 ▶).
FIGURE 1.

(A) Structures of guanine mono- and dinucleotide cap analogs methylated at N(7) and (B) the capped triribonucleotide, m7GpppAm2′pUm2′pAm2′. Protons that partially dissociate at pH 7.2 are marked with asterisks.
A new technique in protein engineering, that is, intein-mediated protein ligation (IPL; Holford and Muir 1998; Xu and Evans 2001), has been applied to generate pure eIF4E, selectively phosphorylated at Ser 209 (Fig. 2 ▶). This method allows us to join a synthetic peptide and a recombinant protein via a peptide bond in a direct chemical reaction in vitro. The key to this process is the generation of a recombinant protein possessing a C-terminal α-thioester that can react with an N-terminal cysteine of the synthetic peptide to form the peptide bond. The recombinant protein with the reactive α-thioester can be obtained by exploiting intein, an element of the natural protein splicing. Protein splicing is a posttranslational process, during which a protein undergoes an intramolecular rearrangement, that is, excision of an intein from the precursor protein, and the connection of the flanking regions, exteins. It has been shown that protein splicing involves thioester intermediates (Xu and Perler 1996). It is possible to chemically intercept the splicing process with a thiol reagent by appending the recombinant fragment to the N terminus of the modified intein, thereby generating the corresponding recombinant protein α-thioester (Chong et al. 1997; Severinov and Muir 1998).
FIGURE 2.

Pattern for obtaining a phosphorylated eIF4E via the IPL method. In the first step, the cDNA of truncated eIF4E (28–204) was cloned to pTXB1 vector (New England Biolab). Then it was expressed in E. coli as a fusion protein consisting of the eIF4E (28–204)–Mxe GryA intein–chitin binding domain. The fusion was reconstituted from inclusion bodies by one-step dialysis. In the next step, the fusion protein was absorbed on chitin beads and incubated with ethanethiol overnight to generate stable C-terminal thioester on the target protein. After purification by HPLC, ethyl α-thioester derivative of eIF4E (28–204) was ligated with synthetic fragment of eIF4E (205–217) to yield the protein, either phosphorylated at Ser 209 or unphosphorylated.
Here we studied the cap affinity for the phosphorylated eIF4E using eIF4E obtained by the IPL method and selected synthetic cap analogs.
RESULTS
Strategy for obtaining phosphorylated eIF4E by the IPL method
The efficiency of protein semisynthesis by means of the IPL method depends greatly on the proper choice of the ligation site. The ligation reaction occurs exclusively between the C-terminal thioester and the N-terminal cysteine, thus requiring a cysteine in the sequence of a target protein. The target protein needs to be divided into two fragments: the first containing the region of the protein to be readily modified by chemical synthesis, and the second being expressed successfully in the intein system. Serine 209 being the ninth amino acid from the C terminus of eIF4E provides for a relatively short synthetic peptide. However, there is no naturally occurring cysteine in the wild-type eIF4E in the position appropriate for efficient ligation. Therefore, to make the ligation feasible, a conservative Thr 205 amino acid was changed to a cysteine. According to the crystal structure (Marcotrigiano et al. 1997), Thr 205 is located at a flexible surface loop remote from the cap-binding site. Consequently, for the ligation, the eIF4E (28–217) was divided into two fragments, eIF4E (205–217), which was obtained chemically, and eIF4E (28–204), which was expressed in Escherichia coli in an expression system containing Mxe GryA intein. Additionally, the splitting site was chosen in such a way that an alanine was the last residue of the C terminus of eIF4E (28–204). It was shown that the presence of this amino acid at the C terminus of the target protein protects from in vivo cleavage, whereas in vitro cleavage by DTT remains very efficient (Chong et al. 1998).
Generation of thioester-tagged eIF4E (28–204)
An intein expression plasmid, pTXB1, containing the truncated eIF4E (28–204) was generated and expressed in E. coli. First, different induction conditions were tested to achieve successful soluble expression of the fusion protein. When the BL21(DE3) or ER2566 cells transformed with pTXBΔΔ4E were induced by 0.1 or 0.3 mM IPTG at 37°C or at room temperature (20–25°C), practically all of the expressed fusion protein was found in the insoluble fraction. Only at the lower induction temperature (15°C) did the fusion protein appear in the soluble fraction, but the amount was very low (data not shown), a few micrograms of the fusion protein from 1 L of culture. For these reasons, we chose to reconstitute the fusion protein from bacterial inclusion bodies. It was refolded by one-step dialysis, absorbed on chitin beads, and treated with 3% (v/v) ethanethiol overnight to generate a stable C-terminal thioester of eIF4E (see Materials and Methods). The ethanethiol was used instead of 2-mercaptoethanesulfonic acid (MESNA) or thiophenol because the ethyl α-thioester derivative of the target protein is stable enough and can be stored for several months as a lyophilized powder without significant hydrolysis (Ayers et al. 1999). Analysis of the chitin beads after thiol-induced cleavage on SDS-PAGE (Fig. 3B ▶, lanes 1,2) showed that more than 80% of eIF4E was cleaved from intein, which is consistent with correct refolding of the mini-intein. The cleaved eIF4E was eluted from a chitin column along with intein-CBD by 90% acetonitrile, because of poor solubility of eIF4E (28–204). Proteins were separated by HPLC, and the desired product, purified C-terminal thioester of eIF4E, was characterized by ESI-MS; predicted (average isotopic) mass: 21750.0 daltons, observed: 21750.2 ± 0.1 daltons. The reconstruction of eIF4E (28–204) in fusion with intein appeared to be a very efficient method to prepare milligram amounts of pure fusion protein with the active intein.
FIGURE 3.

Characteristic of semisynthetic eIF4Es. (A,B) Expression and ligation reaction visualized on a Coomassie-stained 12% SDS-PAGE gel. (A) (lane 1) Crude extract from uninduced cells, (lane 2) crude extract from cells induced by 0.5 mM IPTG at 37°C for 3 h, (lane 3) insoluble fraction of lysed cells, (lane 4) soluble fraction of lysed cells, (lane 5) fusion protein after refolding from inclusion bodies loaded on chitin column, (lane 6) chitin column flow through, (lane 7) chitin beads before cleavage with ethanethiol, (lane 8) protein marker weight standards (SIGMA); (B) (lane 1) chitin beads before cleavage with ethanethiol, (lane 2) chitin beads after cleavage with ethanethiol, (lane 3) product of ligation reaction of two fragments eIF4E loaded on ion-exchange column, (lane 4) flow through, (lane 5) semisynthetic phosphorylated eIF4E purified on Mono SP column, (lane 6) protein marker weight standards (SIGMA), (lane 7) recombinant murine eIF4E (28–217). (C) Characteristic of semisynthetic proteins by electrospray ionization MS. (Top) Wild-type murine eIF4E (28–217), predicted mass: 22113.0 daltons, (middle) semisynthetic unphosphorylated eIF4E (28–217)T205C, predicted mass: 22115.2 daltons, (bottom) semisynthetic phosphorylated eIF4E (28–217)T205C/S209phos, predicted mass: 22195.0 daltons.
Protein ligation
The joining reaction of ethyl α-thioester derivative of eIF4E (28–204) with the synthetic phosphorylated or unphosphorylated peptide of eIF4E fragment (205–217) was performed under denaturing conditions (6 M guanidine hydrochloride) at pH 8.0, in the presence of the thiol reagent MESNA (for details, see Materials and Methods). The reaction time (72 h) and an excess of peptide (∼10-fold) strongly influence the efficiency of ligation, which was nearly 100% at pH 8.0–9.0 (Fig. 3B ▶, lane 3). At lower pH and in the absence of the thiol reagent, the reaction did not proceed. Both the phosphorylated and unphosphorylated semisynthetic eIF4E (28–217)T205C proteins were refolded by one-step dialysis and subsequently purified by ion-exchange chromatography on Mono SP column (shown for phosphorylated eIF4E in Fig. 3B ▶, lane 5). Molecular masses of the obtained proteins were in good agreement with those predicted theoretically (Fig. 3C ▶).
Phosphorylation of eIF4E attenuates binding of cap analogs
Phosphorylation at Ser 209 (Flynn and Proud 1995; Joshi et al. 1995) occurs in response to various extracellular stimuli (Kleijn et al. 1998; Gingras et al. 1999). The comparison of the cap-binding affinity for the phosphorylated versus unphosphorylated forms of eIF4E by means of fluorescence titration experiments (Niedzwiecka et al. 2002) could elucidate the role of such protein modification in the translation initiation mechanism, and also resolve the contradictions in literature (Minich et al. 1994; Scheper et al. 2002). A fast and accurate time-synchronized fluorescence titration method (TST) was recently successfully applied to measure the affinity of the cap analogs for eIF4E (Niedzwiecka et al. 2002), yielding accurate equilibrium association constants (Kas). The method takes into account all the factors that influence the correctness and accuracy of the data: emission of the free ligand, the inner filter effect, and the concentration of the active protein (Pact) as a free parameter of the numerical fitting (see Materials and Methods). Neglect of some or usually all of these factors in previous communications led to erroneous (Carberry et al. 1989, 1990; Ueda et al 1991; Minich et al. 1994) or qualitative results (Scheper et al. 2002).
Comparison of the fluorescence quenching experiments for the wild-type eIF4E and its phosphorylated and unphosphorylated, semisynthetic counterpart is shown in Figure 4 ▶ for a few selected cap analogs: m7GMP, m7GTP, m7GpppG, and m7GpppAm2′pUm2′pAm2′. The titration curves perfectly fit the experimental data points, testifying to the high accuracy of the Kas values (Table 1 ▶). As can be deducted from the titration results, replacement of Thr 205 by a cysteine had no effect on the protein affinity for the cap. The wild-type and the mutated eIF4E exhibit similar binding affinities for the cap analogs within three standard deviations (Table 1 ▶), which is crucial for our analysis of the effects of the phosphorylation.
FIGURE 4.

Titration curves for binding of unphosphorylated eIF4E (28–217) (open square), semisynthetic unphosphorylated eIF4E (28–217)T205C (closed circle), and semisynthetic phosphorylated eIF4E (28–217)T205C/S209phos (closed triangle) to the cap analogs. Both forms of eIF4E, each one at a concentration of 0.1 μM, were titrated with m7GTP and m7GMP (top), m7GpppG (middle), and m7GpppAm2′pUm2′pAm2′ (bottom), at 20°C in 50 mM HEPES/KOH at pH 7.2, containing 100 mM KCl, 0.5 mM EDTA, and 1 mM DTT. Protein fluorescence was excited at 280 nm and observed at 337 nm. The shift of the titration curve to a higher concentration of the cap analog for phosphorylated versus unphosphorylated eIF4E results from weaker binding affinity of the cap analogs for the protein.
TABLE 1.
Equilibrium association constants (Kas) for interactions of phosphorylated and unphosphorylated eIF4E with various cap analogs at 20°C
| Kas [μM−1] | ||||
| Cap analogs | eIF4E (28–217) | eIF4E (28–217) T205C | eIF4E (28–217) T205C/S209phos | Kas (wt)/Kas (phos) |
| m7GTP | 119.7 ± 5.7 | 111.6 ± 3.7 | 42.6 ± 1.8 | 2.8 |
| m7GDP | 20.9 ± 1.5 | 17.8 ± 0.6 | 10.5 ± 0.3 | 2.0 |
| m7GMP | 1.5 ± 0.1 | 1.5 ± 0.1 | 1.1 ± 0.1 | 1.4 |
| m7GpppG | 12.5 ± 0.3 | 10.5 ± 0.3 | 5.8 ± 0.3 | 2.2 |
| m27,3′OGpppG | 10.2 ± 0.3 | 7.9 ± 0.2 | 4.33 ± 0.15 | 2.4 |
| m7Gpppm2′OG | 7.5 ± 0.2 | 7.8 ± 0.2 | 3.2 ± 0.2 | 2.3 |
| m7GpppC | 6.1 ± 0.3 | 5.9 ± 0.2 | 2.8 ± 0.2 | 2.2 |
| m7GpppA | 5.0 ± 0.2 | 4.9 ± 0.1 | 2.26 ± 0.10 | 2.2 |
| m7Gpppm6A | 3.9 ± 0.2 | 4.25 ± 0.20 | 1.99 ± 0.70 | 2.0 |
| m7Gpppm7G | 5.0 ± 0.2 | 4.83 ± 0.20 | 2.84 ± 0.10 | 1.8 |
| m7GppppG | 110.9 ± 6.0 | 97.5 ± 3.2 | 27.3 ± 1.1 | 4.1 |
| m7Gppppm7G | 49.4 ± 1.5 | 50.8 ± 1.6 | 20.1 ± 1.0 | 2.5 |
| m7GpppAm2′pUm2′pAm2′ | 43.0 ± 1.8 | 44.0 ± 2.6 | 9.6 ± 0.4 | 4.5 |
Phosphorylation of eIF4E at Ser 209 reduces its affinity for the cap to different extents, depending on the structure of the cap analog (Fig. 5 ▶; Table 1 ▶). The association constants of phosphorylated eIF4E for m7GTP and m7GMP are, respectively, ∼3- to 1.5-fold lower, as compared to the unphosphorylated protein. The dinucleotide triphosphate analogs bind to the phosphorylated eIF4E about twofold more weakly, analogous to m7GDP. Replacing the second guanine base by adenine or cytosine in the dinucleotide cap analog only slightly changes the affinity for the proteins. The addition of a δ-phosphate group into the 5′-5′ phosphate bridge, as in the tetraphosphate dinucleotide m7GppppG, causes more than a fourfold decrease of the affinity for phosphorylated eIF4E, whereas the extension of the RNA chain to the tetranucleotide gives the highest, up to a 4.5-fold reduction of Kas (see below). Introduction of a 2′-methyl group at the second guanosine moiety only slightly reduces the Kas value for the phosphorylated eIF4E.
FIGURE 5.

Decreasing affinity for phosphorylated eIF4E Kas(phos)/Kas(wt) [%] versus negative charge of the cap analogs. The resultant charges at given pH were estimated from the experimental pKa1 values for dissociation of the N1 proton at of 7-methyloguanosine (Rhoads et al. 1983; Wieczorek et al. 1995), and from the experimental pKa2 values for dissociation of the second proton of the terminal phosphate group in the mononucleotide cap analogs (Dawson et al. 1969).
Elongation of the phosphate chain in both classes of analogs, that is, mononucleotides and dinucleotides, results in clear enhancement of the binding strength to eIF4E. It is most probably due to the increase of the resultant negative charge of the cap molecule (Niedzwiecka et al. 2002). The introduction of the second nucleotide reduces the number of exchangeable protons and hence the electric charge, thus resulting in smaller Kas. This effect is less marked in the case of phosphorylated eIF4E as compared to the unphosphorylated protein. The longer the negatively charged phosphate chain, the more pronounced is the increase in Kas(wt)/ Kas(phos): For m7GMP, both Kas are similar; for m7GDP and the dinucleotide triphosphates, the ratio is ∼2 to 2.4; and for m7GTP and m7GppppG, they are 2.8 and 4.1, respectively. Positive charge in the second base of the tetraphosphate m7Gppppm7G results in the reduction of Kas as compared to m7GppppG, and restoring of the Kas(wt)/ Kas(phos) value to that characteristic for the dinucleotide triphosphates.
To summarize, the results suggest that the destabilization effect of the eIF4E phosphorylation on the formation of the complexes with cap analogs arises from the electrostatic repulsion between the negatively charged parts/residues of eIF4E and the cap: the phosphate group at Ser 209 of eIF4E and the 5′-to-5′ phosphate chain of the cap (see Discussion).
Capped triribonucleotide interacts with eIF4E more strongly than dinucleotide cap analogs
The capped triribonucleotide, m7GpppAm2′pUm2′pAm2′, binds to eIF4E four-fold more strongly than m7GpppG, and about eight-fold more strongly than m7GpppA, which possesses the same second base as the capped trimer (Table 1 ▶). Phosphorylation of eIF4E reduces its affinity for this cap analog by ∼4.5-fold. This effect is twice as large as that observed for all other dinucleotide cap analogs, except for the highly negatively charged tetraphosphate m7GppppG. This suggests that some additional charge-related interactions are disrupted by the presence of the phosphate group at Ser 209, and raises the notion that nucleotides situated next to the cap in m7GpppAm2′pUm2′pAm2′ and/or their backbone phosphates are involved in the formation of additional stabilizing contacts with eIF4E (see Discussion). However, the total eIF4E fluorescence quenching Q (Q = F0 − F∞) is very similar, ∼61 ± 5%, for different mono- and dinucleotide cap analogs as well as for capped triribonucleotide. This proves the lack of explicit differences in the eIF4E conformational changes upon interaction with the mRNA oligomer and with the cap structure alone.
DISCUSSION
To derive quantitative data on the effect of serine phosphorylation on eIF4E interaction with the cap, we have generated, by the unique IPL technique (Holford et al. 1998), an eIF4E protein that is specifically phosphorylated at Ser 209 and used it in a novel fluorometric time-synchronized titration method (Niedzwiecka et al. 2002). The uncertainty of the determined Kas values does not exceed 5%, except for the weak binding m7GMP (less than 10%). The selection of the cap analogs possessing different negative charges clearly shows an electrostatic repulsion between the phosphate group of eIF4E and the phosphate chain of cap that causes a reduction in the affinity of eIF4E for the cap. This is consistent with our previous data that anchoring of the cap phosphates is the first step of the specific recognition in the eIF4E cap-binding center (Niedzwiecka et al. 2002). Thus, our results are in accord with the fluorometric and SPR measurements of Scheper et al. (2002), and at variance with the previous report of Minich et al. (1994). The latter data were also based on fluorescence quenching experiments, in which the phosphorylated and unphosphorylated forms of eIF4E, separated by affinity chromatography, were titrated by m7GTP, m7GpppG, and capped globin mRNA. However, as noted before (Scheper et al. 2002), the purity of eIF4E was not documented and it could have contained eIF4G or 4E-BP.
The influence of eIF4E phosphorylation on the interaction with the cap is dependent on the interaction of the mRNA chain with the surface of eIF4E. Extending the chain from di- to tetranucleotide enhances the binding with the unphosphorylated eIF4E, whereas phosphorylating the protein reduces this effect. The mode of interaction of the second and subsequent nucleotides with the unphosphorylated eIF4E, which may be deduced from the known structures of the complexes, is highly speculative. According to the fluorescence results (Table 1 ▶) m7GpppG binds to eIF4E two times more tightly than m7GpppA. No interaction between the guanosine and the truncated eIF4E is observed in the crystal structure (Niedzwiecka et al. 2002), contrary to the crystallographic data for the full-length human eIF4E bound with m7GpppA (Tomoo et al. 2002). In the latter complex, a conformational change of the C-terminal protein loop stabilizes the adenosine by hydrogen bonding with Thr 205 and Thr 211. Some intermolecular contacts between the second adenosine and the protein were also observed by NOE measurements for yeast eIF4E bound with 13C/15N double-labeled adenosine containing m7GpppA (Matsuo et al. 2000). Moreover, no NOE contacts with the yeast protein were detected for the double-labeled cytosines of m7GpppACC (Matsuo et al. 2000), and the Gibbs free energy change (ΔΔG) due to additional interactions of the pUm2′pAm2′ fragment with murine eIF4E is −5.24 kJ/mole (calculated from the Kas values in Table 1 ▶). Our results point to some stabilizing, electrostatic interactions between the unphosphorylated protein and the RNA chain, which are diminished by +1.7 kJ/mole due to the introduction of an additional negative charge at the protein surface by the phosphorylation.
Recently, genetic data demonstrated that eIF4E phosphorylation is required for cell growth and proliferation, because mutation of the Drosophila eIF4E phosphorylation site causes slow growth and retarded development (Lachance et al. 2002). However, the mechanism by which phosphorylation of eIF4E at Ser 209 regulates the translation is poorly understood. Two molecular models of regulation of the eIF4E activity by phosphorylation at Ser 209 were proposed. The “clamping” model, based on the crystallographic structure of the murine eIF4E complex with m7GDP, was proposed by Marcotrigiano et al. (1997). It postulates the formation of a salt bridge between the phosphorylated Ser 209 and Lys 159, which are 7 Å apart and are situated across the entrance to the cap-binding slot. Such a clamp could stabilize the mRNA chain at the protein surface. Although the new spectroscopic results contradict the earlier observations (Minich et al. 1994) that phosphorylated eIF4E exhibits an increase in the cap-binding activity, the phosphorylation and clamping event could occur but only after and not prior to mRNA binding to eIF4E. Thus, it is possible that phosphorylation occurs only after mRNA binding. A different model was proposed by Scheper et al. (2002). The association and dissociation rates for binding of capped mRNA to eIF4E, as roughly estimated by SPR, are at odds with the clamping model, inasmuch as an increase of the dissociation rate (10-fold) is observed upon phosphorylation of eIF4E. The authors proposed two possible mechanisms responsible for the effect of eIF4E phosphorylation on translation initiation. By decreasing the affinity for cap, phosphorylation of eIF4E could facilitate the release of tethered eIF4F from the 5′ end of mRNA to promote ribosome scanning. However, it is not known yet whether any of the eIF4 factors is associated with the scanning ribosome (Dever 2002). Another possible role of the phosphorylation (Scheper et al. 2002) might be “reprogramming” of the translational machinery by the release of initiation factors from existing translational complexes, allowing other mRNAs to become translated.
Our data stress the importance of electrostatic interactions between three charged components of the eIF4E-mRNA complex, that is, the negatively charged Ser 209, the cap phosphate chain, and the positively charged Lys 159. Depending on the type of eIF4E and/or the bound cap analog, the mutual orientation of Ser 209 and Lys 159, both located at the flexible polypeptide loops, is totally different, giving rise to at least two possible dynamic models of the intermolecular interactions in the eIF4E–cap complexes (Fig. 6 ▶). Phosphorylation at Ser 209 after the formation of the eIF4E–mRNA complex can either attract Lys 159 to form a salt bridge, with a concomitant induced fit that would bury the phosphate chain of the cap deeper into eIF4E (in accordance with the clamping model), or the phosphate group at Ser 209 can repulse the cap phosphate chain out of the protein surface toward the Lys 159 side chain (in accordance with Scheper’s model). In the former case, the proposed formation of the salt bridge fastens together the flexible parts of the eIF4E, thus stabilizing the mRNA chain in the complex. In the latter mechanism, the positively charged Lys 159 acts like a positively charged molecular “angle” to pull away the negatively charged triphosphate chain of the capped mRNA from the protein, thus facilitating dissociation of the eIF4E–cap complex. Both mechanisms could be conditioned by the energy of electrostatic interactions. Our results point to the significance of the electrostatic forces for the processes of formation, stabilization, and dissociation of the eIF4E–cap complex. These interactions are crucial for the regulation of the translation initiation step, implicating the importance of protein phosphorylation.
FIGURE 6.

Possible modes of conformation changes in the eIF4E–cap complex upon phosphorylation at Ser 209 (for detailed descriptions, see Discussion). The Lys 159 side chain is either marked in plain gray or filled with a wavy broken pattern to reflect the ordered or the conformationally free states, respectively. Hydrogen bonds and stacking contacts of the m7G moiety are represented by solid dashes in the tightly bound complex or by dotted dashes after dissociation of the phosphate chain.
MATERIALS AND METHODS
Cloning, expression, and purification of eIF4E (28–204)–intein–CBD fusion
The DNA clone for the truncated fragment of eIF4E (28–204 amino acid) was constructed by the polymerase chain reaction (PCR) from eIF4E (28–217) cDNA. The PCR product was recloned into NdeI and SapI sites of the intein vector pTXB1 (New England Biolabs), yielding pTXB1–4EΔΔ. ER2566 cells (New England Biolabs), transformed with pTXB1–4EΔΔ, were grown in LB medium at 37°C until O.D.600 reached ∼1, and induced by 0.5 mM IPTG for 3 h at 37°C to yield the fusion protein in inclusion bodies. The cells were harvested by centrifugation, resuspended in lysis buffer (20 mM HEPES/KOH at pH 7.5, 100 mM KCl, 0.5 mM EDTA, 10% glycerol) and lysed by sonication. After centrifugation of the lysate, the supernatant was removed and the pellet was washed three times with 20 mM HEPES/KOH (pH 7.2) containing 1 M guanidine hydrochloride, 10% glycerol, and 1 mM Tris-(2-carboxyethyl)phosphine (TCEP). The inclusion bodies were dissolved in 20 mM HEPES/KOH (pH 7.2), 6 M guanidine hydrochloride, 10% glycerol, 1 mM TCEP, and cell debris was removed by centrifugation. The supernatant containing pure fusion protein, eIF4E (28–204)–intein–CBD, was diluted in 20 mM HEPES/KOH (pH 7.2), 6 M guanidine hydrochloride, 10% glycerol, 1 mM TCEP to a protein concentration of less than 0.1 mg/mL, and dialyzed against 50 mM HEPES/KOH (pH 7.2), 100 mM KCl, 0.5 mM EDTA. The solution of the refolded fusion protein was concentrated on a chitin column equilibrated with 20 mM HEPES/KOH (pH 7.2), 250 mM KCl, 0.1 mM EDTA, at 4°C.
Preparation of eIF4E (28–204)-Ethyl-α-thioester
eIF4E–intein–CBD fusion protein bound to chitin beads was suspended in 20 mM HEPES/KOH (pH 7.5), 0.25 M KCl, 0.1 mM EDTA, 1 mM TCEP, to which 3% (v/v) ethanethiol was added to generate a C-terminal thioester on the target protein (Ayers et al. 1999). Beads were incubated for 20 h at room temperature with gentle stirring to equilibrate the reagents. After incubation, the chitin beads were washed with 90% acetonitrile to elute the cleaved protein and the intein–CBD from chitin. The ethyl-α-thioester derivative of eIF4E was purified from the intein by use of semipreparative HPLC (Waters 600E Multisolvent Delivery system). The protein was lyophilized and stored at −20°C. The purified protein was analyzed by ESI-MS, predicted (average isotopic) mass: 21750.0 daltons, observed: 21750.2 ± 0.1 daltons.
Synthesis of 205–217 eIF4E fragments
Two peptides corresponding to 205–217 fragment of murine eIF4E (CKSGS[p]TTKNRFVV), phosphorylated and unphosphorylated at Ser 209, were synthesized chemically on a Wang resin (Novabiochem) using Fmoc strategy (Pennington and Dunn 1994). Phosphoserine was directly incorporated during the synthesis as the mono-protected N-α-Fmoc-O-benzyl phosphoserine derivative (Novabiochem; Wakamiya et al. 1994). The peptides were deprotected and cleaved from the resin using the mixture of trifluoroacetic acid:trisopropylsilane:water at 95:2.5:2.5. The peptides were purified by semipreparative HPLC and analyzed by ESI-MS: the unphosphorylated eIF4E peptide, predicted mass: 1426.6 daltons, observed: 1426.7 ± 0.1 daltons; the phosphorylated peptide, predicted mass: 1506.7 daltons observed: 1507.0 ± 0.1 daltons.
Ligation reaction
The ligation of eIF4E (28–204)-ethyl-α-thioester with the synthetic fragment of eIF4E (205–217) was performed in 50 mM HEPES/KOH (pH 9.0), 6 M guanidine hydrochloride, 100 mM KCl, 0.5 mM EDTA, 1 mM TCEP, and 100 mM MESNA. The lyophilized protein and peptide were dissolved at a final concentration of 0.1 to 0.3 mM and 1 to 2 mM, respectively. The final pH of the mixture was adjusted to 9.0 by adding KOH. The ligation reaction was performed for 72 h at 4°C with occasional stirring. After completing the ligation reaction, the proteins designated as eIF4E(28–217)T205C and eIF4E(28–217)T205C/S209phos, for unphosphorylated and phosphorylated forms of eIF4E, respectively, were purified by analytical HPLC. The masses of the obtained products were confirmed by ESI-MS.
For fluorescence assays, the ligated proteins were refolded by one-step dialysis from 6 M guanidine hydrochloride and purified by ion-exchange chromatography on a Mono SP column (Pharmacia) as described previously (Edery et al. 1998). Concentrations of the proteins were determined spectrophotometrically assuming ɛ280 = 53,400 M−1cm−1.
Expression and purification of murine eIF4E (28–217)
The murine eIF4E (28–217) cDNA was cloned into the pET3b vector (Novagen). The vector was ransformed into the BL21(DE3) E. coli strain and expressed as inclusion bodies. The protein was dissolved in 6 M guanidine hydrochloride, renatured by one-step dialysis, and purified by ion-exchange chromatography on a Mono SP column (Edery et al. 1998). The concentration of eIF4E was determined from absorption at 280 nm as stated above.
Chemical syntheses of cap analogs
Syntheses and purification of mono- and dinucleotide cap analogs were performed as described previously (Darzynkiewicz et al. 1985, 1990). The cap analogs concentrations were calculated from weighed amounts (±5%) and confirmed spectrophotometrically (Cai et al. 1999).
The capped triribonucleotide m7GpppAm2′pUm2′pAm2′ was prepared from m7GDP and triribonucleotide pAm2′pUm2′pAm2′ (m denotes 2′-O-methyl group within the nucleotide units). The latter one was obtained by a conventional phosphoramidite solid-phase synthesis using 5′-O-(4,4′dimethoxytrityl)-2′-O-methyladenosine-3′-O-succinate immobilized to LCAA-CPG (2′-OMe-A-RNA 500 from Glen Research) as a support, appropriate 5′-O-(4,4′-dimethoxytrityl)-2′-O-methylribonucleoside-3′-O-(2cyanoethyl-N,N-diisopropylphosphoramidities) as building blocks and tetrazole as an activator. The last coupling was performed with (2-cyanoethyl) [2,2-bisethoxycarbonyl-3-(4,4′-dimethoxytrityloxy)propyl-N,N-diisopropylphosphoramidite] followed by oxidation with iodine. The oligonucleotide conjugate cleaved from the support with concentrated aqueous (conc. aq.) ammonia exhibited the expected mass number (m/z 1527.5 by ESI-MS). After preparative HPLC purification, the product was treated with 80% aq. acetic acid, then with 40% aq. methylamine and purified by preparative HPLC again to give 5′-phosphorylated trinucleotide (m/z 1023.5 by ESI-MS). The subsequent reaction with imidazole in DMF in the presence of 2,2′-dithiodipyridine and triphenylphosphine yielded imidazolide of the oligoribonucleotide, which was coupled with 7-methylguanosine 5′-diphosphate in DMF in the presence of ZnCl2. After preparative HPLC purification, the final product was obtained as ammonium salt with the predicted formula peak in the mass spectrum.
Fluorescence binding assay
Fluorescence measurements were run on a LS-50B spectrofluorometer (Perkin Elmer Co.), in a quartz semimicro cuvette (Hellma) with optical lengths of 4 mm and 10 mm for absorption and emission, respectively. The sample was thermostatted at the temperature of 20.0 ± 0.3°C, controlled with a thermocouple inside the cuvette. Protein fluorescence was excited at 280 nm (slit 2.5 nm bandwidth, auto cutoff filter) and the fluorescence intensity was monitored by the registration of the whole spectrum in the range of 295 to 450 nm (with emission slit 4.0 nm and 290 nm cutoff filter). Alternatively, continuous time-synchronized titration (TST) at a single wavelength of 337 nm was applied, with the integration time of 30 sec and the gap of 30 sec for adding the ligand, using magnetic stirring to mix the added ligand and to keep the temperature constant. Titration was performed in 50 mM HEPES/KOH (pH 7.20), 100 mM KCl, 0.5 mM EDTA, and 1 mM DTT by adding 1 μL aliquots of the cap solution to 1400 μL of 0.1 μM eIF4E solution. Each titration consisted of 35–45 data points. Fluorescence intensities, when necessary, were corrected for dilution (less than 4%) and for inner filter effect. Association equilibrium constants (Kas) were obtained by fitting the theoretical curve for fluorescence intensity (F) upon total concentration of cap analogs ([L]) to the titration data according to the equation:
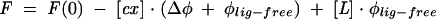 |
with the concentration of the cap-eIF4E complex [cx] given by
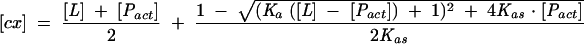 |
where Δφ stands for the difference between the fluorescence efficiencies of the apo- and cap-bound proteins; φlig-free is the fluorescence efficiency of the free cap analog; F(0) is the initial fluorescence, and Pact is the total concentration of the active protein (Niedzwiecka et al. 2002). The final Kas was calculated as a weighted average from 5 to 10 independent titration series. Numerical least-squares nonlinear regression analysis was performed using ORGIN 6.0 from Microcal Software Inc.
Acknowledgments
This work was supported by Human Frontier Science Program RG-0303/198-M and by Polish Committee for Scientific Research: KBN 6 P04A 055 17, KBN 3 P04A 071 22, BST-718/BF, BW-1525/BF, and PBZ-KBN-059/T09/10.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CBD, chitin binding domain from Bacillus circulans
CPG, controlled pore glass
IPTG, isopropyl-β-D-thiogalactopyranoside
eIF, eukaryotic initiation factor
4E-BP, 4E binding protein
LCAA, long chain alkylamine
MESNA, 2-mercaptoethanesulfonic acid
Mxe GryA intein, intein from Mycobacterium xenopi gryA gene
m7GMP, 7-methylguanosine 5′-monophosphate
m7GDP, 7-methylguanosine 5′-diphosphate
m7GTP, 7-methylguanosine 5′-triphosphate
m7GpppG, P1-7-methylguanosine-5′ P3-guanosine-5′ triphosphate
TCEP, tris-(2-carboxyethyl)phosphine
TST, time-synchronized titration
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2133403.
REFERENCES
- Ayers, B., Blaschke, U.K., Camarero, J.A., Cotton, G.J., Holford, M., and Muir, T.W. 1999. Introduction of unnatural amino acids into proteins using expressed protein ligation. Biopolymers 51: 343–354. [DOI] [PubMed] [Google Scholar]
- Cai, A.-L., Jankowska-Anyszka, M., Centers, A., Chlebicka, L., Stepinski, J., Stolarski, R., Darzynkiewicz, E., and Rhoads, R.E. 1999. Quantitative assessment of mRNA cap analogues as inhibitors of in vitro translation. Biochemistry 38: 8538–8547. [DOI] [PubMed] [Google Scholar]
- Carberry, S.E., Rhoads, R.E., and Goss, D.J. 1989. A spectroscopic study of the binding of m7GTP and m7GpppG to human protein synthesis initiation factor 4E. Biochemistry 28: 8078–8083. [DOI] [PubMed] [Google Scholar]
- Carberry, S.E., Darzynkiewicz, E., Stepinski, J., Tahara, S.M., Rhoads, R.E., and Goss, D.J. 1990. A spectroscopic study of the binding of N-7-substituted cap analogues to human protein synthesis initiation factor 4E. Biochemistry 29: 3337–3341. [DOI] [PubMed] [Google Scholar]
- Chong, S., Mersha, F.B., Comb, D.G., Scott, M.E., Landry, D., Vence, L.M., Perler, F.B., Benner, J., Kucera, R.B., Hirvonen, C.A., et al. 1997. Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene 192: 271–281. [DOI] [PubMed] [Google Scholar]
- Chong, S., Williams, K.S., Wotkowicz, C., and Xu, M.Q. 1998. Modulation of protein splicing of the Saccharomyces cerevisiae vacuolar membrane ATPase intein. J. Biol. Chem. 273: 10567–10577. [DOI] [PubMed] [Google Scholar]
- Darzynkiewicz, E., Ekiel, I., Tahara, S.M., Selinger, L.S., and Shatkin, A.J. 1985. Chemical synthesis and characteristic of 7-methylguanosine cap analogues. Biochemistry 24: 1701. [Google Scholar]
- Darzynkiewicz, E., Stepinski, J., Tahara, S.M., Stolarski, R., Ekiel, I., Haber, D., Neuvonen, K., Lehikoinen, P., Labadi, I., and Lonnberg, H. 1990. Synthesis, conformation and hydrolytic stability of P1, P3-dinucleotide triphosphate related to mRNA 5′-cap, and comparative kinetic studies on their nucleoside and nucleotide monophosphate analogs. Nucleosides & Nucleotides 9: 599–618. [Google Scholar]
- Dawson, R.M., Elliott, D.C., Elliott, W.H., and Jones, K.M. 1969. Data for Biochemical Research, vol. 2, Clarendon Press, Oxford, UK.
- Dever, T.E. 2002. Gene-specific regulation by general translation factors. Cell 108: 554–556. [DOI] [PubMed] [Google Scholar]
- Duncan, R., Milburn, S.C., and Hershey, J.W. 1987. Regulated phosphorylation and low abundance of HeLa cell initiation factor eIF-4F suggest a role in translational control. Heat shock effects on eIF-4F. J. Biol. Chem. 262: 380–388. [PubMed] [Google Scholar]
- Edery, I., Altmann, M., and Sonenberg, N. 1998. High-level synthesis in Escherichia coli of functional cap-binding eukaryotic initiation factor eIF-4E and affinity purification using a simplified cap-analog resin. Gene 74: 517–525. [DOI] [PubMed] [Google Scholar]
- Flynn, A. and Proud, C.G. 1995. Serine 209, not serine 53, is the major site of phosphorylation in initiation factor eIF4E in serum-treated Chinese hamster ovary cells. J. Biol. Chem. 270: 21684–21688. [DOI] [PubMed] [Google Scholar]
- Gingras, A.-C., Raught, B., and Sonenberg, N. 1999. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68: 913–963. [DOI] [PubMed] [Google Scholar]
- Gingras, A.-C., Raught, B., Gygi, S.P., Niedzwiecka, A., Miron, M., Burley, S.K., Polakiewicz, R.D., Wyslouch-Cieszynska, A., Aebersold, R., and Sonenberg, N. 2001. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes & Dev. 15: 2852–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holford, M. and Muir, T.W. 1998. Adding “splice” to protein engineering. Structure 6: 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imataka, H., Gradi, A., and Sonenberg, N. 1998. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J. 17: 7480–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Barve, S., Rychlik, W., and Rhoads, R.E. 1990. Alteration of the major phosphorylation site of eukaryotic protein synthesis initiation factor 4E prevents its association with the 48 S initiation complex. J. Biol. Chem./ITL 265: 2979–2983. [PubMed] [Google Scholar]
- Joshi, B., Cai, A.-L., Keiper, B.D., Minich, W.B., Mendez, R., Beach, C.M., Stepinski, J., Stolarski, R., Darzynkiewicz, E., and Rhoads, R.E. 1995. Phosphorylation of eukaryotic protein synthesis initiation factor 4E at Ser-209. J. Biol. Chem. 270: 14597–14603. [DOI] [PubMed] [Google Scholar]
- Kleijn, M., Scheper, G.C., Voorma, H.O., and Thomas, A.A. 1998. Regulation of translation initiation factors by signal transduction. Eur. J. Biochem. 253: 531–544. [DOI] [PubMed] [Google Scholar]
- Lachance, P.E., Miron, M., Raught, B., Sonenberg, N., and Lasko, P. 2002. Phosphorylation of eukaryotic translation initiation factor 4E is critical for growth. Mol. Cell. Biol. 22: 1656–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamphear, B.J. and Panniers, R. 1990. Cap binding protein complex that restores protein synthesis in heat-shocked Ehrlich cell lysates contains highly phosphorylated eIF-4E. J. Biol. Chem. 265: 5333–5336. [PubMed] [Google Scholar]
- Lamphear, B.J., Kirchweger, R., Skern, T., and Rhoads, R.E. 1995. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap-dependent and cap-independent translational initiation. J. Biol. Chem. 270: 21975–21983. [DOI] [PubMed] [Google Scholar]
- Marcotrigiano, J., Gingras, A.-C., Sonenberg, N., and Burley, S.K. 1997. Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell 89: 951–961. [DOI] [PubMed] [Google Scholar]
- Matsuo, H., Moriguchi, T., Takagi, T., Kusakabe, T., Buratowski, S., Sekine, M., Kyogoku, Y., and Wagner, G. 2000. Efficient synthesis of 13C, 15N-labeled RNA containing the cap structure m7GpppA. J. Am. Chem. Soc. 122: 2417–2421. [Google Scholar]
- Mathews, M.B., Sonenberg, N., and Hershey, J.W.B. 2000. Origins and principles of translation control. In Translational control of gene expression (eds. N. Sonenberg et al.), pp. 1–32. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Minich, W.B., Balasta, M.L., Goss, D.J., and Rhoads, R.E. 1994. Chromatographic resolution of in vivo phosphorylated and nonphosphorylated eukaryotic translation initiation factor eIF-4E: Increased cap affinity of the phosphorylated form. Proc. Natl. Acad. Sci. USA 91: 7668–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedzwiecka, A., Marcotrigiano, J., Stepinski, J., Jankowska-Anyszka, M., Wyslouch-Cieszynska, A., Dadlez, M., Gingras, A.-C., Mak, P., Darzynkiewicz, E., Sonenberg, N., et al. 2002. Biophysical studies of eIF4E cap-binding protein: Recognition of mRNA 5′ cap structure and synthetic fragments of eIF4G and 4E-BP proteins. J. Mol. Biol. 319: 615–635. [DOI] [PubMed] [Google Scholar]
- Pennington, M.W. and Dunn, B.M. 1994. Peptide synthesis protocols. In Methods in molecular biology, vol. 35, pp. 91–169. Humana Press, Totowa, NJ.
- Pyronnet, S., Imataka, H., Gingras, A.-C., Fukunaga, R., Hunter, T., and Sonenberg, N. 1999. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 18: 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads, R.E., Hellmann, G.M., Remy, P., and Ebel, J.P. 1983. Translational recognition of messenger ribonucleic acid caps as a function of pH. Biochemistry 22: 6084–6088. [DOI] [PubMed] [Google Scholar]
- Scheper, G.C., Morrice, N.A., Kleijn, M., and Proud, C.G. 2001. The mitogen-activated protein kinase signal-interating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol. Cell. Biol. 21: 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper, G.C., van Kollenburg, B., Hu, J., Luo, Y., Goss, D.J., and Proud, C.G. 2002. Phosphorylation of eukaryotic initiation factor 4E markedly reduces its affinity for capped mRNA. J. Biol. Chem. 277: 3303–3309. [DOI] [PubMed] [Google Scholar]
- Severinov, K. and Muir, T.W. 1998. Expressed protein ligation, a novel method for studying protein–protein interactions in transcription. J. Biol. Chem. 273: 16205–16209. [DOI] [PubMed] [Google Scholar]
- Sonenberg, N. 1996. mRNA 5′ cap binding protein and control of cell growth. In Translational control (eds. J.W.B. Hershey et al.), pp. 245–269. Cold Spring Laboratory Press, Cold Spring Harbor, NY
- Svitkin, Y.V., Ovchinnikov, L.P., Dreyfuss, G., and Sonenberg, N. 1996. General RNA binding proteins render translation cap dependent. EMBO J. 15: 7147–7155. [PMC free article] [PubMed] [Google Scholar]
- Tomoo, K., Shen, X., Okabe, K., Nozoe, Y., Fukuhara, S., Morino, S., Ishida, T., Taniguchi, T., Hasegawa, H., Terashima, A., et al. 2002. Crystal structure of 7-methylguanosine 5′-triphosphate (m7GTP)- and P1-7-methylguanosine P3-adenosine-5′,5′-triphosphate (m7 GpppA)-bound human full length eukaryotic initiation factor 4E: Biological importance of the C-terminal flexible region. Biochem. J. 362: 539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda, H., Maruyama, H., Doi, M., Inoue, M., Ishida, T., Morioka, H., Tanaka, T., Nishikawa, S., and Uesugi, S. 1991. Expression of a synthetic gene for human cap binding protein (human IF-4E) in Escherichia coli and fluorescence studies on interaction with mRNA cap structure analogues. J. Biochem. (Tokyo) 109: 882–889. [DOI] [PubMed] [Google Scholar]
- Wakamiya, T., Saruto, K., Yasuoka, J., and Kusumoto, S. 1994. An efficient procedure for solid-phase synthesis of phosphopeptides by Fmoc strategy. Chem. Lett. 1099–1102.
- Waskiewicz, A.J., Johson, J.C., Penn, B., Mahalingam, M., Kimball, S.R., and Cooper. JA. 1999. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase mnk1 in vivo. Mol. Cell. Biol. 19: 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek, Z., Stepinski, J., Jankowska, M., and Lonnberg, H. 1995. Fluorescence and absorption spectroscopic properties of RNA 5′-cap analogues derived from 7-methyl-, N2,7-dimethyl- and N2,N2,7-trimethyl-guanosines. J. Photochem. Photobiol. B 28: 57–63. [DOI] [PubMed] [Google Scholar]
- Xu, M.-Q. and Perler, F.B. 1996. The mechanism of protein splicing and its modulation by mutation. EMBO J. 15: 5146–5153. [PMC free article] [PubMed] [Google Scholar]
- Xu, M.-Q. and Evans, T.C. 2001. Intein-mediated ligation and cyclization of expressed proteins. Methods 24: 257–277. [DOI] [PubMed] [Google Scholar]