

Abstract
Nucleotide analog interference mapping (NAIM) is a powerful method for identifying RNA functional groups involved in protein–RNA interactions. We examined particles assembled on modified U1 small nuclear RNAs (snRNAs) in vitro and detected two categories of interferences. The first class affects the stability of two higher-order complexes and comprises changes in two adenosines, A65 and A70, in the loop region previously identified as the binding site for the U1 small nuclear ribonucleoprotein (snRNP)-specific U1A protein. Addition of an exocyclic amine to position 2 of A65 interferes strongly with protein binding, whereas removal or modification of the exocyclic amine at position 6 makes little difference. Modifications of A70 exhibit the opposite effects: Additions at position 2 are permitted, but modification of the exocyclic amine at position 6 significantly inhibits protein binding. These interactions, critical for U1A-U1 snRNA recognition in the context of in vitro snRNP assembly, are consistent with previous structural studies of the isolated protein with the RNA hairpin containing the U1A binding site. The second category of interferences affects all partially assembled U1-protein complexes by decreasing the stability of Sm core protein associations. Interestingly, most strong interferences occur at phosphates in the terminal stem-loop region of U1, rather than in the Sm binding site. These data argue that interactions with the phosphate backbone of the terminal stem loop are essential for the stable association of Sm core proteins with the U1 snRNA. We suggest that the stem loop of all Sm snRNAs may act as a clamp to hold the ring of Sm proteins in place.
Keywords: U1A protein, U1 snRNA, NAIM, RNA-protein interactions, Sm snRNPs
INTRODUCTION
The U1 small nuclear ribonucleoprotein (snRNP) is an essential spliceosome component, functioning not only in recognition of the 5′ ends of introns, but also in communicating with other spliceosome participants to identify an intron as a splicing substrate (for review, see Nilsen 1998). It acts in the earliest stages of spliceosome assembly, using the 5′ end of the U1 snRNA to form a short duplex with sequences immediately downstream of the 5′ splice site. Once the branch point and 3′ splice site are recognized by the U2 snRNP and its associated proteins, the pre-spliceosomal complex recruits the U4/U5/U6 tri-snRNP. U1 then hands off the identified 5′ splice site to U5 and U6, and the first step of splicing can occur: U2 positions the 2′-hydroxyl of the branch point adenosine to attack the phosphate at the junction between the upstream exon and 5′ end of the intron. Exon ligation and intron excision subsequently occur when the 3′ hydroxyl of the upstream exon, which is the leaving group in the first reaction, attacks the phosphate at the downstream intron–exon junction.
Because the U1 snRNP is a well characterized RNP, it is an excellent test subject for applying new methodology such as the one presented here. In many eukaryotes, including human, U1 is the most abundant of the splicing snRNPs, which has made the identification and characterization of its protein components relatively straightforward (Will et al. 1994). In addition to a wealth of biochemical and genetic data on the U1 RNA and proteins, structural studies have provided insights into the architecture of the U1 snRNP. First, a high-resolution crystal structure of the U1A protein and its binding site in stem loop II of the U1 snRNA (Fig. 1 ▶) revealed molecular interactions important for specific binding (Nagai et al. 1990; Oubridge et al. 1994). Second, the structure of the entire U1 snRNP has been surveyed by cryo-electron microscopy (EM: Stark et al. 2001). This EM map provides a three-dimensional picture of how the parts of the U1 snRNP fit together. A dominant feature of the EM structure is the ring of Sm core proteins, deduced from the high-resolution X-ray structures of two heterodimers containing four of the seven proteins that make up the Sm ring (Kambach et al. 1999).
FIGURE 1.

Summary of site-specific interferences in the in vitro assembly of U1 snRNA–protein complexes. The U1 stem regions are numbered with Roman numerals. The bold “A”s in the loop closing stem II are primary points of interaction for U1A protein binding. The open and gray nucleotides represent different degrees of phosphorothioate interference (kappa values) with core protein assembly. The gray box highlights the Sm binding site sequence.
The Sm proteins (B [or B′], D1, D2, D3, E, F, and G) are central components of four of the five snRNPs (U1, U2, U4, and U5) that function in the major spliceosome (for review, see Will and Lührmann 2001). Assembly of these seven Sm proteins onto the conserved Sm binding site of each of these RNAs is an essential step in snRNP biogenesis. In vertebrate cells, assembly of Sm proteins and trimethylation of the RNA 5′ cap must occur in the cytoplasm prior to nuclear reentry (Fischer and Lührmann 1990; Hamm and Mattaj 1990). The core Sm proteins thus provide the framework on which to build mature snRNPs containing additional proteins specific to each particle. The core Sm proteins also comprise the antigen recognized by anti-Sm autoantibodies produced by Lupus patients (Lerner and Steitz 1979).
Nucleotide analog interference mapping (NAIM) is a powerful tool for rapid screening of RNA functional groups (Ryder et al. 2000). NAIM relies on the susceptibility of phosphorothioate diesters to strand scission in the presence of iodine and ethanol (Gish and Eckstein 1988). The Strobel lab has developed a series of 5′-O-(1-thio)-nucleoside analog triphosphates that can be incorporated into RNA transcripts by the wild-type or a mutant form of T7 RNA polymerase (Ryder et al. 2000). These nucleotide analogs introduce small perturbations into the chemical makeup of an RNA and enable a more detailed functional group analysis than that obtainable with the naturally occurring four nucleotides (A, C, G, and U). The random incorporation of a single phosphorothioate-tagged analog into a transcript generates a population of modified RNAs that can be simultaneously examined for function.
We adapted NAIM to provide information on functional groups contributing to the in vitro assembly of the U1 snRNP particle. To examine the simultaneous binding of multiple proteins to the snRNA, we exploited protein dissociation, rather than equilibrium binding or association. By documenting changes in the interference pattern as one or more proteins dissociates from the complex, we identified not only groups anticipated to participate in the specific binding of U1A protein to the RNA, but also an unexpected set of interactions involving the backbone of the 3′ stem loop of U1 snRNA. These contacts are likely to be a common feature that maintains RNP integrity in all members of the Sm snRNP family.
RESULTS
Preparation of partially assembled U1 snRNP complexes
To apply NAIM, it is necessary to compare a starting population with an active fraction, here naked U1 snRNA compared to U1 snRNA that has assembled with core snRNP proteins. SnRNP proteins purified by a two-step method were previously shown to be sufficient to reconstitute U1, U2, and U5 core snRNP particles that can be activated upon introduction into splicing reactions (Ségault et al. 1995; Will et al. 1996), even though stoichiometric amounts of all the proteins are not present in the core protein preparation. The core protein preparation contains the seven Sm proteins and is enriched in the U1 snRNP-specific A and C proteins, but not 70K (Will et al. 1994), which is removed along with snRNAs on DE53 resin, presumably because of its highly phosphorylated state.
Despite the simplified nature of the core protein mixture, we observed that its binding to radioactive U1 snRNA, as assessed by native gel electrophoresis, showed strong sigmoidal dependence with respect to protein concentration (data not shown) in forming the two slowest migrating species (Fig. 2 ▶). This is apparently due to a complex order of protein binding, making it difficult to establish conditions where partially bound and free snRNA could be easily fractionated.
FIGURE 2.


Reconstituted U1 snRNPs dissociate into distinct faster-mobility species when challenged with excess cold U1 snRNA or other RNA fragments. (A) 32P-labeled U1 snRNA (10 nM) assembled with the core protein preparation to form two partial U1 snRNP species (lane 1). These U1 subparticles were then challenged with excess 4.6 μM unlabeled U1 snRNA (lane 2), 4.6 μM U5 snRNA (lane 3), 4.6 μM U1A stem loop II fragment, binding U1A (lane 4), 4.6 μM U1 Sm site fragment (lane 5), 4.6 μM U1 70K stem loop I fragment, binding U170K protein (lane 6), 4.6 μM each of pairs of U1 RNA fragments (lanes 7–9), or all three fragments (lane 10). Lane 11: 4.6 μM tRNA was also used as chase RNA. The U1 RNA fragments are described in Materials and Methods. Mock-treated 32P -labeled U1 RNA in the absence of protein is shown in lane 12. CPLX = complex. (B) The mobility of complexes 1–5 was retarded by the addition of Y12, an Sm protein-specific antibody.
In contrast, when reconstitution reactions containing core proteins and labeled U1 snRNA were challenged with excess unlabeled U1, the dissociation of proteins from the two most slowly migrating RNA–protein species (Fig. 2A,B, ▶ complexes 4,5) showed single exponential decay, with a relatively slow rate of dissociation (∼0.01–0.02 min−1; data not shown) to the combined faster mobility species (Fig. 2A ▶, lane 2, complexes 1–3 and free U1 snRNA). The free U1 snRNA band was identified by its comigration with the U1 RNA in the absence of protein (lane 12). The appearance of complexes of faster mobility was dependent on challenge with unlabeled U1 snRNA, because neither U5 snRNA (lane 3) nor tRNA (lane 11) could effectively compete radioactive U1 out of complexes 4 and 5.
To assign proteins present in the various complexes, we performed chase experiments with unlabeled U1 snRNA fragments known to bind specific proteins. Chasing with a U1 fragment containing stem loop II, which binds the U1A protein (Fig. 2A ▶, lane 4), resulted in a decrease in complexes 4 and 5 and an increase in complexes 1 and 2. Complex 3, which has been variable in our experiments, is completely absent in lane 4, suggesting that complexes 3, 4, and 5, but not complexes 1 and 2, contain the U1A protein. The appearance of complex 3 was dependent on the presence of the Sm binding site in the chase RNA (data not shown), further arguing that complex 3 contains U1A protein but may lack a full complement of Sm proteins.
In similar chase experiments with U1 snRNA fragments containing the U1 Sm binding site (Fig. 1A ▶, lane 5) or stem loop I that binds the U1 70K protein (lane 6), very little complex 1–3 or free U1 snRNA was generated. Although U5 snRNA (lane 3) has an Sm binding site, its sequence is different from that of U1, perhaps explaining the reproducible but slight difference between lanes 3 and 5. When U1 snRNA fragments were combined for the chase, stem loop II that binds U1A and the U1 Sm binding site produced an additive effect, yielding free labeled U1 snRNA (lane 7). Stem loop I combined with stem loop II (lane 8) or with the Sm binding site (lane 9) mimicked the chase with U1A-binding stem loop II (lane 4) or the Sm binding site alone (lane 5). A combination of all three RNA fragments (lane 10) produced a dissociation pattern resembling that seen with full-length U1 snRNA used in the chase (lane 2) or with the U1A-binding stem loop II and Sm binding site combination (lane 7).
From these results, we conclude that the U1A and Sm proteins (but not the U1 70K protein) are major components of RNA–protein complexes generated in our in vitro system. Although complexes 4 and 5 contain U1A protein and complexes 1 and 2 do not, we are not able to explain why these complexes appear as pairs. Differences between complexes 1 and 2 and between 4 and 5 could be due to protein representation, for instance in the number of Sm proteins bound or the presence of U1C protein (present in our core protein preparation). This would be consistent with the Sm proteins being stably associated in smaller heteromeric forms in solution compared to the full ring structure seen by EM (Raker et al. 1996; Raker et al. 1999; Stark et al. 2001). Alternatively, different conformations of the same complex could yield gel mobility differences.
All complexes (1–5) contained the Sm core proteins as confirmed by super-shifting with the anti-Sm antibody Y12 (Fig. 2B ▶, lane 1), leaving only the free U1 RNA band. Unfortunately, we could not identify the various complexes further because available antibodies specific for U1-specific proteins likewise super-shifted free U1 snRNA, making the results uninterpretable (data not shown).
NAIM analysis of assembled complexes
The generation of several RNP complexes containing U1 snRNA, as described above, provided a sensitive, reproducible method for assaying the effects of functional group modification on the dissociation of U1 proteins from the U1 snRNA. NAIM employs phosphorothioate chemistry to efficiently measure the consequences of single-nucleotide substitutions in RNAs (Ryder et al. 2000). Nucleoside triphosphate analogs with sulfur substitutions of the non-bridging Rp oxygen at the α-phosphate were therefore randomly incorporated into U1 snRNA, and then assayed for their representation in each U1-protein complex resolved by native gel analysis, as shown in Figure 2 ▶. The recovered snRNAs were cleaved at the phosphorothioate linkage with iodine in ethanol to yield a ladder on a sequencing gel (Fig. 3A ▶). A substitution that interferes with binding of the proteins present in a particular complex yields a band of disproportionately low intensity in the sequencing ladder. This effect is reflected in a high Kappa (κ) value (Table 1 ▶).
FIGURE 3.


U1A protein interactions with U1 snRNA defined by NAIM. (A) The sequencing gel shows iodine cleavage of RNAs isolated from native gel bands like those in Figure 2 ▶. The phosphorothioate-tagged analog incorporated into each U1 snRNA is noted above each set of lanes: DAP, 2,6-diaminopurine; m6A, N-methyl adenosine; Pur, purine; 2AP, 2-aminopurine. The numbers immediately below each nucleotide indicate the complex (1, 2, 4, or 5) from which the RNA was extracted. The A lanes (1–5) represent the control phosphorothioate isolated from each complex, and the S lanes are sample control lanes demonstrating phosphorothioate incorporation. For recognition by the U1A protein, an amino group at the 6-position is required on A70, but not on A65; an amino group at the 2-position is permitted on A70, but not on A65. (B) Proposed interactions of A65 and A70 with U1A residues. The U1A residue (Arg 52 or Asp 90) is shown interacting with a preferred position on the adenosine base (N1 of purine 65 or the exocyclic amine at the 6-position of purine 70). Allowed modifications are shown in bold, and those that interfere are in open letters. The NHR representation of the amine at the 6-position indicates that the absence of this amine or its modification (when R is a methyl group) is tolerated; the NH2 at the 2-position indicates that the presence of an amine is tolerated.
TABLE 1.
Kappa (κ) values for analogs of three adenosines in the U1A stem-loop binding region
| Complex 1 | Complex 2 | Complex 3 | Complex 4 | Complex 5 | |
| A65 | 1.2 ± 0.3 | 1.3 ± 0.3 | 1.1 ± 0.2 | 1.3 ± 0.5 | 1.2 ± 0.2 |
| 2AP | 0.4 ± 0.3 | 0.7 ± 0.4 | 2.6 ± 0.9 | 3.3 ± 1.5 | 6.0 ± 2.5 |
| DAP | 0.4 ± 0.2 | 1.2 ± 1.0 | 2.2 ± 0.5 | 3.7 ± 1.9 | 5.7 ± 3.5 |
| m6A | 1.1 ± 0.4 | 1.5 ± 1.0 | 1.3 ± 0.3 | 1.5 ± 0.6 | 2.2 ± 1.3 |
| Pur | 1.2 ± 0.7 | 1.0 ± 0.2 | 1.2 ± 0.4 | 1.0 ± 0.2 | 1.3 ± 0.3 |
| A70 | 0.6 ± 0.3 | 0.7 ± 0.2 | 1.0 ± 0.3 | 1.1 ± 0.2 | 1.1 ± 0.2 |
| 2AP | 0.6 ± 0.3 | 0.7 ± 0.4 | 1.3 ± 0.5 | 1.6 ± 0.6 | 2.9 ± 1.4 |
| DAP | 1.0 ± 0.4 | 1.0 ± 0.3 | 2.3 ± 1.0 | 1.6 ± 1.2 | 2.2 ± 1.3 |
| m6A | 0.4 ± 0.3 | 0.7 ± 0.4 | 2.5 ± 0.7 | 3.8 ± 0.9 | 4.0 ± 2.6 |
| Pur | 0.6 ± 0.3 | 0.6 ± 0.2 | 1.9 ± 0.5 | 2.4 ± 1.0 | 5.5 ± 2.8 |
| A77 | 1.0 ± 0.4 | 1.2 ± 0.4 | 1.0 ± 0.2 | 1.0 ± 0.3 | 1.2 ± 0.3 |
| 2AP | 1.5 ± 1.0 | 2.0 ± 1.0 | 1.6 ± 0.5 | 1.5 ± 0.7 | 1.2 ± 0.3 |
| DAP | 0.8 ± 0.3 | 1.2 ± 0.5 | 1.4 ± 0.5 | 1.0 ± 0.3 | 1.4 ± 0.8 |
| m6A | 1.0 ± 0.4 | 0.9 ± 0.2 | 1.1 ± 0.2 | 1.4 ± 0.7 | 1.0 ± 0.4 |
| Pur | 1.3 ± 0.8 | 1.2 ± 0.7 | 1.0 ± 0.2 | 1.2 ± 0.6 | 1.2 ± 0.3 |
Experiments were performed in at least triplicate and, for some values, data sets were collected on both 5′- and 3′-labeled U1 snRNA to control for labeling effects. Errors represent the range of values seen in replicate experiments.
Adenosine analogs provided information regarding U1A protein binding to its cognate stem loop II in the context of the entire U1 snRNA (Fig. 3A ▶, Table 1 ▶). Each complex (complex 3 was underrepresented in the experiment in Fig. 3A ▶) was examined for the effect of replacing the A with N6-methyl adenosine (m6A), purine, 2,6-diaminopurine (DAP), or 2-amino purine (2AP). For complexes 1 and 2, modifications at position 65 or 70 were represented to the same extent as in the original transcript (κ values <2, Table 1 ▶). In contrast, complexes 4 and 5 showed site-specific effects (κ values > 2). At position 65, methylating the exocyclic amine (m6A, lanes 13,14) or removing the exocyclic amine (Pur, Fig. 3A ▶, lanes 17,18) had no effect, but moving the amine to the 2-position (2AP, lanes 21,22) significantly lowered its representation in complexes 4 and 5 (Table 1 ▶). Placing an amine at both the 2- and 6-positions (DAP) also destabilized complexes 4 and 5 (lanes 9,10; Table 1 ▶). At position 70, opposite effects were observed: Purine and m6A interfered with complexes 4 and 5, whereas 2AP and DAP produced minimal interference (Fig. 3A ▶, Table 1 ▶). In contrast, purine interfered strongly at A70, and 2AP interfered only weakly, suggesting that there may be some compensation from having an amine on the 2-postion of A70. Note that high κ values (Table 1 ▶) for an analog in the slower mobility complexes (4 and 5) correlated with κ values below 1 in the faster complexes (1 and 2), indicating that these variant nucleotides are overrepresented in complexes lacking the U1A protein (see Fig. 2 ▶). Moreover, high κ values were not due to degradation of RNAs containing these particular substitutions, but rather to the dissociation of protein(s) required to resolve the various complexes. Our results are compatible with what is seen in the cocrystal structure of U1A and its target stem loop (Oubridge et al. 1994), where the N1 of A65 and exocyclic amine of A70 make primary interactions with the U1A protein (Fig. 3B ▶).
We also examined uridine interactions using pseudouridine and 5-methyl uridine, as well as 2′-deoxyuridine, and 2′-fluorouridine (data not shown). None of these modifications affected protein binding to stem loop II (the U1A binding site) or to the Sm binding site.
One group of modifications that did affect all U1 RNA–protein complexes were phosphorothioate backbone substitutions (Fig. 4 ▶) in the terminal stem-loop IV and in stem III, both neighboring the Sm binding site (see Fig. 1 ▶). Some of the interferences in the terminal stem loop (positions 141–165) and at G110 in stem III were very strong, producing high κ values (> 5, see Fig. 1 ▶). Strikingly, the pattern of interference across the terminal stem loop followed the backbone surface of a helical region: As the helix twists, the phosphorothioate interferences were less intense, consistent with weaker protein interactions. This interference pattern is similar to the pattern seen in the packing of RNA helices in the stable P4-P6 domain of the group I intron (Murphy and Cech 1993).
FIGURE 4.

Iodine cleavage of RNAs containing phosphorothioates of A, C, G, or U shows interference at numerous positions near the 3′ end of U1. Numbers below each nucleotide indicate the complex (as in Fig. 2 ▶) from which the RNA is extracted. White balls indicate the stronger sites of interference in stem loop IV (see Fig. 1 ▶ for κ values). The numbers on the side represent nucleotide position. The Sm binding site sequence (nucleotides 125–133) is also indicated. S lanes are sample control lanes demonstrating phosphorothioate incorporation. Note that although some lanes may appear more intense because more RNA was loaded in the lane, these loading differences are accounted for in the calculation of κ. Occasionally, a common degradation band is seen (such as at position 145), excluding this nucleotide from analysis in this data set.
Because all in vitro-assembled complexes exhibited interference in stem loops III and IV upon phosphorothioate substitution, we propose that these modifications weaken core Sm protein interactions with the U1 snRNA. Interestingly, phosphorothioates within the Sm binding site itself produced no interference, as observed for the base-specific modifications described above (Fig. 4 ▶). Thus, important functional groups within the U1 Sm binding site have thus far eluded identification by NAIM. Because we cannot assess U1A bound alone to the RNA, we also can draw no conclusions about whether phosphate modifications indirectly affect U1A protein binding.
DISCUSSION
Although the U1 particle is the best characterized of the splicing snRNPs, we have uncovered previously unknown aspects of its RNA–protein interactions. Most intriguing is the finding that backbone phosphates, predominantly in the terminal stem loop, are important for core protein assembly. Curiously, no interferences from either base or backbone substitutions were observed in the Sm binding site sequence, even though Sm proteins are clearly present in the reconstituted particles. We also observed that particular functional groups within the U1A binding site play essential roles, consistent with interactions visualized in the crystal structure of the U1A-U1 stem loop II complex.
Functional groups contributing to U1A binding
A collection of biochemical, genetic, and structural data point to A65 in stem loop II of U1 snRNA as making important contacts with Arg52, a member of the RNP1 motif within the RRM domain of the U1A protein (see Fig. 3B ▶). Mutating Arg52 to Gln was found to abolish RNA binding, whereas mutating it to Lys has only a small effect, suggesting that this amino acid forms a salt bridge with the RNA (Nagai et al. 1990; Jessen et al. 1991). Accordingly, mutating A65 to C decreased U1A binding by three orders of magnitude (Stump and Hall 1995), whereas an A65 to G mutation reduced binding ∼10-fold (Jessen et al. 1991). The high-resolution crystal structure of U1A and its cognate stem loop places Arg52 in close proximity to residue A65 (Oubridge et al. 1994). Further computational analysis has modeled a water-mediated interaction between the gamma-NH2 of Arg52 and N1 of A65, with Arg52 forming a central link in a hydrogen-bonding network (Reyes and Kollman 2000). Placing an exocyclic amine at the 2-position of A65 would be expected to cause steric clash with the gamma-NH2 of Arg52, consistent with the interference seen with DAP and 2AP at this position (Table 1 ▶, Fig. 3 ▶). In contrast, methylating the amine of A65 (m6A) does not cause a clash because the amine is pointing away from Arg52 and the free rotation of the methyl group likely allows a stable fit with the U1A protein.
Although Arg52 is a critical residue, amino acids within the entire loop 3 region of the U1A protein make multiple contacts with U1 snRNA (Jessen et al. 1991; Oubridge et al. 1994). The high-resolution model also suggests a hydrogen bond between Glu19 and the exocyclic amine of A65 (Oubridge et al. 1994). Lack of interference by purine and m6A (albeit a trace in complex 5; see Table 1 ▶) supports the conclusion that removal or modification of the exocyclic amine at the 6 position of A65 only marginally affects the association of U1A with the partially assembled snRNP complex. This suggests that the proposed A65-Glu19 interaction contributes less to the binding energy than A65-Arg52.
In the U1A protein-U1 hairpin crystal structure (Oubridge et al. 1994), A70 of U1 RNA also interacts with the U1A protein (see Fig. 3B ▶). Both its exocyclic amine and N1 are in close proximity to the backbone amide at residues Asp90 and Ser91. In addition, N7 of A70 is coordinated to a water molecule that has the C2 carbonyl of C69 and the backbone amide of Thr89 as ligands. The base of A70 stacks between Phe56 and C71. We observed (Table 1 ▶) that an amine can be accommodated at the 2-position of A70 (as in DAP), but that modification or deletion of the amine at the 6-position is detrimental to U1A binding (exemplified by purine and m6A, which produce high κ values in the higher complexes). While addition of a methyl group or deletion of the N6 from A70 would clearly affect its ability to hydrogen bond with the backbone at Asp90, these modifications may subsequently affect the ability of the base to properly stack on Phe56 and affect the N7 water-mediated ligand network. The available space at the 2-position of A70 permits modification that is not allowed at A65. Although our examination of interactions focused on modified adenosines and uridines in the U1A binding region of U1 snRNA, we identified no new interactions, nor did we deduce the lack of any interactions proposed by the cocrystal structure. Because the critical positions that we deduce from NAIM are comparable to those seen in the crystal structure of the U1A protein and its stem-loop ligand (Oubridge et al. 1994), the nature of U1 snRNA-U1A protein interactions is likely unchanged in the context of the entire snRNP.
A stem-loop clamp for the Sm ring
When embarking on this study, we had hoped to identify RNA functional groups within the Sm consensus sequence of U1 snRNA that would provide insights into how an Sm site is recognized by the Sm proteins. Previously, ethylation of the phosphate backbone was observed not to interfere with binding of an excess of Sm proteins, whereas the Sm site and part of stem III were protected from Fe(II)-EDTA hydroxy-radical cleavage (at the 1′ and 4′ positions; Hartmuth et al. 1998). These observations, in addition to the finding that A126 and A135 were unusually sensitive to methylation by DMS in the native U1 snRNP, suggested that proteins recognize and order the Sm site nucleotides (Hartmuth et al. 1998). Nonetheless, even using an extremely sensitive method, we found no interferences in the Sm region for changes in either base or backbone moieties.
Instead, the main interferences we identified resulted from phosphorothioates incorporated into the terminal stem loop of U1. Almost all Sm protein-bound snRNAs have a terminal stem loop, with the few exceptions being low-abundance variants (Patterson and Guthrie 1987; Xu et al. 1997), perhaps indicative of poor stability. The secondary structure, but not the sequence, of this stem loop is conserved. Interestingly, deletion of the terminal stem of U1 produced a stable RNA in Xenopus oocytes that was not Sm-precipitable (Jarmolowski and Mattaj 1993), whereas deletion of the terminal stem loop in HeLa extracts altered the sedimentation behavior of the resulting U1 particles (Patton et al. 1987). Here, we observed that several phosphates within the terminal stem loop of U1 significantly affect RNP stability when changed to phosphorothioates. Moreover, the pattern of the interference mapped onto the stem-loop structure suggests that just one face of the stem loop is being recognized.
By comparing our results with the recent cryo-EM structure of the U1 snRNP (Stark et al. 2001), we propose that the interference pattern arises because the stem loop acts as a clamp on the Sm core structure. In the EM model, the terminal stem loop contacts one side of the Sm ring, whereas the cloverleaf structure of the 5′ portion of U1 snRNA rests on the other side. Introducing phosphorothioates into the contact surface of the terminal stem loop would be expected to be disruptive, weakening the overall affinity of the Sm core complex for the snRNA. The EM structure also provides a physical explanation for the phosphorothioate interferences observed in stem III of U1 (see Fig. 1 ▶). This helix resides close to the Sm ring, sandwiching it against the terminal stem loop.
The Sm core protein structure was determined by X-ray analysis of two Sm protein dimers, as well as by EM (Kambach et al. 1999; Stark et al. 2001). The seven proteins each interact with a specific set of neighbors to form stable heteromers, helping to establish the order of proteins around the ring (Raker et al. 1999). The seven proteins, D1, D2, D3, B (or B′), E, F, and G, form a closed ring structure with a central hole that is highly basic. By considering crosslinking data and the protein structures, Urlaub et al. (2001) concluded that the single-stranded Sm sequence likely runs through the hole; the EM structure of the U1 snRNP is consistent with this idea (Stark et al. 2001). Since the hole is insufficient in diameter to accommodate a double-stranded RNA, it seems unlikely that the terminal stem loop of an snRNA threads through the heptamer. Thus, our phosphorothioate interference data predict that the heptamer assembles directly on the Sm binding site, with the terminal stem loop acting to stabilize the association.
Understanding how functional groups of the U1 snRNA affect snRNP assembly is a prerequisite to studying higher-order U1-containing structures. Although it is conceivable that the details of the interactions resulting from in vivo assembly of the Sm proteins, which is known to involve the SMN complex (for reviews, see Will and Lührmann 2001; Paushkin et al. 2002), differ from those in our in vitro-assembled U1 subparticles, the correspondence that we see with the EM model derived from native U1 snRNPs (Stark et al. 2001) argues for the relevance of our conclusions. This report therefore lays the groundwork to enable further NAIM analyses of the involvement of U1 snRNA in the assembly of active spliceosomal complexes.
MATERIALS AND METHODS
Preparation of snRNAs
U1 snRNAs were transcribed using T7 polymerase from EcoRI linearized plasmid pT7U1, producing a full-length human U1 RNA with a 5′GGG leader for efficient transcription. Phosphorothioate analogs were randomly incorporated into U1 snRNA, with concentrations of individual NTPs and analog NTPs, depending upon the incorporation efficiency of each analog (Ryder et al. 2000). After purification on a 6% denaturing polyacrylamide gel, samples of each U1 snRNA were splint-labeled on their 3′ ends with 4 Cs, using (α-32P)-dCTP and the oligonucleotide GGG GAATTCAGGGGAAAGCGCGAACGCAGT (Hausner et al. 1990). Ten pmole U1 transcript was combined with 10 pmole oligonucleotide in 6 μL of 80 mM Tris, pH 7.5 and 0.2 mM EDTA. The solution was heated at 95°C for 1 min and immediately cooled to 70°C. The components were annealed by allowing them to cool to 45°C over 10 min, and were then kept at room temperature for an additional 10 min. Two μL containing 50 mM MgCl2, 5 mM DTT, and 2 μL Sequenase (USB), as well as 4 μL (α-32P)-dCTP were then added to make a final volume of 15 μL and incubated at 37°C for 30 min. For 5′ end labeling, U1 transcripts were synthesized with a fivefold excess of guanosine over GTP and subsequently treated with T4 polynucleotide kinase and (γ-32P)-ATP. The labeled RNAs were denatured at 90°C for 1 min, and purified on a 6% denaturing polyacrylamide gel. The U1 snRNA fragments were made by standard RNA oligonucleotide synthesis (Keck Facility, Yale University). The stem I RNA (containing the binding site for U1 70K) included positions 17 to 47 of U1. The stem II RNA (the U1A binding site) contained nucleotides 60 to 79 of U1. The Sm binding site RNA was made from a PCR construct containing a T7 promoter; the sequence contained two guanosines to initiate transcription, nucleotides 11 to 15 of U1 to provide half of stem I, a single A linker, and nucleotides 92 to 164 of U1 containing the complete Sm binding site. U1 fragment RNAs were purified on a 10% gel.
Protein binding and NAIM
The U1 snRNAs were bound to a preparation of total HeLa snRNP proteins (TPs) obtained as described (Will et al. 1994). The mixture includes the seven U1 Sm proteins, as well as the U1A and U1C proteins, and a lower concentration of U1 70K protein (Will et al. 1996). U1 snRNA (1.5 × 106 cpm, 10 nM) was denatured at 95°C for 30 sec, and a solution containing 24 U RNase inhibitor, 1.7 mM ATP, 8.7 mM MgCl2, 3.4 mM DTT, 7.3 mM HEPES, 18 mM KCl, 2% glycerol, and 72 μM EDTA in a 10 μL volume was added. TPs were then added in excess (∼2.5 pmole of each protein) and allowed to bind to U1 snRNA for 10 min at 30°C. Next, the reaction was brought to a final concentration of 25 mM creatine phosphate, 30 mM KCl, 0.04 mg/mL carrier RNA in 25 μL. Finally, unlabeled, unmodified U1 snRNAs were added in 10-fold excess over TPs (∼25 pmole) and allowed to incubate at 30°C for 35 min. To establish the protein components in each U1 complex, the unlabeled U1 snRNA was replaced with an excess of other RNAs, as described in Figure 2 ▶. After adding loading buffer to a final concentration of 0.10 mg/mL heparin, the samples were fractionated on a nondenaturing 8% polyacrylamide (19:1 acrylamide: bisacrylamide) gel for 1.5 h at 500V. Protein-snRNA complexes were excised and eluted from the gel into 10 mM Tris, pH 7.0, 1 mM EDTA, 0.23 M NaCl, 0.1% SDS. RNAs were isolated from each eluted complex by PCA extraction and ethanol precipitation. The U1 snRNAs, resuspended in 9 μL H2O, were then cleaved at sites of phosphorothioate incorporation by adding 1 μL of 2 mM I2 solution in ethanol, and the resulting fragments were resolved to single nucleotide resolution on a 10% denaturing polyacrylamide gel. Band intensities were quantified by PhosphorImager (Molecular Dynamics) analysis.
For each nucleotide analog tested, the interference value (κ) at each position represents the relative amount of each modified RNA bound in an RNP particle versus the parent phosphorothioate in the same particle, with both values normalized for incorporation with an unselected sample of the input RNA. Kappa was calculated as follows:
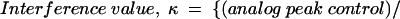 |
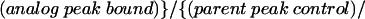 |
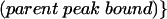 |
For phosphorothioate interference, where the oxygen to sulfur change creates the analog, κ was simply the ratio of phosphorothioate incorporated (control) over the phosphorothioate bound in the particle. The interference values of each band in a given lane were averaged after discounting bands with intensities outside two standard deviations. The interference values were then divided by the average to normalize for differences in amount loaded in each lane and for the extent of the iodine cleavage reaction in each lane. The resulting value, defined as κ, represents the interference factor that results from the substitution of an analog at each position, relative to that of the parental nucleotide at that position. κ values > 2 indicate interference, and values < 0.5 indicate enhancements (Ryder et al. 2000).
Acknowledgments
We thank Scott Strobel for his gift of 5′-O-(1-thio)-nucleoside analog triphosphates and J. Cortes for preparing the pT7U1 plasmid used in this study. We thank Therese Yario and Ingrid Sprinz for experimental assistance, Lara Weinstein Szewczak and Kazio Tycowski for critical reading of the manuscript, and the members of the Steitz laboratory for helpful discussions. This work was supported by the NIH (grant GM26154 to J.A.S.). J.A.S. is an investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2136103.
REFERENCES
- Fischer, U. and Lührmann, R. 1990. An essential signaling role for the m3G cap in the transport of U1 snRNP to the nucleus. Science 249: 786–790. [DOI] [PubMed] [Google Scholar]
- Gish, G. and Eckstein, F. 1988. DNA and RNA sequence determination based on phosphorothioate chemistry. Science 240: 1520– 1522. [DOI] [PubMed] [Google Scholar]
- Hamm, J. and Mattaj, I.W. 1990. Monomethylated cap structures facilitate RNA export from the nucleus. Cell 63: 109–118. [DOI] [PubMed] [Google Scholar]
- Hartmuth, K., Rakar, V.A., Huber, J., Branlant, C., and Lührmann, R. 1998. An unusual chemical reactivity of Sm site adenosines strongly correlates with proper assembly of core U snRNP particles. J. Mol. Biol. 285: 133–147. [DOI] [PubMed] [Google Scholar]
- Hausner, T.P., Giglio, L.M., and Weiner, A.M. 1990. Evidence for base-pairing between mammalian U2 and U6 small nuclear ribonucleoprotein particles. Genes & Dev. 4: 2146–2156. [DOI] [PubMed] [Google Scholar]
- Jarmolowski, A. and Mattaj, I.W. 1993. The determinants for Sm protein binding to Xenopus U1 and U5 snRNAs are complex and non-identical. EMBO J. 12: 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen, T.H., Oubridge, C., Teo, C.H., Pritchard, C., and Nagai, K. 1991. Identification of molecular contacts between the U1 A small nuclear ribonucleoprotein and U1 RNA. EMBO J. 10: 3447–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambach, C., Walke, S., Young, R., Avis, J.M., de la Fortelle, E., Raker, V.A., Lührmann, R., Li, J., and Nagai, K. 1999. Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell 96: 375–387. [DOI] [PubMed] [Google Scholar]
- Lerner, M.R. and Steitz, J.A. 1979. Antibodies to small nuclear RNAs complexed with proteins are produced by patients with systemic lupus erythematosus. Proc. Natl. Acad. Sci. 76: 5495–5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, F.L. and Cech, T.R. 1993. An independently folding domain of RNA tertiary structure within the Tetrahymena ribozyme. Biochemistry 32: 5291–5300. [DOI] [PubMed] [Google Scholar]
- Nagai, K., Oubridge, C., Jessen, T.H., Li, J., and Evans, P.R. 1990. Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A. Nature 348: 515–520. [DOI] [PubMed] [Google Scholar]
- Nilsen, T.W. 1998. RNA-RNA interactions in nuclear pre-mRNA splicing. In RNA structure and function (eds. R. Simmons and M. Grunberg-Manago), pp. 279–307. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Oubridge, C., Ito, N., Evans, P.R., Teo, C.H., and Nagai, K. 1994. Crystal structure at 1.92 A resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature 372: 432–438. [DOI] [PubMed] [Google Scholar]
- Patterson, B. and Guthrie, C. 1987. An essential yeast snRNA with a U5-like domain is required for splicing in vivo. Cell 49: 613–624. [DOI] [PubMed] [Google Scholar]
- Patton, J.R., Patterson, R.J., and Pederson, T. 1987. Reconstitution of the U1 small ribonucleoprotein particle. Mol. Cell. Biol. 7: 4030–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushkin, S., Gubitz, A.K., Massenet, S., and Dreyfuss, G. 2002. The SMN complex, an assemblyosome of ribonucleoproteins. Curr. Opin. Cell Biol. 14: 305–312. [DOI] [PubMed] [Google Scholar]
- Raker, V.A., Plessel, G., and Lührmann, R. 1996. The snRNP core assembly pathway: Identification of stable core protein heteromeric complexes and an snRNP subcore particle in vitro. EMBO J. 15: 2256–2269. [PMC free article] [PubMed] [Google Scholar]
- Raker, V.A., Hartmuth, K., Kastner, B., and Lührmann, R. 1999. Spliceosomal U snRNP core assembly: Sm proteins assemble onto an Sm site RNA nonanucleotide in a specific and thermodynamically stable manner. Mol. Cell Biol. 19: 6554–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes, C.M. and Kollman, P.A. 2000. Investigating the binding specificity of U1A-RNA by computational mutagenesis. J. Mol. Biol. 295: 1–6. [DOI] [PubMed] [Google Scholar]
- Ryder, S.P., Ortoleva-Donnelly, L., Kosek, A.B., and Strobel, S.A. 2000. Chemical probing of RNA by nucleotide analog interference mapping. Methods Enzymol. 317: 92–109. [DOI] [PubMed] [Google Scholar]
- Ségault, V., Will, C.L., Sproat, B.S., and Lührmann, R. 1995. In vitro reconstitution of mammalian U2 and U5 snRNPs active in splicing: Sm proteins are functionally interchangeable and are essential for the formation of functional U2 and U5 snRNPs. EMBO J. 14: 4010–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark, H., Dube, P., Lührmann, R., and Kastner, B. 2001. Arrangement of RNA and proteins in the spliceosomal U1 small nuclear ribonucleoprotein particle. Nature 409: 539–542. [DOI] [PubMed] [Google Scholar]
- Stump, W.T. and Hall, K.B. 1995. Crosslinking of an iodo-uridine-RNA to a single site on the human U1A N-terminal RNA binding domain. RNA 1: 55–63. [PMC free article] [PubMed] [Google Scholar]
- Urlaub, H., Raker, V.A., Kostka, S., and Lührmann, R. 2001. Sm protein-Sm site RNA interactions within the inner ring of the spliceosomal snRNP core structure. EMBO J. 20: 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will, C.L. and Lührmann, R. 2001. Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 13: 290–301. [DOI] [PubMed] [Google Scholar]
- Will, C.L., Kastner, B., and Lührmann, R. 1994. Analysis of ribonuclear protein interactions. In RNA Processing (eds. S.J. Higgins and B.D. Hames), pp. 141–177. IRL Press, New York.
- Will, C.L., Rumpler, S., Klein Gunnewiek, J., van Venrooij, W.J., and Lührmann, R. 1996. In vitro reconstitution of mammalian U1 snRNPs active in splicing: The U1-C protein enhances the formation of early (E) spliceosomal complexes. Nucleic Acids Res. 24: 4614–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y., Ben-Shlomo, H., and Michaeli, S. 1997. The U5 RNA of trypanosomes deviates from the canonical U5 RNA: The Leptomonas collosoma U5 RNA and its coding gene. Proc. Natl. Acad. Sci. 94: 8473–8478. [DOI] [PMC free article] [PubMed] [Google Scholar]