

Abstract
We have previously described an example of extensively A-to-G edited cDNA derived from adult heads of the fruitfly Drosophila melanogaster. In that study, the source of the predicted antisense RNA pairing strand for template recognition by dADAR editase was not identified, and the biological significance of the observed hyperediting was not known. Here, we address each of these questions. 4f-rnp and sas-10 are closely adjacent X-linked genes located on opposite DNA strands that produce convergent transcripts. We show that developmentally regulated antisense sas-10 readthrough mRNA arises by activation of an upstream promoter P2 during the late embryo stage of fly development. The sas-10 readthrough transcripts pair with 4f-rnp mRNA to form double-stranded molecules, as indicated by A-to-G editing observed in both RNA strands. It would be predicted that perfect RNA duplexes would be targeted for modification/degradation by enzyme pathways that recognize double-stranded RNAs, leading to decline in 4f-rnp mRNA levels, and this is what we observe. The observation using quantitative RT-PCR that sas-10 readthrough and 4f-rnp transcript levels are inversely related suggests a role for the antisense RNA in posttranscriptional regulation of 4f-rnp gene expression during development. Potential molecular mechanisms that could lead to this result are discussed, one of which is targeted transcript degradation via the RNAi pathway. Insofar as the dADAR editase and RNAi pathways are known to be constitutive in this system, it is likely that control of antisense RNA transcription is the rate-limiting factor. The results provide insight into roles of naturally occurring antisense RNAs in regulation of eukaryotic gene expression.
Keywords: ADAR, antisense gene regulation, antisense RNA, RNA editing, RNAi
INTRODUCTION
RNA editing by double-stranded RNA-binding enzymes in the ADAR (adenosine deaminases that act on RNA) family constitutes an interesting and important example of posttranscriptional modification that occurs throughout multicellular animals, and has recently been reviewed (Emeson and Singh 2001; Hough and Bass 2001). The reaction involves deamination of targeted adenosine to inosine, which pairs with cytidine during first-strand cDNA synthesis in a reverse transcription reaction. In the subsequent PCR step during a typical reverse transcription-polymerase chain reaction (RT-PCR), cytidine pairs with guanosine, and thus the targeted sites are visualized during DNA sequencing as “A-to-G” conversions, which is how we will refer to these nucleotide changes here. We have previously described an example of extensively A-to-G edited 4f-rnp cDNA derived from adult heads of the fruitfly Drosophila melanogaster (Petschek et al. 1996, 1997). In that study, the source of the predicted antisense RNA pairing strand for template recognition by dADAR was not identified, and the biological significance behind the observed hyperediting was not explored. In this report, we describe results that address each of these questions.
Two classes of ADAR-targeted substrates have been recognized: specific and promiscuous. The first example of specific class A-to-G editing was described in pre-mRNA transcripts encoding glutamate-gated ion channel proteins in the mammalian central nervous system (Sommer et al. 1991), and has been reviewed (Seeburg 1996). Editing here changes particular gene-encoded amino acids, and constitutes a new kind of genetic regulation mechanism. The specificity of this edit class appears to depend on a particular combination of local bulges and loops owing to mismatches in the duplex RNA substrate, in addition to the nucleotide sequence itself (Aruscavage and Bass 2000). The duplex RNA substrate for ADAR in the case of R/G (arginine codon conversion to glycine codon) editing of gluR-B, -C, and -D pre-mRNAs arises by pairing between introns and exons, a requirement that has been shown to result in a remarkable degree of nucleotide sequence conservation within intron 13 of gluR-C in a comparison of human and chicken (Aruscavage and Bass 2000). In Drosophila, specific class A-to-G RNA editing was first reported in nervous system calcium channel α1 cDNAs (Smith et al. 1996), and this editing class has been well characterized in the example of the para gene, which encodes a brain Na+-ion channel protein (Palladino et al. 2000a, b; Reenan et al. 2000). Here, comparison between D. melanogaster and Drosophila virilis species, which diverged about 60 million years ago (Beverley and Wilson 1984), revealed pairing between complementary regions of an evolutionarily conserved intron and exon in combination with a characteristic pattern of loops and bulges to create the site-specific structure (Hanrahan et al. 2000). The extent of sequence conservation observed in the cited study is another clear example wherein pairing potential for subsequent ADAR recognition is maintained by selective pressure.
The promiscuous class of A-to-G RNA editing, which has also been termed hypermutation, was first detected as an unwinding/modifying activity when duplex RNAs were microinjected into Xenopus oocytes (Bass and Weintraub 1987, 1988; Wagner et al. 1989), and typically results in cDNAs containing from 10%–50% deaminated As (reviewed in Bass 1997). The first example of hypermutation to be described in an animal RNA was seen in oocyte cDNA clones of the bFGF transcript in Xenopus (Kimelman and Kirschner 1989), later shown to occur in only a small fraction of the transcripts (Saccomanno and Bass 1999). Here, the bFGF gene overlaps a gene located on the opposite DNA strand, and the two converging transcripts form a long RNA duplex, which serves as an excellent ADAR substrate. Several examples of hyperediting have been reported in the 3′-UTR of Caenorhabditis elegans (Morse and Bass 1999) and human brain mRNAs (Morse et al. 2002), where long hairpin structures serve as ADAR substrates. Recently, it has been shown that nuclear hyperediting antagonizes duplex RNA degradation via the cytoplasmic RNAi pathway (Scadden and Smith 2001). Hyperedited RNAs are retained in the nucleus in a Xenopus oocyte model system owing to binding by the protein p54nrb (Zhang and Carmichael 2001). The biological significance of the hypermutation examples thus far discovered has remained largely a matter of speculation, often invoking some function in control of gene expression.
Here, we report discovery of developmentally regulated readthrough transcripts arising from a Drosophila gene identified as sas-10, residing on the DNA strand opposite that of 4f-rnp. Transcripts from these two genes are convergent, but do not overlap in all developmental stages. Overlapping transcripts are shown to form double-stranded RNAs, which are detected as early as the late embryo stage of development, as evidenced by identification of edited cDNA clones interpreted as having arisen due to dADAR-catalyzed A-to-G conversions in both the 4f-rnp and sas-10 readthrough pre-mRNA strands. The observation that sas-10 readthrough and 4f-rnp transcript levels are inversely related, as in the bFGF example, suggests a role for antisense RNA in posttranscriptional regulation of 4f-rnp gene expression during development. Potential molecular mechanisms that could lead to this result are discussed.
RESULTS
The Drosophila CG4202 gene is a sas-10 homolog
The recently completed Drosophila genome project (reviewed in Celniker 2000) shows that the nearest neighbor to the 4f-rnp gene is CG4202, and that the two genes which are separated by an intergenic spacer of only 95-bp utilize opposite DNA strands for synthesis of their converging transcripts. We attempted to identify CG4202 by looking for potential homologs in other species using a BLAST search, and discovered that this gene is a homolog to yeast and mammalian sas-10 “something-about-silencing” (Kamakaka and Rine 1998), as shown in Figure 1 ▶. In common with its yeast homolog, fly sas-10 is predicted to code for a nuclear protein that contains extensive poorly conserved tracts rich in both acidic and basic amino acid residues, and a highly conserved C-terminal basic domain that includes a classical bipartite nuclear localization signal. A conceptual translation predicts that the encoded protein contains 428 amino acids, of which about 40% are charged.
FIGURE 1.

Identification of the fly CG4202 gene as a sas-10 homolog. A BLAST search of the GenBank/EMBL database showed best fit to the yeast and mammalian sas-10 “something-about-silencing” gene. Amino acid sequence alignment shows Drosophila (D.M.) compared to human (H.S.). Shared amino acid identities and conservative changes (+) are shown in center. Putative bipartite nuclear localization signal (NLS) is bracketed. About 40% of the fly amino acid residues are charged.
Quantitative RT-PCR of 4f-rnp and sas-10 gene transcript levels during development
We previously hypothesized (Petschek et al. 1997) that extensive 4f-rnp mRNA editing, which extended throughout the transcript, may have involved transcription of a long (>5 kb) antisense pairing strand. Such a molecule could conceivably have arisen via sas-10 transcriptional readthrough. To test this hypothesis, a series of nested primer sets was utilized to detect and quantify sas-10 transcripts of differing length throughout development. Each successive primer set employed (Fig. 2 ▶) reached further downstream from one located within the 3′-end of the sas-10 gene coding region (sas-LFT), the longest span extending for 251 nt into the complement of the 4f-rnp coding region. One primer set specific for 4f-rnp transcripts was also utilized (Dros E-2/PX-2), in addition to a control primer set specific for rp49 transcripts. It was found (Fig. 2A ▶) that every sas-10 primer set utilized detected transcripts, which were characterized by DNA sequencing. The sas-LFT/sas-3 primer set transcripts are complementary to the 4f-rnp 3′-UTR for 182 nt, and the sas-LFT/sas-4 set could pair with 4f-rnp mRNA for 467 nt (Fig. 3 ▶). These results show that long readthrough transcripts partially complementary to 4f-rnp mRNA are indeed produced, but do not reveal whether or not these molecules extend to the full length of 4f-rnp. It is possible that the sas-LFT/sas-3 and sas-LFT/sas-4 primer sets are detecting a long ∼6000 nt sas-10 readthrough transcript previously observed as a faint band in Northern gels, following hybridization with a strand-specific riboprobe (Hunt-Smith 1999). It is apparent that 4f-rnp and sas-10 expressions are developmentally regulated.
FIGURE 2.

Quantitative RT-PCR of 4f-rnp and sas-10 transcript levels during development. Orientation diagram shows locations of genes and polarities of their corresponding transcripts. Locations of nested primer sets numbered 1–4 used for PCR are at each end of the arrows. Gene leader (L), trailer (T), and intergenic spacer (S) locations are shown. (A) Representative gels after SYBR Green I staining of cDNA bands. (B) Shorter length sas-10 transcript levels during development, relative to rp49 reference. Numbers of repeat determinations per developmental stage are shown in brackets and standard error bars are indicated. (C) 4f-rnp transcript levels during development, relative to rp49 reference. Labeling is as in (B). (D) Long sas-10 readthrough transcript levels during development, relative to rp49 reference. Labeling is as in (B). Note the roughly 200-fold difference in sas-10 signal intensities between (B) and (D) and also that levels of 4f-rnp and sas-10 readthrough transcripts (C) and (D) are inversely related at late embryo stages of development (15–21 h).
FIGURE 3.

Diagrammatic alignment of compiled genomic sequence organization and sas-10 transcription unit characteristics. Orientation diagrams at top and bottom show relative locations of genes sas-10 and 4f-rnp, separated by 95-bp intergenic spacer (S), with their corresponding leader (L) and conventional trailer (T) tracts. Within poly(A) cDNA clones 4R1#6 (Hunt-Smith 1999) and GH08670 (Drosophila EST database), which both initiate transcription at promoter P1, locations of 5′-termini relative to start codon AUG, putative poly(A) signals (AAUAAA; AAUAUA) relative to 3′-termini, and transcript lengths relative to the start codon (+1) are indicated. Sas-10 transcripts determined by RT-PCR as arising from promoter P2 are labeled with the PCR primers utilized. Their numbering scheme is as for the cDNA clones, and the extent of their final length in vivo is unknown. cDNA clone GH08670 and the shorter RT-PCR transcript depicted overlap with the 4f-rnp trailer to varying extents, while the longer RT-PCR transcript depicted also overlaps the final 251 nt of the 4f-rnp coding region.
The results to this point are qualitative, but were quantified by comparing SYBR Green I-stained cDNA band intensities to the constitutively expressed rp49 reference gene transcripts, previously shown to be present at comparable levels throughout Drosophila development (O’Connell and Rosbash 1984). 4f-rnp transcripts are detected at moderate levels during embryonic stages of development, but then decline in late stage embryos (15–21 h), to plateau at diminished levels until adulthood (Fig. 2C ▶). This gene has previously been shown to be constitutively expressed throughout development, as also reported here, but the earlier study (Hess et al. 1996) did not attempt to quantify mRNA levels. Recently, an extensive analysis measuring transcript levels of 4028 wild-type Drosophila genes using DNA microarrays (Arbeitman et al. 2002) revealed a similar 4f-rnp developmental expression profile to the one shown here (accessible at: http://flygenome.yale.edu/Lifecycle).
Quantification of sas-10 mRNA levels using primer set sas-LFT/sas-1 (Fig. 2B ▶) would not distinguish between conventional length transcripts terminating at the trailer, such as polyadenylated cDNA clone 4R1#6 (Hunt-Smith 1999) and long readthrough transcripts, but would detect both together. This primer set produced an intensely staining cDNA band, which showed a profile similar to that of 4f-rnp, reflecting presence of sas-10 transcripts at relatively high levels during embryonic stages of development, but then declining in late-stage embryos to plateau at diminished levels until adulthood.
Utilization of primer sets sas-LFT/sas-2 through sas-LFT/sas-4 enabled detection and quantification of sas-10 readthroughs to the exclusion of conventional length transcripts. The two primer sets designated sas-LFT/sas-3 and sas-LFT/sas-4 produced relatively faintly staining cDNA bands (compare intensity scales; Fig. 2B,D) indicative of their lower frequency compared to the conventional length transcripts. The mRNA profile across development as exemplified by primer set sas-LFT/sas-3 (Fig. 2D ▶) is clearly different from that seen for conventional length sas-10 mRNAs, with few transcripts detectable during early embryo stages followed by a peak at second instar in the former.
The Drosophila sas-10 gene utilizes dual promoters
Dual promoters are fairly common for developmentally regulated Drosophila genes, and our discovery (above) of two sas-10 transcript classes (conventional length and long readthrough), the latter first appearing while the former is at its peak, led us to hypothesize that these two transcript classes might utilize different promoters. During screening of a 0–4 h Drosophila embryo cDNA library for the longest sas-10 gene transcripts (J. Petschek, unpubl.), the 1389 nt polyadenylated cDNA clone designated 4R1#6 was isolated and sequenced (Hunt-Smith 1999). Subsequently, our search of the Drosophila EST database revealed a somewhat longer polyadenylated adult head cDNA clone designated GH08670, having the same 5′-terminus as 4R1#6 but a longer 3′-UTR, which analysis showed is complementary to the final 149 nt of the 4f-rnp 3′-UTR. These two transcripts differ in length by 242 nt, as shown by direct sequencing, and it is not known if they occur in identical cells. Both transcripts contain a potential poly(A) signal (Fig. 3 ▶). The similarity in position of their 5′-termini suggested that these two transcripts could have arisen from the same promoter, which we have designated P1. Transcripts corresponding to cDNA clone GH08670 would not have been detected by primer sets sas-LFT/sas-3 or sas-LFT/sas-4, because these downstream primers fall outside the sequence of GH08670 (Fig. 3 ▶). In preliminary Northern blot experiments utilizing poly(A+) RNAs isolated from different developmental stages, sas-10 transcripts thought to represent short conventional mRNAs on the basis of their estimated size were detected in 0–4 h embryos but then diminish to very low levels as they are replaced by transcripts corresponding to GH08670 (N.T. Peters and J.C. Vaughn, unpubl.), reappearing in adult body RNAs (Hunt-Smith 1999).
To test the hypothesis that conventional length and long readthrough transcripts may originate from different promoters, we designed two different primer sets. One set (Alt-LS/Alt-R) amplified a 343-bp fragment beginning just inside the 5′-terminus of the sas-10 cDNA clones 4R1#6 and GH08670, visualized as an intensely staining band after RT-PCR using RNAs from early developmental stages (Fig. 4A ▶). In the other set (Alt-LL/Alt-R), the forward primer was located 170 nt upstream of these two cDNA 5′-termini. Amplifications using this primer set produced faintly staining bands using late-embryo RNAs after RT-PCR (Fig. 4B ▶), interpreted to demonstrate existence of an alternative promoter P2, transcribing a less abundant sas-10 transcript class. Identities of amplified fragments were verified by sequencing. It is our view that the relatively faintly staining RT-PCR bands observed (Fig. 2D ▶) for the long sas-10 readthrough transcripts correspond to the faintly staining band (Fig. 4B ▶), which originates from promoter P2 (Fig. 3 ▶). Decline in promoter P1 utilization would then be correlated with onset of promoter P2 activation, which could explain how a single-copy sas-10 gene could produce different transcript classes (conventional length and long readthrough) during development.
FIGURE 4.

The Drosophila sas-10 gene utilizes dual promoters. Orientation diagram shows locations of gene-specific nested primer sets (brackets) utilized for RT-PCR. In primer set #1 (primers Alt-LS/Alt-R), upstream primer is located just inside 5′-end of cDNA clone 4R1#6, which produces transcripts that terminate at the trailer (T). In primer set #2 (primers Alt-LL/Alt-R), upstream primer is located 170 nt further upstream. (A) Following RT-PCR, gel electrophoresis of products, and cDNA band staining with SYBR Green I, primer set #1 gave intensely staining bands relative to rp49 (Fig. 2A ▶) when using early developmental stage RNA templates including 6–12 h (1a) and 12–18 h (1b). These bands derive from promoter P1, previously known. (B) In contrast, primer set #2 gave a faintly staining band (composition verified by sequencing) relative to rp49 when using later developmental stage RNA templates including 15–21 h embryo (2, short arrow). The faint band is interpreted to derive from a second, distal promoter P2. Densely staining, rapidly migrating bands are due to primer dimers. Sizing standard (S) used during agarose gel electrophoresis was plasmid pBR322 DNA cut with AluI.
RNA editing within double-stranded 4f-rnp and readthrough sas-10 transcripts
We previously described a single hyperedited adult head 4f-rnp transcript, designated cDNA clone H8.3, which contained 263 A-to-G edits distributed randomly throughout both coding and noncoding regions (Petschek et al. 1997). Given the requirement of the dADAR editase for double-stranded RNA templates, it was proposed that a separately transcribed pairing strand of unknown origin could exist. We have now shown that long sas-10 readthroughs extending at least as far as the complement of the 3′-terminal 467 nt in 4f-rnp transcripts (Figs. 2 ▶, 3 ▶) begin to appear in late embryo stages. We tested the hypothesis that 4f-rnp and long sas-10 readthrough transcripts exist in the same tissues, a condition expected to result in double-stranded RNA which would be a potential target for modification via dADAR editase, by screening for A-to-G edits in 4f-rnp transcripts during development. The plethora of edited sites previously observed in adult head cDNA clone H8.3 (Petschek et al. 1997) made it appear likely that a screen for frequency of editing across development could most easily be carried out using a restriction enzyme approach upon isolated cDNAs, as has been utilized successfully by others (Saccomanno and Bass 1999; Hanrahan et al. 2000). However, this was not the result we obtained (Rohrbach 2001). With one exception, not a single cDNA clone from among the 168 examined from a variety of fly developmental stages, including 83 from adult heads, showed a clear evidence of abundant sequence changes attributable to RNA editing.
It is possible that these disappointing results reflect a lower-than-expected frequency of RNA editing per molecule, which would be difficult to detect using the restriction enzyme approach. This prompted us to explore a direct sequencing assay for RNA editing. The pooled results (Fig. 5A ▶) show that A-to-G editing occurs at random positions throughout the 4f-rnp 3′-UTR test sequence in many cDNA clones, whereas numerous other clones showing T-to-C conversions are detected. In no case did we observe both classes of conversion in the same clone.
FIGURE 5.

RNA editing in 4f-rnp trailer region and its sas-10 readthrough complement. (A) Compilation of edit site locations (heavy dots). The 23 edits observed are randomly distributed over 12 different sites. (B) Percent edited transcripts from different developmental stages and different locations in the adult fly. Numbers of clones sequenced are indicated for each.
The frequency of RNA editing detected (Fig. 5B ▶) is 7.8% of the 51 transcripts assayed in late embryo cDNAs, but this rises to 24.4% in adult heads (45 cDNA clones) and 16.7% in adult bodies (48 cDNA clones). Pooling the results for editing in adults, 20.4% of the 93 cDNA clones sequenced are edited, a 2.6-fold increase during development. These results are comparable to those reported by Hanrahan et al. (2000) in an assay for A-to-G editing at the Fsp site in the fly para gene transcript, which encodes a brain Na+ ion channel protein, interpreted as consequences of the very high dADAR levels observed in adult flies (Palladino et al. 2000b). In para mRNAs, about 15% of the transcripts were edited in late embryo, in contrast to about 68% in adults, a 4.5-fold increase. It is likely that greater numbers of edited transcripts would have been detected in our study than reported here, had we targeted more than the 162 nt of 4f-rnp 3′-UTR located between the PCR primers employed.
We suggest that the observed T-to-C conversions arise from A-to-G edits in the bottom strand sas-10 readthrough transcripts of a double-stranded RNA/RNA duplex. After second-strand cDNA synthesis during PCR, these sites would then be visualized as top-strand T-to-C conversions. This interpretation was originally suggested in another system during study of a human measles patient, when 50% of the uridines appeared as cytidines in cDNAs synthesized from viral matrix mRNA (Cattaneo et al. 1988). Subsequently, it was suggested that these hypermutations had arisen as A-to-I conversions in the paired RNA genome (Bass et al. 1989). Our results are interpreted to show that 4f-rnp and sas-10 readthrough transcripts coexist in the same tissues and form double-stranded molecules in some cells, which are then targeted for editing by the dADAR editase. Existence of A-to-G editing represents an assay for double-stranded RNAs in cells, and avoids the potential for artifactual RNA duplex formation inherent in other more direct experimental approaches, such as RNase protection assay.
Assays for Taq polymerase fidelity determination
The RT-PCR method we have employed for detection of RNA editing utilizes Taq DNA polymerase, considered a low-fidelity polymerase because it lacks 3′ → 5′ exonuclease (proofreading) activity (reviewed in Gelfand and White 1990). We therefore addressed the possibility that a fraction of the apparent edits observed in our study was due to errors by Taq polymerase. At one extreme, Saiki et al. (1988) reported 17 errors among 6692 nt sequenced, for a calculated 1.7 × 10−4 errors per base per PCR cycle. Utilizing different systems, comparable error rates were reported in studies by Tindall and Kunkel (1988) and Keohavong and Thilly (1989). Strikingly, errors observed were all attributed to A-to-G changes in the latter study. In each of these cited studies, nonoptimal PCR reaction conditions utilizing relatively high concentrations of each dNTP and also MgCl2 were employed, conditions subsequently shown to promote Taq polymerase infidelity (Gelfand and White 1990). In contrast, others have reported substantially lower error rates when using 1.5 mM MgCl2 and lower dNTP concentrations during PCR, as in our study. For example, Goodenow et al. (1989) observed no errors among 5400 nucleotides sequenced from 34 clones (81,000 total nucleotides), for a calculated error rate of <1.2 × 10−5. Fucharoen et al. (1989) detected no errors during sequencing of clones totaling 14,990 nucleotides, for a calculated error rate of <6.6 × 10−5.
To control for possible errors due to Taq infidelity, we performed the following two analyses (data not shown). First, utilizing the same Taq batch number as for our editing sequencings, we sequenced each of two different genomic clones, twice, for a 1840-nt DNA tract spanning the 4f-rnp and sas-10 genes obtained from our lab stock of D. melanogaster strain Oregon R. We then sequenced two corresponding genomic clones, twice, for this DNA tract obtained from a population of this species living near Oxford, OH. All molecules were completely sequenced on both strands, and enzyme errors were attributed to base changes detected at a site in one clone of a population but not in the other clone, relative to the published Celera fly genome sequence (Celniker 2000). Aside from two changes due to polymorphisms (identical changes at the same site in both clones of the Oxford, OH population), we detected a total of only one change among the 7360 nucleotides sequenced, which could have arisen from Taq error, for a calculated error rate of <1.0 × 10−5, comparable to the cited results obtained by others using similar reaction conditions. Finally, we prepared and sequenced 50 randomly picked genomic DNA clones following PCR of the identical tract utilized in our editing sequencings, using a different enzyme batch number. Within the 8100 nt sequenced, we detected one change attributed to Taq error, an error rate slightly lower than that observed in our first analysis.
In contrast to the relatively low Taq error rates determined in the two control experiments, the numbers of base changes observed during our editing sequencings were higher by about fourfold (late embryo) to 11-fold (adult heads). We conclude that base changes attributed to RNA editing in our study are not due to errors by the Taq polymerase procedure utilized. As previously mentioned, the increase in frequency of base changes attributed to editing during development in our study parallels that reported by others (Hanrahan et al. 2000), lending support to our conclusion.
DISCUSSION
Important roles for naturally occurring antisense RNAs in regulatory processes involving transcription and DNA replication among prokaryotes are well established (reviewed in Inouye 1988). In both prokaryotes and eukaryotes, “sense” mRNA three-base codons pair with three-base “antisense” anticodons in tRNAs to direct amino acids into proper sequence during the translation process on ribosomes. Among eukaryotes, the role of antisense RNAs during spliceosomal excision of introns has been extensively studied (reviewed in Adams et al. 1996). Eukaryotic cells contain a large and complex population of small RNAs that are primarily found associated with the nucleolus, referred to as snoRNAs. These RNAs are complementary to specific sites on pre-rRNAs, where their associations guide specific 2′-O-methylation and pseudouridine modifications in rRNAs (reviewed in Filipowicz and Pogacic 2002).
Naturally occurring antisense RNAs in eukaryotes, and their potential role in posttranscriptional regulation of gene expression, have been reviewed (Dolnick 1997; Knee and Murphy 1997; Eddy 1999; Brantl 2002). In C. elegans, the 22 nt-long Lin-4 antisense RNA has been shown to regulate developmental timing of larval molts by binding to the 3′-UTRs of target mRNAs encoded by the lin-14 and lin-28 genes (Lee et al. 1993; Moss et al. 1997). The mammalian Xist RNA, which coats and is required for inactivation of the X-chromosome (reviewed in Panning and Jaenisch 1998) may be regulated by the antisense RNA transcript Tsix (Lee et al. 1999). The studies most relevant to ours are those in which 3′-UTRs are required for the regulation of mRNA stability (reviewed in Lipman 1997). A well-characterized example is seen in the vertebrate basic fibroblast growth factor-2 (bFGF) RNAs. Here, exon III and the 3′-UTR of bFGF transcripts overlap and pair with the 3′ end, including exons III and IV, of a converging antisense mRNA termed bFGF-AS, coded on the opposite DNA strand and originally described in Xenopus oocytes (Kimelman and Kirschner 1989). It has been shown that bFGF-AS encodes a 217 amino acid protein in Xenopus (Volk et al. 1989) and a larger protein in rat (Li et al. 1996). In Xenopus unfertilized oocytes, the antisense transcript is present in 20-fold excess over the sense transcript, and the two mRNAs pair over about 900 nt in the cytoplasm, where they are largely protected from modification by ADAR reaching the cytoplasm after germinal vesicle breakdown due to the presence of a cytosolic protein factor, cyto-dsRBP (Saccomanno and Bass 1999). During oocyte maturation, a small fraction of bFGF mRNAs undergo extensive A-to-G RNA editing as detected in a restriction enzyme-based assay, although assay by direct sequencing as in the present study may have detected larger numbers of less extensively edited RNAs. Onset of editing is accompanied by bFGF mRNA degradation beginning at oocyte maturation and completely by 4 h into development, whereas bFGF-AS mRNA persists into the early cleavage stages (Kimelman et al. 1988). RNA/RNA pairing thus appears to play a role in vertebrate bFGF mRNA stability, possibly owing to nucleotide modifications derived from ADAR activity, as in Xenopus. Inversely proportional amounts of bFGF sense and antisense transcripts have also been observed in several adult organs of chicken (Zuniga et al. 1993). Further, it has been reported that in the developing rat brain abundance of sense and antisense bFGF transcripts are inversely related, suggesting a role for the antisense transcript in posttranscriptional regulation of bFGF expression in this tissue (Knee et al. 1997), although it is not yet known whether or not RNA editing of transcripts accompanies these effects in that system.
Comparisons of human and Xenopus bFGF-AS exons III and IV, which are complementary to the bFGF 3′-UTR, have shown that their nucleotide sequences are 73% identical (Murphy and Knee 1994). More impressively, BLAST sequence alignment shows that the rat bFGF-AS exons III and IV (Knee et al. 1997) are 98% identical over a length of 301 nt with the human homolog (Murphy and Knee 1994), species that diverged about 100 million years ago. It has been reported that rat bFGF-AS is complementary to 464 nucleotides located in two widely separated regions of the human 6.7-kb bFGF 3′-UTR: a 60-bp region including the most distal poly(A+) signal motif of bFGF mRNA and 404-bp lying just downstream of the proximal bFGF poly(A+) site (Knee et al. 1997). This observation prompted the suggestion that physical interaction between the two mRNAs could play a role in the regulation of bFGF mRNA polyadenylation site usage. Interestingly, differential poly(A) signal usage is also observed between different sas-10 transcripts in our study (Fig. 3 ▶).
The long antisense RNA deriving from sas-10 readthrough transcription described in our study is similar to, yet distinct from, the bFGF system. In our example, sas-10 also codes for a protein, and the formation of double-stranded RNAs is accompanied by A-to-G RNA editing. As in the bFGF example, the paired sense and antisense RNAs are present in very different relative abundance, although in our case it is the 4f-rnp sense strand population which declines in abundance and then persists into later development. Long sas-10 antisense readthrough transcription is developmentally regulated, this transcript class not appearing until the late embryo stage of development, potentially owing to activation of upstream promoter P2. In common with the bFGF system, we find that long sas-10 antisense readthrough and 4f-rnp transcript levels are inversely related, rising levels of the antisense transcript at late embryo stage being accompanied by decline in 4f-rnp levels. This observation suggests a role for antisense RNA in posttranscriptional regulation of 4f-rnp gene expression during development. Synchrony of 4f-rnp editing onset and the inferred subsequent partial degradation of these transcripts, which both occur in the late embryo stage of development, are not explainable by a sudden increase in dADAR editase activity, which has been shown to be at relatively low levels until about the pupal stage, when it dramatically increases (Hanrahan et al. 2000; Ma et al. 2002). Rather, onset of 4f-rnp editing during development appears to be triggered by the availability of dADAR substrate in the form of RNA/RNA duplexes, owing to the developmentally regulated activation of the sas-10 upstream P2 promoter, and subsequent long sas-10 readthrough transcription. The reason for the dramatic difference in extent of 4f-rnp editing in adult head cDNA clone H8.3 (Petschek et al. 1997) and the results in this study are not known. Clone H8.3 contained an unspliced intron IV owing to an edit AG→GG at the 3′-splice junction, and although polyadenylated, may have never exited the nucleus. If so, its retention there may have permitted additional editing to occur.
The molecular mechanism behind antisense regulation of 4f-rnp gene expression is currently under investigation in our laboratory. It is possible that multiple A-to-I nucleotide conversions, which we have shown increase in frequency beginning at late embryo stage of development at a time when 4f-rnp mRNA levels begin to decline, leads to retention of edited transcripts in the nucleus. This result has been reported for polyoma viral early-strand mRNAs in mammalian cells (Kumar and Carmichael 1997), and in a Xenopus model system (Zhang and Carmichael 2001), and shown to be dependent on an evolutionarily conserved nuclear protein that has a homolog in Drosophila. No difference in signal intensity was observed between adult wild-type and dADAR null mutant 4f-rnp mRNA levels in a recent cDNA microarray analysis (E. Ma, pers. comm.), but such an analysis of cDNAs across embryonic development might be revealing. Formation of mRNA/mRNA duplexes may interfere with binding of nuclear protein factors required for splicing of introns, or assembly of the exon junction complex required for mRNAs to exit the nucleus (Reichert et al. 2002). There are reportedly ribonucleases specific for degradation of inosine-containing RNAs (Scadden and Smith 1997). If RNA/RNA duplexes do enter the cytoplasm, they may be unable to bind to ribosomes, and are subsequently degraded. Even short perfect RNA/RNA duplexes are an excellent substrate for recognition by the RNAi pathway in eukaryotic cells (reviewed in Carthew 2001; Brantl 2002), shown to be especially active in the Drosophila embryo cytoplasm (Tuschi et al. 1999; Zamore et al. 2000). However, modification of perfect RNA duplexes leading to base-pair mismatches by nuclear dADAR, shown to be present at high levels in Drosophila neural tissue (Palladino et al. 2000a), would be predicted to diminish the subsequent potential effects of cytoplasmic RNAi (Bass 2000; Scadden and Smith 2001). It has been reported that the Drosophila testis-expressed Suppressor-of-Stellate gene on the Y-chromosome, arising by paralogous tandem repetition, transcribes from both DNA strands. This results in antisense RNA which posttranscriptionally silences Stellate gene expression from the X-chromosome via an RNAi-like pathway, ensuring male fertility by a novel negative feedback pathway (Aravin et al. 2001). Gene expression control involving interaction between oppositely oriented mRNAs, as in our study, may turn out to be common. In a preliminary search of transcripts arising from merely the final 5 × 106 bp at the distal tip of the Drosophila X-chromosome utilizing FlyBase (http://flybase.bio.indiana.edu), we identified over 30 different examples that are comparable to what we have observed for the 4f-rnp/sas-10 interaction.
Discovery of naturally occurring antisense RNA that is coupled to 4f-rnp and sas-10 RNA editing in Drosophila, a species for which a considerable database exists including a completely sequenced genome, and one that is amenable to powerful molecular genetic experimental approaches, should enable rapid progress in determining mechanisms controlling antisense regulation of gene expression and the role of RNA editing in this process.
MATERIALS AND METHODS
Fly stocks and their propagation
The lab stock of Drosophila melanogaster used in these studies was wild-type strain Oregon R, obtained from the Miami University stocks but originally derived from the National Drosophila Stock Center. Specimens from a D. melanogaster population were also trapped in the vicinity of Oxford, OH. Flies were cultured on standard media consisting of cornmeal, dark corn syrup, Brewer’s yeast, propionic acid, agar, and mold inhibitor. For collection of fly developmental stages to be used for RNA isolations, short plastic chimneys were fitted to 60 mm-diameter Petri dishes containing either standard media or a recipe especially useful for encouragement of egg laying consisting of dextrose, sucrose, apple juice, propionic acid, phosphoric acid, and agar (H. Keshishian, pers. comm. via J. Fernandes). Adult flies (mixed sexes and ages) and fly developmental stages used for DNA and RNA isolations were stored at −70°C until use.
Nucleic acid isolations
Genomic DNAs were isolated from large numbers of adult flies using the procedure described by Jowett (1986), with modifications. Total cell RNA isolations were carried out as previously described (Koller et al. 1987), using a phenol-based procedure that included treatment with RNase-free DNase I. RNA quality was verified by electrophoresis in 1% agarose gels (Ausubel et al. 1994–1997), using integrity of the rRNA bands as the criterion.
Quantitative RT-PCR of 4f-rnp and sas-10 transcripts during development
Conventional PCR amplifications were run in 25–100 μL reaction volumes containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.05% (v/v) NP-40, 200 μM each dNTP, 1 μM each primer, 0.5–1.0 μL cDNA or 0.25–1.0 μg genomic DNA, and 0.5–2.0 μL Taq polymerase (Promega). DNA fragment amplifications employed the following thermocycler parameters: one cycle of 2-min denaturation at 97°C, followed by 30 cycles of 30 sec denaturation at 95°C, 1 min annealing at optimized temperature determined by a preliminary gradient block run, and 1 min extension at 72°C.
Reverse transcription (RT) of total cell RNAs (Kawasaki 1990) isolated from fly developmental stages was carried out in 60 μL reaction volumes containing 3 μg RNA template, using random hexamer primers and 400 U M-MLV enzyme (Promega) per reaction tube. Products were stored at −70°C until used for polymerase chain reaction (PCR). PCR (Mullis and Faloona 1987) was carried out using sas-10 gene strand-specific primer sets designed to detect not only conventional length transcripts (primer set sas-LFT/sas-1), but also readthrough transcripts extending downstream into the intergenic spacer (primer set sas-LFT/sas-2), into the complement of the 4f-rnp gene 3′-UTR (primer set sas-LFT/sas-3) and into about 200 nt of the complement of the 4f-rnp gene 3′-coding region (primer set sas-LFT/sas-4). 4f-rnp transcription was followed using primer set Dros E-2/PX-2, which amplified a fragment of about 400 bp.
We patterned the quantitative RT-PCR procedure utilized on that described by Horikoshi and Sakakibara (2000), but with many significant and important modifications. We utilized SYBR Green I (Molecular Probes) DNA band staining, where DNA bands are laser-excited, gels scanned in the Molecular Dynamics Storm 860 phosphorimager using the blue fluorescence mode, and band intensities directly imported into the computer for quantification using version 5.2 of the ImageQuant software program. The range over which intensity is linearly proportional to DNA amount was estimated utilizing serial dilutions of plasmid pBR322 cut with AluI, and scoring each of the four largest DNA bands individually (910, 657 doublet, 521 and 403 bp). Electrophoresis was in 1.5% Separide (Life Technologies)/1% agarose composite gels or 2% agarose gels and poststaining in a 1:10,000 dilution of stock SYBR Green I in TAE buffer at pH 7.9 (40 mM Tris-HCl, 5 mM sodium acetate, 1 mM disodium EDTA) for 20 min, with continuous gentle rocking. The linear intensity range was found to be between about 1 and 200 ng, although the limits of lower and upper linearity were not further explored. The PCR cycle numbers that were in the linear range were then determined for each primer set utilized, by programming the thermocycler to stop every two cycles so that a reaction tube could be removed. The midrange for each primer set was utilized in subsequent work. Finally, differing cDNA template quantities were run for each primer set, to assure that band intensities were proportional to quantities of cDNA template. Expression levels for genes in this study were compared to that of the constitutively expressed rp49 ribosomal protein reference gene (O’Connell and Rosbash 1984). Results were expressed as quantity of PCR product within the linear amplification range using the equation (Horikoshi and Sakakibara 2000):
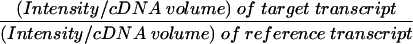 |
DNA fragment isolation, cloning, and sequence analysis
The following applies to treatment of PCR samples intended for subsequent cloning and sequencing. After PCR, reaction products were resolved in low-melting Sea Plaque (FMC) agarose gels, followed by band excision, agar-ACE (Promega) digestion and ethanol precipitation of DNAs. Gel-isolated fragments were cloned into either the pCRII plasmid vector (Invitrogen) or the pGem-T Easy vector (Promega) and transformed into competent DH5αF′ Escherichia coli cells. Following selection of white colonies from ampicillin-drugged, XGAL-treated LB plates, recombinant plasmid DNAs were recovered from cell aliquots as described (Davis et al. 1980). Successful clones were recognized after fly DNA fragment insert excision with EcoRI and agarose gel electrophoresis. The resulting DNA was directly utilized for input into an ABI 3100 automated DNA sequencer (PE Applied Biosystems), using the primers that had been employed for PCR and also additional internal primers, if appropriate. Automated sequencer output analysis was achieved using the EditView program, followed by manual inspection of the data. Sequence alignments were done with reference to the published D. melanogaster 4f-rnp gene (Petschek et al. 1997) and also the Drosophila total genome sequence (Celniker 2000) available via GenBank, using the Genetics Computer Group, University of Wisconsin, software package program Gap. Sequence homology searches were done using BLAST (http://www.ncbi.nlm.nih.gov/BLAST) and the Drosophila expressed sequence tags (EST) database (http://www.fruitfly.org/EST/oldEST.html).
Assays for 4f-rnp transcript RNA editing using restriction enzyme digestions
Given the abundance of edited sites in adult head cDNA clone H8.3 (Petschek et al. 1997), it appeared likely that a screen for frequency of editing across development could most easily be accomplished using a restriction enzyme approach, as has been utilized by others (Saccomanno and Bass 1999; Hanrahan et al. 2000). We selected enzymes known to have recognition sites within 4f-rnp, and each containing both an A and a G within the recognition site. It was predicted that such sites could be destroyed by an A-to-G edit, or created by such an edit, in either case revealing a clone with altered sequence as revealed by band mobility change during gel electrophoresis. Variant clones were then sequenced to reveal the nature of the alteration. We examined cDNA clones from different developmental stages that had been PCR-amplified in two different screens. In the first screen, the amplified DNA fragment extended from top strand primer #15 to bottom strand primer #24, containing a 1350 nt fragment. Enzymes used in this screen, which was utilized for 85 largely preadult developmental stages, were: AluI, Hsp92-II, PvuII, Sau3A-I, and TaqI. In the second screen, the amplified DNA fragment also originated at primer #15 and extended for about 350 nt to the bottom strand primer #16. Enzymes used in this screen, which was utilized for 83 adult head cDNA clones, were: AluI, RsaI, PstI, and TaqI.
Direct sequencing assay for RNA editing in 4f-rnp and sas-10 transcripts
If 4f-rnp and sas-10 gene readthrough transcripts do coexist in the same cells, these RNAs would be expected to pair and in the presence of dADAR, A-to-G conversions would be expected to arise on both RNA strands. We generated cDNA clones via RT-PCR from late embryo (12–21 h development) total cell RNA (51 clones), adult head total cell RNA (45 clones), and adult body total cell RNA (48 clones). Each clone was sequenced on both strands, utilizing primer set 4F-TR-L/4F-TR-R, expected to produce 199-bp fragments (162 bp between primers) derived from the 4f-rnp gene 3′-UTR and its sas-10 readthrough complement. The results were scored for numbers of A-to-G conversions arising due to edits on the 4f-rnp RNA strand and also numbers of T-to-C conversions arising originally due to A-to-G edits on the paired sas-10 RNA bottom strand. To control for possible errors in interpretation of results due to Taq enzyme infidelity, we performed two extensive genomic DNA sequencing analyses, described in Results.
Primers utilized
Primers for RT-PCR analysis of two alternative sas-10 gene promoters:
Alt-LS fwd (5′ end of clone 4R1#6 cDNA): 5′-[GTTGTGTTTGAT TTAATTTAGT]-3′
Alt-LL fwd (5′ of primer #Alt-LS): 5′-[ATCTTGCTTTGCATC CTG]-3′
Alt-R rev (3′ of sas-10 start codon): 5′-[ACGTGCTCCGTTTGC TG]-3′
Primers for quantitative RT-PCR analysis of sas-10 and 4f-rnp transcripts during development:
Sas-LFT fwd (5′ of sas-10 stop codon): 5′-[GCTCCGATGATGAT GATAATGAT]-3′
Sas-1 rev (within sas-10 trailer): 5′-[GATCTTGGCGGATTTAT TGGAT]- 3′
Sas-2 rev (within sas-10/4f-rnp spacer): 5′-[CATATGACAGCTAG TTGACTAAT]-3′
Sas-3 rev (within 4f-rnp trailer): 5′-[AGCGGCCATGGTTTAGT TTGT]- 3′
Sas-4 rev (within 4f-rnp coding region): 5′-[CCATCAAGCGCG AGGCTAAT]-3′
Dros E-2 fwd (5′ of 4f-rnp intron IV): 5′-[ATG(A/G)GC(A/G)GC AGGTCGCC(A/G)]-3′
PX-2 rev (5′ of 4f-rnp intron V): 5′-[AGATCGAGGGCCTGGC AG]-3′
RP49-L fwd (5′ of rp49 gene intron): 5′-[CCAAGGACTTCATCC GCCACC]-3′
RP49-R rev (3′ of rp49 gene intron): 5′-[GCGGGTGCGCTTGTT CGATCC]-3′
Primers for restriction enzyme-based RT-PCR 4f-rnp edited transcript assay:
15 fwd (5′ of 4f-rnp intron IV): 5′-[TGCCGCCTTCAATCATGC]- 3′
24 rev (described below)
16 rev (5′ of 4f-rnp intron V): 5′-[CAATGGACTCGTAGGTGC]-3′
Primers for RT-PCR analysis of RNA editing in overlapping 4f-rnp and sas-10 gene transcripts:
4F-TR-L fwd (at 4f-rnp gene trailer 5′ end): 5′-[CAAATGCCGGC GGCTGA]-3′
4F-TR-R rev (at 4f-rnp gene trailer 3′ end): 5′-[CTTGGTTCATCA AGAAACCA]-3′
Primers for Taq fidelity determination around 4f-rnp/sas-10 junction:
Int-4 fwd (just 5′ of 4f-rnp intron IV): 5′-[CCGTGAGATTTGG CGC]-3′
Int-8 rev (just 3′ of 4f-rnp intron VIII): 5′-[GGTGGGTTTGAA ATGGC]-3′
19 fwd (3′ of 4f-rnp intron V): 5′-[ACAAACGAATCGTGAGCC]-3′
18 rev (3′ of 4f-rnp intron V): 5′-[ACGTAGCTCCACCAGATG]-3′
21-A fwd (just 5′ of 4f-rnp intron VII): 5′-[CGCCTGGTGCACA (A/G)GCT]-3′
PLY-AR rev (5′ of sas-10 stop codon ): 5′-[ATCAAGGCCGGCGT CAC]-3′
29 fwd (3′ of 4f-rnp stop codon): 5′-[CGGCCATGGTTTAGTT TG]-3′
24 rev (3′ of 4f-rnp stop codon): 5′-[CAAACTAAACCATGGC CG]-3′
Acknowledgments
We especially thank Jane Petschek for introducing us to 4f-rnp, for gifts of 4f-rnp and sas-10 gene clones, and for numerous helpful discussions during the formative stages of this research. We also thank Chris Wood for his help in running the many ABI 3100 sequencings, Justin Thomas for his work in cloning and sequencing cDNAs, Janaki Rangarajan for her work on Taq fidelity determinations, Dang-Khoa Nguyen for his work in performing restriction enzyme-based RNA editing assays of cDNA clones, Greg Gaski for collection of wild-type D. melanogaster from Oxford, OH, and genomic DNA isolations, and Tom Gregg for his help in trapping and identifying wild-type Drosophila specimens. We are grateful to the anonymous reviewers whose constructive comments helped to make this a much better manuscript. This work was in part supported by Miami University USS summer research internship awards to C. Byrkett and B. Zalewski, a grant to Miami University from the Howard Hughes Biomedical Research Institute, and also a State of Ohio Research Challenge grant to J. Vaughn. A preliminary report of these results was given at the 44th Annual Drosophila Research Conference, Chicago, IL, in March 2003. The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
NOTE ADDED IN PROOF
We have previously regarded Drosophila 4f-rnp as a novel gene, insofar as no homolog had been identified in other organisms using BLAST database searches. Following transmission of this manuscript to the printer, it was brought to our attention by Jane Petschek that two recent reports have now identified 4f-rnp homologs (Bell et al. 2002; Rader and Guthrie 2002). The human homolog p110/SART3 and Saccharomyces cerevisiae Prp24 function as snRNP recycling factors which are shown to play a direct role in assembly of U4 and U6 snRNPs into the di-snRNP required for subsequent pre-mRNA splicing. Homologs thus far identified include representatives from vertebrates, invertebrates, plants, and fungi.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2120703.
REFERENCES
- Adams, M.D., Rudner, D.Z., and Rio, D.C. 1996. Biochemistry and regulation of pre-mRNA splicing. Curr. Opin. Cell Biol. 8: 331–339. [DOI] [PubMed] [Google Scholar]
- Aravin, A.A., Naumova, N.M., Tulin, A.V., Vagin, V.V., Rozovsky, Y.M., and Gvozdev, V.A. 2001. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr. Biol. 11: 1017–1027. [DOI] [PubMed] [Google Scholar]
- Arbeitman, M.N., Furlong, E.E.M., Imam, F., Johnson, E., Null, B.H., Baker, B.S., Krasnow, M.A., Scott, M.P., Davis, R.W., and White, K.P. 2002. Gene expression during the life cycle of Drosophila melanogaster. Science 297: 2270–2275. [DOI] [PubMed] [Google Scholar]
- Aruscavage, P.J. and Bass, B.L. 2000. A phylogenetic analysis reveals an unusual sequence conservation within introns involved in RNA editing. RNA 6: 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel, F., Brent, R., Kingston, R., Moore, D., Seidman, J., Smith, J., and Struhl, K. (eds. F. Ausubel et al.), 1994. –1997. Current protocols in molecular biology. John Wiley & Sons, New York, NY.
- Bass, B.L. 1997. RNA editing and hypermutation by adenosine deamination. Trends Biochem. Sci. 22: 157–162. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Double-stranded RNA as a template for gene silencing. Cell 101: 235–238. [DOI] [PubMed] [Google Scholar]
- Bass, B.L. and Weintraub, H. 1987. A developmentally regulated activity that unwinds RNA duplexes. Cell 48: 607–613. [DOI] [PubMed] [Google Scholar]
- ———. 1988. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55: 1089–1098. [DOI] [PubMed] [Google Scholar]
- Bass, B.L., Weintraub, H., Cattaneo, R., and Billeter, M.A. 1989. Biased hypermutation of viral RNA genomes could be due to unwinding/modification of double-stranded RNA. Cell 56: 331. [DOI] [PubMed] [Google Scholar]
- Bell, M., Schreiner, S., Damianov, A., Reddy, R., and Bindereif, A. 2002. p110, a novel human U6 snRNP protein and U4/U6 snRNP recycling factor. EMBO J. 21: 2724–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beverley, S.M. and Wilson, A.C. 1984. Molecular evolution in Drosophila and the higher diptera. II. A time scale for fly evolution. J. Mol. Evol. 21: 1–13. [DOI] [PubMed] [Google Scholar]
- Brantl, S. 2002. Antisense RNA regulation and RNA interference. Biochim. Biophys. Acta 1575: 15–25. [DOI] [PubMed] [Google Scholar]
- Carthew, R.W. 2001. Gene silencing by double-stranded RNA. Curr. Opin. Cell Biol. 13: 244–248. [DOI] [PubMed] [Google Scholar]
- Cattaneo, R., Schmid, A., Eschle, D., Baczko, K., ter Meulen, V., and Billeter, M.A. 1988. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell 55: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celniker, S.E. 2000. The Drosophila genome. Curr. Opin. Genet. Dev. 10: 612–616. [DOI] [PubMed] [Google Scholar]
- Davis, R.W., Botstein, D., and Roth, J.R. 1980. A manual of genetic engineering: Advanced bacterial genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Dolnick, B.J. 1997. Naturally occurring antisense RNA. Pharmacol. Ther. 75: 179–184. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. 1999. Noncoding RNA genes. Curr. Opin. Genet. Dev. 9: 695–699. [DOI] [PubMed] [Google Scholar]
- Emeson, R.B. and Singh, M. 2001. Adenosine-to-inosine RNA editing: Substrates and consequences. In: RNA editing (ed. B.L. Bass), pp. 109–138. Oxford University Press, New York, NY.
- Filipowicz, W. and Pogacic, V. 2002. Biogenesis of small nucleolar ribonucleoproteins. Curr. Opin. Cell Biol. 14: 319–327. [DOI] [PubMed] [Google Scholar]
- Fucharoen, S., Fucharoen, G., Fucharoen, P., and Fukumaki, Y. 1989. A novel ochre mutation in the B-thalassemia gene of a Thai. J. Biol. Chem. 264: 7780–7783. [PubMed] [Google Scholar]
- Gelfand, D.H. and White, T.J. 1990. Thermostable DNA polymerases. In: PCR protocols (eds. M.A. Innis et al.), pp. 129–141. Academic Press, New York, NY.
- Goodenow, M., Huet, T., Saurin, W., Kwok, S., Sninsky, J., and Wain-Hobson, S. 1989. HIV-1 isolates are rapidly evolving quasispecies: Evidence for viral mixtures and preferred nucleotide substitutions. J. Acquir. Immune Defic. Syndr. 2: 344–352. [PubMed] [Google Scholar]
- Hanrahan, C.J., Palladino, M.J., Ganetzky, B., and Reenan, R.A. 2000. RNA editing of the Drosophila para Na+ channel transcript: Evolutionary conservation and developmental regulation. Genetics 155: 1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, K.A., Simone, A.A., and Petschek, J.P. 1996. Spatial and temporal expression of 4f-rnp gene in Drosophila melanogaster. Differentiation 61: 103–111. [DOI] [PubMed] [Google Scholar]
- Horikoshi, T. and Sakakibara, M. 2000. Quantification of relative mRNA expression in the rat brain using simple RT-PCR and ethidium bromide staining. J. Neurosci. Methods 99: 45–51. [DOI] [PubMed] [Google Scholar]
- Hough, R.F. and Bass, B.L. 2001. Adenosine deaminases that act on RNA. In: RNA editing (ed. B.L. Bass), pp. 77–108. Oxford University Press, New York, NY.
- Hunt-Smith, D.L. 1999. “Does a natural antisense RNA molecule direct editing of Drosophila 4f-rnp transcripts?” Thesis, Miami University, Ohio.
- Inouye, M. 1988. Antisense RNA: Its functions and applications in gene regulation—A review. Gene 72: 25–34. [DOI] [PubMed] [Google Scholar]
- Jowett, T. 1986. Preparation of nucleic acids. In: Drosophila, a practical approach (ed. D.B. Roberts), pp. 275–286. IRL Press, New York, NY.
- Kamakaka, R.T. and Rine, J. 1998. Sir- and silencer-independent disruption of silencing in Saccharomyces by Sas10p. Genetics 149: 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki, E.S. 1990. Amplification of RNA. In: PCR protocols (ed. M.A. Innis et al.), pp. 21–27. Academic Press, New York, NY.
- Keohavong, P. and Thilly, W.G. 1989. Fidelity of DNA polymerases in DNA amplification. Proc. Natl. Acad. Sci. 86: 9253–9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelman, D. and Kirschner, M.W. 1989. An antisense mRNA directs the covalent modification of the transcript encoding fibroblast growth factor in Xenopus oocytes. Cell 59: 687–696. [DOI] [PubMed] [Google Scholar]
- Kimelman, D., Abraham, J.A., Haaparanta, T., Palisi, T.M., and Kirschner, M.W. 1988. The presence of fibroblast growth factor in the frog egg: Its role as a natural mesoderm inducer. Science 242: 1053–1056. [DOI] [PubMed] [Google Scholar]
- Knee, R. and Murphy, P.R. 1997. Regulation of gene expression by natural antisense RNA transcripts. Neurochem. Int. 31: 379–392. [DOI] [PubMed] [Google Scholar]
- Knee, R., Li, A.W., and Murphy, P.R. 1997. Characterization and tissue-specific expression of the rat basic fibroblast growth factor antisense mRNA and protein. Proc. Natl. Acad. Sci. 94: 4943–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller, H.T., Frondorf, K.A., Maschner, P.D., and Vaughn, J.C. 1987. In vivo transcription from multiple spacer rRNA gene promoters during early development and evolution of the intergenic spacer in the brine shrimp Artemia. Nucleic Acids Res. 15: 5391–5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, M. and Carmichael, G.G. 1997. Nuclear antisense RNA induces extensive adenosine modifications and nuclear retention of target transcripts. Proc. Natl. Acad. Sci. 94: 3542–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J., Davidow, L., and Warshawsky, D. 1999. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet. 21: 400–404. [DOI] [PubMed] [Google Scholar]
- Lee, R., Feinbaum, R., and Ambros, V. 1993. The C. elegens heterochromatic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854. [DOI] [PubMed] [Google Scholar]
- Li, A.W., Too, C.K.L., and Murphy, P.R. 1996. The basic fibroblast growth factor (FGF-2) antisense RNA (GFG) is translated into a MutT-related protein in vivo. Biochem. Biophys. Res. Commun. 223: 19–23. [DOI] [PubMed] [Google Scholar]
- Lipman, D.J. 1997. Making (anti)sense of non-coding sequence conservation. Nucleic Acids Res. 25: 3580–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, E., Tucker, M.C., Chen, Q., and Haddad, G.G. 2002. Developmental expression and enzymatic activity of pre-mRNA deaminase in Drosophila melanogaster. Mol. Brain Res. 102: 100–104. [DOI] [PubMed] [Google Scholar]
- Morse, D.P. and Bass, B.L. 1999. Long RNA hairpins that contain inosine are present in Caenorhabditis elegans poly(A)+ RNA. Proc. Natl. Acad. Sci. 96: 6048–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse, D.P., Aruscavage, P.J., and Bass, B.L. 2002. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc. Natl. Acad. Sci. 99: 7906–7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss, E., Lee, R., and Ambros, V. 1997. The cold shock domain protein LIN-28 controls developmental timing in C. elegens and is regulated by the lin-4 RNA. Cell 88: 637–646. [DOI] [PubMed] [Google Scholar]
- Mullis, K.B. and Faloona, F.A. 1987. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 155: 335–350. [DOI] [PubMed] [Google Scholar]
- Murphy, P.R. and Knee, R.S. 1994. Identification and characterization of an antisense RNA transcript (gfg) from the human basic fibroblast growth factor gene. Mol. Endocrinol. 8: 852–859. [DOI] [PubMed] [Google Scholar]
- O’Connell, P. and Rosbash, M. 1984. Sequence, structure, and codon preference of the Drosophila ribosomal protein 49 gene. Nucleic Acids Res. 12: 5495–5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino, M.J., Keegan, L.P., O’Connell, M.A., and Reenan, R.A. 2000a. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 102: 437–449. [DOI] [PubMed] [Google Scholar]
- ———. 2000b. dADAR, a Drosophila double- stranded RNA-specific adenosine deaminase is highly developmentally regulated and is itself a target for RNA editing. RNA 6: 1004–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panning, B. and Jaenisch, R. 1998. RNA and the epigenetic regulation of X-chromosome inactivation. Cell 93: 305–308. [DOI] [PubMed] [Google Scholar]
- Petschek, J.P., Mermer, M.J., Scheckelhoff, M.R., Simone, A.A., and Vaughn, J.C. 1996. RNA editing in Drosophila 4f-rnp gene nuclear transcripts by multiple A-to-G conversions. J. Mol. Biol. 259: 885–890. [DOI] [PubMed] [Google Scholar]
- Petschek, J.P., Scheckelhoff, M.R., Mermer, M.J., and Vaughn, J.C. 1997. RNA editing and alternative splicing generate mRNA transcript diversity from the Drosophila 4f-rnp locus. Gene 204: 267–276. [DOI] [PubMed] [Google Scholar]
- Rader, S.D. and Guthrie, C. 2002. A conserved LSM-interaction motif in Prp24 required for efficient U4/U6 di-snRNP formation. RNA 8: 1378–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reenan, R.A., Hanrahan, C.J., and Ganetzky, B. 2000. The mlenapts RNA helicase mutation in Drosophila results in a splicing catastrophe of the para Na+ channel transcript in a region of RNA editing. Neuron 25: 139–149. [DOI] [PubMed] [Google Scholar]
- Reichert, V.L., Le Hir, H., Jurica, M.S., and Moore, M.J. 2002. 5′ exon interactions within the human spliceosome establish a framework for exon junction complex structure and assembly. Genes Dev. 16: 2778–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach, J.A. 2001. “The role of antisense transcription in control of 4f-rnp gene expression in Drosophila melanogaster.” Thesis, Miami University, Ohio.
- Saccomanno, L. and Bass, B.L. 1999. A minor fraction of basic fibroblast growth factor mRNA is deaminated in Xenopus stage VI and matured oocytes. RNA 5: 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki, R.K., Gelfand, D.H., Stoffel, S., Scharf, S.J., Higuchi, R., Horn, G.T., Mullis, K.B., and Erlich, H.A. 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239: 487–491. [DOI] [PubMed] [Google Scholar]
- Scadden, A.D.J. and Smith, C.W.J. 1997. A ribonuclease specific for inosine-containing RNA: A potential role in antiviral defense? EMBO J. 16: 2140–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2001. RNAi is antagonized by A→I hyperediting. EMBO Rep. 2: 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg, P.H. 1996. The role of RNA editing in controlling glutamate receptor channel properties. J. Neurochem. 66: 1–5. [DOI] [PubMed] [Google Scholar]
- Smith, L.A., Wang, X., Peixoto, A.A., Neumann, E.K., Hall, L.M., and Hall, J.C. 1996. A Drosophila calcium channel α1 subunit gene maps to a genetic locus associated with behavioral and visual defects. J. Neurosci. 16: 7868–7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer, B., Kohler, M., Sprengel, R., and Seeburg, P.H. 1991. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67: 11–19. [DOI] [PubMed] [Google Scholar]
- Tindall, K.R. and Kunkel, T.A. 1988. Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry 27: 6008–6013. [DOI] [PubMed] [Google Scholar]
- Tuschi, T., Zamore, P.D., Lehmann, R., Bartel, D.P., and Sharp, P.A. 1999. Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev. 13: 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk, R., Koster, M., Poting, A., Hartmann, L., and Knochel, W. 1989. An antisense transcript from the Xenopus laevis bFGF gene coding for an evolutionarily conserved 24 kd protein. EMBO J. 8: 2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, R.W., Smith, J.E., Cooperman, B.S., and Nishikura, K. 1989. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc. Natl. Acad. Sci. 86: 2647–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore, P.D., Tuschi. T., Sharp, P.A., and Bartel, D.P. 2000. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101: 25–33. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. and Carmichael, G.G. 2001. The fate of dsRNA in the nucleus: A p54nrb-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 106: 465–475. [DOI] [PubMed] [Google Scholar]
- Zuniga, A., Borja, M., Meijers, C., and Zeller, R. 1993. Expression of alternatively spliced bFGF first coding exons and antisense mRNAs during chicken embryogenesis. Dev. Biol. 157: 110–118. [DOI] [PubMed] [Google Scholar]