

Abstract
The bacterial tRNA processing enzyme ribonuclease P (RNase P) is a ribonucleoprotein composed of a ∼400 nucleotide RNA and a smaller protein subunit. It has been established that RNase P RNA contacts the mature tRNA portion of pre-tRNA substrates, whereas RNase P protein interacts with the 5′ leader sequence. However, specific interactions with substrate nucleotides flanking the cleavage site have not previously been defined. Here we provide evidence for an interaction between a conserved adenosine, A248 in the Escherichia coli ribozyme, and N(-1), the substrate nucleotide immediately 5′ of the cleavage site. Specifically, mutations at A248 result in miscleavage of substrates containing a 2′ deoxy modification at N(-1). Compensatory mutations at N(-1) restore correct cleavage in both the RNA-alone and holoenzyme reactions, and also rescue defects in binding thermodynamics caused by A248 mutation. Analysis of pre-tRNA leader sequences in Bacteria and Archaea reveals a conserved preference for U at N(-1), suggesting that an interaction between A248 and N(-1) is common among RNase P enzymes. These results provide the first direct evidence for RNase P RNA interactions with the substrate cleavage site, and show that RNA and protein cooperate in leader sequence recognition.
Keywords: RNase P, ribozyme, substrate binding, pre-tRNA
INTRODUCTION
Large RNA enzymes (groups I and II self-splicing introns and RNase P RNA) catalyze phosphoryltransfer and hydrolysis reactions at specific phosphate centers within large, structurally complex RNA substrates. Like group I and group II introns, interactions both proximal and distal to the RNase P cleavage site contribute to binding and catalysis (for example, Narlikar and Herschlag 1997, 1998; Loria and Pan 1999), and the most extensive contacts are expected to occur flanking the reactive phosphate (for example, Knitt et al. 1994; Pyle et al. 1994; Su et al. 2001). RNase P substrate recognition is of particular interest as the biological function of this enzyme requires the correct processing of all cellular pre-tRNAs. In Bacteria, RNase P substrate recognition occurs through the cooperation of the RNA subunit, which is the catalytic subunit of the enzyme (Guerrier-Takada et al. 1983; Guerrier-Takada and Altman 1984), and the RNase P protein subunit, whose principal role is in substrate binding (Frank and Pace 1998; Kurz and Fierke 2000; Christian et al. 2002). The mature tRNA portion of the substrate is bound by RNase P RNA, whereas the protein subunit contacts the 5′ leader sequence (Crary et al. 1998; Kurz et al. 1998; Loria et al. 1998; Niranjanakumari et al. 1998). However, contacts between RNase P and functional groups flanking the scissile phosphate have not been identified, leaving an important gap in our understanding of RNase P substrate recognition and the elements that comprise the enzyme active site.
Our current understanding of RNase P substrate binding and cleavage site recognition is summarized in Figure 1 ▶. Chemical protection and interference studies have shown that RNase P recognizes the geometry of the stacked acceptor and T-loop helices of pre-tRNA, and contacts functional groups proximal to the cleavage site and within the T-stem and loop (Kurz and Fierke 2000; Christian et al. 2002, and references therein). Substrate features proximal to the cleavage site that contribute to affinity and catalysis include the 3′ RCCA sequence, the G(1)-C(72) base pair and the 2′ hydroxyl at N(-1) (nucleotide numbering as in Fig. 1 ▶). In the holoenzyme, the protein subunit makes additional contacts to nucleotides in the 5′ leader sequence (Crary et al. 1998; Niranjanakumari et al. 1998). Mutagenesis and biochemical studies have shown that the 3′ RCCA sequence of pre-tRNA substrates contributes to substrate binding by pairing with the L15 element of RNase P RNA (Kirsebom and Svard 1994; Oh and Pace 1994; Tallsjo et al. 1996). However, the specific interactions involving the 2′ hydroxyl of N(-1) and the G(1)-C(72) base pair remain undefined.
FIGURE 1.

Summary of known RNase P RNA–pre-tRNA interactions. At left is the consensus secondary structure of bacterial RNase P RNAs (Pace and Brown 1995). Nucleotide A248 (E. coli numbering) is boxed. Nucleotides conserved in more than 90% of bacterial RNase P RNAs are indicated. Capital letters indicate 100% conservation. Elements of ribozyme structure referred to in the text are indicated. The stacked acceptor stem and T stem-loop of pre-tRNAASP are shown on the right. The RNase P cleavage site is indicated by an arrow and the region of the 5′ leader sequence that is contacted by C5 protein is circled (Niranjanakumari et al. 1998). Solid lines indicate previously characterized interactions (Kirsebom and Svard 1994; Pan et al. 1995; Tallsjo et al. 1996). Dotted lines indicate short-range intermolecular crosslinking results between the substrate cleavage site and the ribozyme (Kufel and Kirsebom 1996; Christian et al. 1998).
The importance of understanding interactions with functional groups flanking the cleavage site is underscored by the observation that removal or substitution of the 2′-OH at N(-1) decreases the rate of substrate cleavage by 100- to 1000-fold (Smith and Pace 1993; Loria and Pan 1998, 1999). In the context of a substrate that is cleaved at multiple sites, Pan and colleagues showed that a 2′ deoxy modification at N(-1) affects the rate of cleavage at both correct and incorrect sites. In contrast, substitutions of 2′-OH groups at incorrect cleavage sites have no effect. These and other observations lead to a model in which cleavage at correct and incorrect sites involves formation of distinct enzyme–substrate complexes, which differ in their interactions with the 5′ leader, but involve similar recognition of the T stem-loop and the 2′-OH group at the correct cleavage site (Loria and Pan 1998, 1999).
The need to identify interactions between RNase P and nucleotides flanking the cleavage site has also been highlighted by accumulating evidence that the nucleotide base at N(-1) makes an important contact with the enzyme active site. In the context of weakened interactions with the RCCA motif, cleavage site selection is dependent on the identity of N(-1) (Brannvall et al. 1998, 2002). In addition, in the context of a minimal substrate representing only the acceptor stem of a tRNA, changes in N(-1) identity influence the rate of cleavage (Zuleeg et al. 2001). Similarly, changing the identity of N(-1) in a full-length pre-tRNA alters the rate of catalysis by the RNase P holoenzyme and results in miscleavage of certain substrates (Crary et al. 1998; Loria et al. 1998; Loria and Pan 1998).
Several studies suggest that conserved adenosines in the J5/15 region of the ribozyme are likely to be involved in interactions with substrate features proximal to the scissile phosphate. As indicated in Figure 1 ▶, photocrosslinking agents positioned on either side of the scissile phosphate crosslink to J5/15, demonstrating its proximity to the cleavage site (Burgin and Pace 1990; Kufel and Kirsebom 1996; Christian et al. 1998; Christian and Harris 1999). In addition, nucleotide analog interference mapping has shown that nucleotide base and backbone functional groups in J5/15 contribute to binding and catalysis (Hardt et al. 1996; Siew et al. 1999; Heide et al. 2001; Kaye et al. 2002). These observations, together with the high degree of nucleotide base conservation in J5/15 (Brown 1999), made this region an interesting target for further experiments designed to identify enzyme–substrate interactions. Here, we describe the identification of a functionally important pairing interaction between a conserved adenosine in J5/15 and the N(-1) position of pre-tRNA. These findings provide the first direct evidence for a specific contact between the ribozyme active site and pre-tRNA, and show that the protein and RNA subunits of the RNase P holoenzyme both contribute directly to leader sequence recognition.
RESULTS
Conserved residues in J5/15 contribute to ribozyme affinity and specificity
To determine the role of J5/15 in enzyme function, we examined the effects of point mutations at the three nucleotides within this element on substrate binding and catalysis by RNase P RNA from Escherichia coli in combination with a pre-tRNAASP substrate bearing a 5-nucleotide 5′ leader. As shown in Table 1 ▶, mutations in J5/15 cause defects in both substrate binding and catalysis, but have a more pronounced effect on substrate binding. C247, which is an A in some bacterial RNase P RNAs (Brown 1999), is least sensitive to mutation, with none of the mutants showing more than a sixfold effect on substrate affinity. In contrast, mutations at A248 and A249, which are universally conserved among bacterial and archaeal RNase P RNAs, had larger effects on binding affinity and catalysis.
TABLE 1.
Effects of J5/15 and N(−1) mutants on binding and catalysis
| Enzyme | Substrate N(-1) nucleotide | Kd,obs (nM)a | 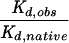 |
kobs (min−1)b | 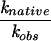 |
| Native | U(-1) (native) | 1.8 ± 0.5 | 5.0 ± 0.7 | ||
| C247A | U(-1) | 10 ± 5.6 | 5.7 | 4.2 ± 0.8 | 1.2 |
| C247G | U(-1) | 10 ± 2.8 | 5.7 | 3.9 ± 0.4 | 1.3 |
| C247U | U(-1) | 7.0 ± 3.5 | 3.8 | 4.5 ± 0.3 | 1.1 |
| A248G | U(-1) | 2.0 ± 0.9 | 1.1 | 4.0 ± 0.7 | 1.3 |
| A248C | U(-1) | 28 ± 0.5 | 16 | 2.4 ± 0.3 | 2.1 |
| A248U | U(-1) | 520 ± 40 | 290 | 0.8 ± 0.1 | 6.2 |
| A249G | U(-1) | >1000 | >560 | — | |
| A249C | U(-1) | >1000 | >560 | — | |
| A249U | U(-1) | >1000 | >560 | — | |
| Native | A(-1) | 9.4 ± 2.1 | 5.2 | 3.2 ± 0.1 | 1.6 |
| Native | G(-1) | 6.0 ± 2.2 | 3.3 | 4.9 ± 1.0 | 1.0 |
| Native | C(-1) | 430 ± 50 | 240 | 4.2 ± 1.4 | 1.2 |
| A248C | G(-1) | 2.9 ± 0.6 | 1.6 | — | |
| A248U | A(-1) | 80 ± 29 | 45 | — |
—, values not determined.
aKd,obs values were determined by gel mobility shift assay under standard reaction conditions (see Materials and Methods). Indicated uncertainties represent the standard deviation of three or more trials.
bkobs values indicate single turnover rate constants at saturating enzyme concentrations (see Materials and Methods). Uncertainties are the standard deviation of three or more trials.
Mutation of A249 results in large (>560-fold) increases in the observed dissociation constant for substrate binding (Kd,obs), but comparatively small effects on catalysis, as monitored by the single turnover rate constant at saturating enzyme concentrations (kobs, see Materials and Methods). Although the high Kd,obs values made it difficult to achieve saturating enzyme concentrations in single turnover experiments, rates observed at subsaturating concentrations indicate that these mutants have catalytic defects no more than sevenfold (data not shown). In addition, these mutants exhibit biphasic reaction kinetics, where only a fraction (10%–50%) of the substrate is cleaved in a fast phase. This observation is consistent with a population of misfolded ribozyme, and precludes straightforward interpretation of the kinetic and thermodynamic data. The large increase in the apparent Kd,obs values for the mutants may also be due to misfolding. Nevertheless, the data do indicate that A249 is important for function, and likely contributes to ribozyme structure.
Unlike other mutations in J5/15, mutations at A248 had significant effects on both ribozyme binding and catalysis (Fig. 2A,B). Although the A248G mutation results in relatively minor changes in binding and catalysis, A248C and A248U mutations result in twofold and sixfold decreases in kobs, as well as 16- and 290-fold increases in Kd,obs, respectively. Despite these relatively large changes in Kd,obs, gel shifts for A248C and A248U mutants were qualitatively similar to that of the native enzyme and did not exhibit increased smearing or other artifacts due to weak binding (see Fig. 2B ▶). Importantly, single turnover reaction kinetics for these mutants fit a single exponential function and proceed to the same extent as that observed with the native ribozyme. Although kinetics only provide a low resolution picture of the conformational complexity of an enzyme population, these data suggest that mutation of A248 does not lead to significant conformational heterogeneity.
FIGURE 2.

Effects of A248 mutants on binding and catalysis by the RNase P ribozyme. (A) Binding of pre-tRNA by A248 mutant ribozymes as assayed by gel mobility shift. Normalized plots show the fraction of substrate shifted to the bound complex as a function of enzyme concentration. Reactions contained 1 M ammonium acetate, 25 mM CaCl2, 20 mM PIPES at pH 6.0, and 2 nM ligated substrate. The data are fit to Equation 1 (see Materials and Methods). (Diamonds) Data for the native ribozyme; (triangles) A248G; (squares) A248C; and (circles) A248U. (B) Primary data from which the curves in A were obtained. Enzyme concentrations increase from left to right. (C) Normalized plots of fraction cut versus time for single turnover reactions at saturating enzyme concentrations (see Materials and Methods). Reactions were carried out under standard conditions (see Materials and Methods) with 4 nM substrate. Data are fit to a single exponential function, and are labeled as in A.
In addition to engendering defects in substrate binding and catalysis, mutation of substrate or RNase P RNA features involved in cleavage site selection can result in substrate miscleavage within one to two nucleotides of the authentic tRNA 5′ end (for example, Kirsebom and Svard 1994; Brannvall et al. 1998). Accordingly, we examined whether mutations in J5/15 result in miscleavage. Initial experiments using an unmodified pre-tRNA substrate showed that none of the mutations result in any change in the pattern of substrate cleavage (Fig. 4 ▶, see below). However, we observed that when the substrate contained a 2′-deoxy modification at N(-1), there was a marked increase in substrate miscleavage by the A248 mutants. As shown in Figure 3 ▶, the majority of J5/15 mutants cleave the deoxy substrate at precisely the same phosphodiester bond as the native enzyme. However, A248 mutations all result in some degree of miscleavage that generates a shorter leader fragment. As shown in Figure 4 ▶, this leader fragment has the same gel mobility as a synthetic oligonucleotide of the sequence AAAA, indicating that the site of miscleavage is one nucleotide 5′ of the correct cleavage site. Strikingly, the A248U mutant cleaved the substrate to yield a product population where only 13% of the input substrate was cleaved at the correct phosphodiester bond.
FIGURE 4.

Rescue of A248U miscleavage by N(-1) mutation. Annealed substrates (see Materials and Methods) with U, A, dU, or dA at N(-1) were cleaved overnight with the indicated A248 mutant ribozymes (lanes 8–23). Lanes 1 and 24 are size standards produced by partial ribonuclease T1 digestion of substrates. Lanes 2 and 25 are products from partial alkaline hydrolysis. Lane 3 contains oligonucleotides corresponding to products of correct (AAAAU) and incorrect (AAAA) cleavage. Lanes 4–7 show uncut substrate oligonucleotides. Variations in mobility of the correctly cut fraction are due to differences in nucleotide composition.
FIGURE 3.

Cleavage of pre-tRNA bearing a 2′ deoxy modification at N(-1) by the native ribozyme and J5/15 mutants. Mobility of the correct cleavage product (AAAAU) is indicated. A248 mutants (lanes 6–8) show miscleavage to yield shorter 5′ products. A249G and A249C mutant enzymes do not cut to completion, but when quantified are observed to choose the correct cleavage site at the same frequency as the native enzyme. The presence of the band marked by an asterisk varied between substrate preparations and was not observed with annealed substrates (see Materials and Methods).
Compensatory mutations at the substrate N(-1) position suppress miscleavage and rescue defects in binding affinity caused by A248 mutations
Reasoning that miscleavage could arise from the formation of non-native interactions, we considered models in which A248U forms an interaction similar to that of the native enzyme but shifted one nucleotide in the 5′ direction, as observed in the pattern of substrate cleavage. One simple model for a J5/15 interaction with the cleavage site, consistent with the mutational data, would be that A248 interacts with the U at N(-1) and that mutation of A248 to U results in an interaction with the A at the N(-2) position, bringing about a shift of the cleavage site in the 5′ direction.
The most simple and direct test of pairing between A248 and U(-1) is to examine whether compensatory changes at these positions can rescue enzyme specificity. As shown in Figure 4 ▶, we compared the pattern of cleavage products obtained for all three mutations at A248 and the native enzyme with substrates containing either a 2′ deoxyuridine or 2′ deoxyadenosine at position N(-1) (referred to as dU(-1) and dA(-1), respectively). As shown above, miscleavage is observed for the A248U ribozyme with the dU(-1) substrate. In contrast, we observed a dramatic decrease in the fraction of substrate miscleaved by the A248U ribozyme when the dA(-1) substrate was used. In addition, an increase in miscleavage was observed for the A248C ribozyme, whereas a small decrease in miscleavage was observed with A248G. Significantly, all of the changes in substrate cleavage pattern due to changes at A248 and the substrate base at N(-1) required the deletion of the N(-1) 2′ hydroxyl. We found that incorporation of this modification through RNA ligation or through the use of an annealed substrate (see Materials and Methods) give quantitatively identical results. In addition the N(-1) 2′ hydroxyl has been demonstrated to contribute to enzyme activity, but has been proposed to function in a cleavage site independent manner (Loria and Pan 1998, 1999). Our results show that the contributions of N(-1) base and backbone functional groups are interrelated, and suggest a model in which the N(-1) 2′ hydroxyl selectively stabilizes enzyme–substrate complexes with the correct phosphodiester bond docked into the active site.
The ability to rescue miscleavage induced by ribozyme mutations at A248 through compensatory changes in base identity at N(-1) strongly supports the hypothesis of a pairing interaction between these two nucleotides in the native enzyme-substrate complex. To investigate the nature of this pairing interaction, we determined the fraction of product resulting from correct cleavage for all possible combinations of nucleotides at A248 and N(-1). As shown in Figure 5A ▶, mutant ribozymes show different extents of correct cleavage depending on the identity of the substrate nucleotide base at N(-1). As discussed above, the A248U mutant interacts with the dU(-1) substrate to cleave almost entirely at the incorrect N(-2) position, but this same ribozyme cleaves the dA(-1) substrate primarily at the correct phosphodiester bond. In addition, the dG(-1) substrate was miscleaved by the native enzyme as well as by the A248U and A248G ribozymes; however, 80% of cleavage occurred at the correct site when there was a C residue at position 248. Similarly, the dC(-1) substrate also was primarily cleaved at the incorrect N(-2) site by both the native ribozyme and the A248 mutants. Nonetheless, the A248G mutant gave rise to the greatest extent of correct cleavage of the dC(-1) substrate. On the basis of these observations we conclude that docking of the correct phosphodiester bond into the active site of the ribozyme is influenced by pairing between A248 and the N(-1) nucleotide base in the substrate.
FIGURE 5.

A248 mutant cleavage specificity as a function of substrate base composition. (A) Fraction correct cleavage by native and A248 mutant ribozymes for substrates that were varied at N(-1). The fraction correct cleavage of dA(-1) is shown with white bars; dG(-1) with black bars; dC(-1) with gray bars; and dU(-1) with cross-hatched bars. Error bars indicate the standard deviation of three or more independent measurements. (B) Miscleavage as a function of N(-2) base identity, for substrates with dA at N(-1). White bars indicate the fraction of miscleavage for A(-2), black bars G(-2), gray bars C(-2), and cross-hatched bars U(-2).
Another aspect of the proposed model is that the observed miscleavage is due to inappropriate pairing with the base at N(-2) in the 5′ leader. Thus, mutation of N(-2) should engender miscleavage in a manner that is dependent on the A248 mutant. Accordingly, we measured the extent of miscleavage of substrates with N(-2) mutations in the context of a substrate with an A at N(-1). As shown in Figure 5B ▶, the greatest extent of miscleavage by the native enzyme was observed with an U at position N(-2). Similarly, the greatest extent of miscleavage for the A248C mutant was observed with a substrate containing a G at N(-2). Interestingly, miscleavage was induced for the A248U ribozyme by changing the N(-2) position to G or C, but not A. Here it is important to note that the A(-2) substrate also contains an A at N(-1), thus it is anticipated that the extent of miscleavage will be reduced by formation of an equivalent interaction between position U248 and A(-1). Similarly, only a small amount of miscleavage was observed for all of the mutant substrates in combination with the A248G ribozyme, possibly indicating a favorable interaction between a G at position 248 and A(-1). Nonetheless, miscleavage can be engendered by N(-2) mutations and the identity of N(-2) mutations leading to miscleavage is dependent on the identity of the ribozyme nucleotide at position 248.
An additional test of the proposed interaction is to ask whether compensatory mutations rescue defects in binding affinity. With the native ribozyme we find that N(-1) mutations result in defects in ground state binding, but have little effect on kobs (Table 1 ▶). Notably, a C(-1) pre-tRNA showed a profound increase in Kd,obs, which is likely due to the ability of the C at N(-1) to pair with G(73). Because G(73) is known to contact RNase P RNA (Tallsjo et al. 1996), this binding defect almost certainly represents the disruption of additional contacts. Importantly, the A(-1) substrate binds fivefold weaker to the native ribozyme than the U(-1) substrate. However, in the context of the A248U mutant ribozyme, the A(-1) substrate binds sixfold tighter than the U(-1) substrate (Fig. 6A ▶; Table 1 ▶). Similarly, the G(-1) substrate shows a threefold binding defect relative to U(-1) in the native enzyme, but binds the A248C ribozyme 10-fold tighter than U(-1) (Fig. 6B ▶; Table 1 ▶). Thus, consistent with the proposed interaction, A248 mutations alter the N(-1) base preference of the ribozyme for both binding and cleavage site selection.
FIGURE 6.

Rescue of A248 mutant binding defects by N(-1) mutation. (A) The plot shows binding data for the native RNase P RNA with U(-1) substrate (diamonds); A248U binding U(-1) substrate (triangles); native RNase P RNA binding A(-1) substrate (squares); and A248U with A(-1) substrate (circles). (B) The plot shows binding data for native RNase P RNA with U(-1) substrate (diamonds); A248C binding U(-1) substrate (triangles); native RNase P RNA binding G(-1) substrate (squares); and A248C binding G(-1) substrate (circles). All experiments contain 2 nM final substrate concentrations and are fit to Equation 1.
The results presented above strongly suggest the presence of a functionally important interaction between A248 and N(-1) in the RNase P ribozyme reaction. However, these experiments were performed in the absence of the RNase P protein subunit, which also recognizes the 5′ leader sequence. Thus, it is possible that the effects are idiosyncratic to the RNA-alone reaction. To address this issue, we examined the pattern of miscleavage observed in holoenzymes containing A248 mutant RNase P RNAs. As shown in Figure 7A ▶, very little product formation is observed at low ionic strength in reactions containing RNase P RNA, but lacking the protein subunit. Addition of the protein results in formation of cleavage products due to higher affinity of the holoenzyme for substrate under these conditions (Crary et al. 1998), demonstrating that the observed activity is due to the holoenzyme activity and not the RNA alone. Importantly, the miscleavage pattern of dU(-1) and dA(-1) substrates observed with the holoenzyme is essentially identical to that seen with the RNA-alone reactions (cf. Fig. 7B ▶ and Fig. 5A ▶). Specifically, a significant degree of miscleavage was observed only with the A248U mutant and changing the N(-1) position from U to A was able to this suppress miscleavage, as was observed with the RNA-alone reaction. Because the substrate used in these studies contains a leader sequence of sufficient length to interact with the protein subunit in the enzyme–substrate complex, we conclude that the proposed interaction between A248 and N(-1) is also present in the RNase P holoenzyme.
FIGURE 7.

Rescue of the A248U miscleavage phenotype by the RNase P holoenzyme. (A) Resolution of ligated substrate cleavage products. Combinations of enzyme and substrate are as indicated. Correct (AAAAU) and incorrect (AAAA) cleavage products are indicated. Reactions were run under holoenzyme conditions as described in the Materials and Methods. (B) Bar graph showing cleavage site preference for native and A248 mutant enzymes in the holoenzyme reaction. Correct cleavage of dA(-1) substrate is shown in white; dU(-1) bars are cross-hatched. Error bars represent the standard deviation of three or more independent determinations.
Comparative sequence analysis supports a role for the nucleotide base at N(-1) in recognition by RNase P
Previously, it was reported that tRNA genes from E. coli and Bacillus subtilis display a preference for U at position N(-1), and that few tRNA genes from E. coli encode a G at N(-1) (Kufel and Kirsebom 1996; Crary et al. 1998; Brannvall et al. 2002). Because A248 is conserved in bacterial and archaeal RNase P RNAs (Brown 1999), it follows that if this residue interacts with N(-1) in vivo, then this sequence preference should also be conserved in pre-tRNAs from Bacteria and Archaea. To assess the validity of this prediction we determined the N(-1) base composition of pre-tRNAs in 27 bacterial and 12 archaeal genomes (Table 2 ▶).
TABLE 2.
N(-1) composition of bacterial and archael species
| Species | %Aa | %G | %C | %Tb | tRNAsc | log(pT)d |
| Bacteria | ||||||
| Aquifex aeolicus | 84(38) | 2(19) | 5(12) | 9(32) | 44 | −3.5 |
| Bacillus subtilis | 13(35) | 3(8) | 16(17) | 67(40) | 86 | −6.7 |
| Borrelia burgdorferi | 21(24) | 6(22) | 0(31) | 73(24) | 33 | −2.8 |
| Bacillus halodurans C-125 | 13(24) | 5(22) | 43(31) | 40(24) | 63 | −2.6 |
| Buchnera sp. APS | 44(47) | 6(5) | 3(4) | 47(44) | 32 | −0.9 |
| Campylobacter jejuni | 19(29) | 5(7) | 9(14) | 67(51) | 43 | −1.9 |
| Caulobacter crescentus | 20(18) | 10(30) | 29(34) | 41(19) | 51 | −3.8 |
| Chlamydia pneumoniae CWL029 | 13(24) | 11(12) | 21(18) | 55(45) | 38 | −1.2 |
| Chlamydia trachomatis D/UW-3/Cx | 14(15) | 3(13) | 24(23) | 59(49) | 37 | −1.2 |
| Deinococcus radiodurans | 16(19) | 4(24) | 31(30) | 49(27) | 49 | −3.31 |
| Escherichia coli K12 | 13(26) | 7(17) | 20(28) | 60(28) | 88 | −9.51 |
| Haemophilus influenzae Rd | 9(31) | 4(11) | 26(20) | 61(37) | 57 | −3.91 |
| Helicobacter pylori 26695 | 25(31) | 14(16) | 6(6) | 56(48) | 36 | −1.1 |
| Mycobacterium leprae TN | 27(26) | 31(26) | 29(25) | 13(23) | 45 | −1.4 |
| Mycobacterium tuberculosis | 29(23) | 18(26) | 40(31) | 13(21) | 45 | −1.1 |
| Mycoplasma genitalium G37 | 33(39) | 6(10) | 6(18) | 56(33) | 36 | −2.6 |
| Neisseria meningitidis Z2491 | 15(30) | 20(18) | 35(22) | 30(30) | 46 | −0.9 |
| Pasteurella multocida | 12(32) | 2(13) | 30(18) | 56(37) | 57 | −2.9 |
| Pseudomonas aeruginosa PA01 | 13(24) | 5(22) | 43(31) | 40(24) | 63 | −2.6 |
| Rickettsia prowazekii Madrid E | 39(38) | 3(6) | 3(9) | 55(46) | 33 | −1.1 |
| Staphylococcus aureus N315 | 32(39) | 6(6) | 8(11) | 53(44) | 62 | −1.4 |
| Synechocystis PCC6803 | 32(35) | 7(17) | 22(25) | 39(23) | 41 | −1.2 |
| Thermotoga maritima | 13(27) | 22(36) | 11(14) | 54(23) | 46 | −5.6 |
| Treponema pallidum | 20(13) | 16(30) | 24(28) | 40(28) | 45 | −1.6 |
| Ureaplasma urealyticum | 7(28) | 3(8) | 0(17) | 90(47) | 30 | −6.2 |
| Vibrio cholerae | 8(34) | 2(15) | 27(21) | 63(30) | 98 | −11.1 |
| Xylella fastidiosa | 28(25) | 12(20) | 26(21) | 34(35) | 50 | −0.9 |
| Archaea | ||||||
| Aeropyrum pernix | 16(22) | 28(42) | 14(18) | 42(19) | 50 | −4.1 |
| Archaeoglobus fulgidis 2595 | 78(34) | 11(18) | 2(21) | 9(27) | 46 | −2.8 |
| Methanobacterium_thermoautotrophicum_delta H | 15(29) | 21(17) | 3(18) | 62(37) | 39 | −3.0 |
| Methanococcus jannaschii | 35(28) | 8(25) | 5(9) | 51(39) | 37 | −1.4 |
| Pyrobaculum aerophilum | 50(33) | 35(25) | 13(24) | 2(18) | 46 | −3.0 |
| Pyrococcus abyssi | 26(19) | 2(35) | 20(18) | 52(28) | 46 | −3.6 |
| Pyrococcus furiosus DSM 3638 | 17(21) | 2(33) | 15(13) | 65(33) | 46 | −5.1 |
| Pyrococcus horikoshii OT3 | 19(19) | 4(35) | 13(14) | 64(32) | 47 | −5.2 |
| Sulfolobus solfataricus | 15(37) | 13(16) | 11(8) | 61(39) | 46 | −2.8 |
| Sulfolobus tokodaii | 11(32) | 11(14) | 7(11) | 71(43) | 45 | −4.0 |
| Thermoplasma acidophilum | 33(29) | 9(22) | 16(19) | 42(30) | 45 | −1.5 |
| Thermoplasma volcanium | 31(29) | 4(20) | 13(16) | 51(35) | 45 | −2.0 |
aPercent base composition at N(-1). Parenthetical values are the average composition upstream of tRNA genes (see Materials and Methods).
bValues in bold indicate species with enrichment of U at N(-1).
cValues indicate the number of known tRNA genes in the species.
dIndicated values are the log of the probability of finding the given number of U residues in a random sample, based on the average composition (see Materials and Methods). Values in bold indicate species where U enrichment is significant (p < 0.01).
Information from the Genomic tRNA Database (Lowe and Eddy 1997) and from GenBank (Benson et al. 2003) was used to identify N(-1) from known tRNA genes in species of Bacteria and Archaea with sequenced genomes (see Materials and Methods). As shown in Table 2 ▶, we observe a distinct trend in N(-1) composition that is conserved between Bacteria species. In 22 of the 27 species examined, there is a higher percentage of U at N(-1) than would be expected, given the average base composition upstream of tRNA genes in the species. In 13 of these species, there is a probability of less than 1% of finding the given fraction of U in a random sample. In addition, the preference against having a G at N(-1) is also consistent between species. Of the four bacterial species that we examined that do not show a clear enrichment of U at N(-1), the most notable is Aquifex aeolicus, which has a clear preference for A at N(-1). It would, therefore, be of significant interest to know whether J5/15 and A248 are conserved in this species. Unfortunately, no RNase P RNA sequence from A. aeolicus has yet been identified (Willkomm et al. 2002), thus no comparative analysis with this species is possible. As indicated above, the sequence of J5/15 is also conserved in RNase P RNA from Archaea. As shown in Table 2 ▶, a similar conservation is observed for the N(-1) position of pre-tRNAs from this kingdom. The observed conservation of sequence preference in a large number of Bacteria and Archaea is consistent with a biologically relevant interaction between A248 and N(-1).
DISCUSSION
Identification of active site interactions is essential for understanding RNase P catalysis and specificity and is especially intriguing in light of the range of substrates that this enzyme processes. It has been shown that the protein subunit of RNase P contacts the substrate 5′ leader sequence and interactions have been identified between RNase P RNA and the 3′ RCCA motif and T-loop of pre-tRNA substrates. However, interactions involving the nucleotides immediately flanking the scissile phosphate have not been previously established. Here we provide evidence that the substrate base immediately 5′ of the cleavage site interacts with the central conserved adenosine residue in J5/15 (A248 in E. coli numbering) of RNase P RNA.
Our conclusion that A248 and N(-1) form a functional interaction in the RNase P enzyme–substrate complex is supported by a number of experimental observations. First, mutations at A248 lead to defects in binding, catalysis, and cleavage site selection. This combination of phenotypes is often observed when RNase P enzyme-substrate contacts are disrupted (Kirsebom and Svard 1994; Brannvall et al. 1998). Second, both the binding and miscleavage defects of A248 mutants can be rescued by compensatory mutations at N(-1). In each case the N(-1) mutations that lead to rescue are dependent on the identity of the enzyme nucleotide at position 248. Third, miscleavage can be induced by mutating N(-2) to create a favorable, non-native interaction at this position. Finally, miscleavage and rescue are also observed in the holoenzyme reaction. Together, these observations support the model shown in Figure 8A ▶, in which this interaction aids in RNase P cleavage site selection, both in the ribozyme and holoenzyme.
FIGURE 8.

A model for cleavage site recognition by RNase P. (A) Cartoon depicting interactions between RNase P and pre-tRNA in the region of the cleavage site. The substrate cleavage site is indicated with an arrow. Interactions between RNase P RNA and pre-tRNA are indicated by dashed lines. Interactions which this study implicates in cleavage site selection are in red. (B) Schematic representation of the effect of N(-1) deoxy modification, A248 mutation and N(-1) compensatory mutations on relative free energy leading to correct cleavage or miscleavage. Enzyme and substrate combinations are shown at the bottom of each column. Substrate nucleotides A(-2) N(-1) and G(1) are shown; favorable interactions with the enzyme are shown in blue and the cleavage site is indicated with a red arrow. Column 1 shows complexes leading to correct cleavage (bottom) and miscleavage (top) for the native enzyme and the U(-1) substrate. The correct cleavage complex is proposed to be stabilized by interactions with the 2′-OH at N(-1) and the A248-N(-1) interaction. The effect of the A248U mutation is shown in column 2. The correct cleavage complex is destabilized due to the loss of the A248-N(-1) interaction and the miscleavage complex is stabilized by the engendered A248-N(-2) interaction, but correct cleavage remains energetically favorable. Column 3 shows the proposed result of deletion of the 2′-OH of N(-1), which results in the destabilization of the correct cleavage complex, rendering miscleavage favorable. Column 4 shows the effect of the compensatory U to A mutation at N(-1) that returns the cleavage preference to correct cleavage.
To determine the base-pairing geometry of the A248-N(-1) interaction, we undertook a systematic examination of miscleavage, varying A248 in combination with N(-1) or N(-2) to determine which combinations lead to correct cleavage and miscleavage, respectively (Fig. 5 ▶). The interpretation of this analysis, however, is subject to a number of considerations. When varying N(-1) or N(-2), the substrate position that was not varied was maintained as an adenosine. Thus, the degree of miscleavage reflects an internal competition between N(-1) or N(-2) and an adenosine at N(-2) or N(-1), respectively. In addition, mutations can affect multiple enzyme–substrate contacts. For example, in the substrate a C at N(-1) can pair with G(72), leading to the disruption of the contacts involving G(72) (Kirsebom and Svard 1994; Brannvall et al. 1998). Finally, an interaction near the cleavage site is likely to be influenced by other, as yet undefined, constraints that will restrict which pairs may function. Nonetheless, systematic examination of the functional base combinations provides essential insight into any potential pairing interaction.
Based on the pattern of functional nucleotide combinations at A248 and N(-1) we propose that the simplest structural interpretation is cis Watson-Crick / Watson-Crick pairing. When N(-1) and A248 are varied, all A-U, G-C, and G-U combinations yield significant correct cleavage. Note that although all the ribozymes miscleave the dC(-1) substrate, A248G yields the most correct cleavage. Thus, in the correct cleavage assay, canonical Watson-Crick pairs are functionally reversible. These functional combinations are also mirrored in miscleavage results when N(-2) is varied. Large amounts of miscleavage are observed for the native enzyme with U(-2), A248C with G(-2), and A248U with G(-2). As noted above, miscleavage of the A(-2) substrate by A248U may be reduced by the presence of an A at N(-1). The preference for correct cleavage when potential interactions with N(-1) and N(-2) appear to be equivalent suggests that there is an intrinsic preference for correct cleavage. Nonetheless, the preponderance of functional base combinations can be explained by canonical Watson-Crick base pairs.
In addition to the base combinations noted above, several other combinations lead to correct cleavage when N(-1) is varied or miscleavage when N(-2) is varied. In the correct cleavage assay, the dA(-1) substrate is correctly cleaved by the native and A248G ribozymes. The dU(-1) substrate is also correctly cleaved by the A248C ribozyme, and in the miscleavage assay the A248U ribozyme miscleaves the C(-2) substrate. These three combinations, A-A, A-G, and C-U, can all form noncanonical cis Watson-Crick/Watson-Crick base pairs with greater C1′–C1′ distances than those of canonical pairs (Leontis et al. 2002). No positive results were observed for G-G combinations, which are not allowed in cis Watson-Crick/Watson-Crick pairing, or for C-C and U-U, which yield narrower Watson-Crick pairs. In addition, A-G and C-U pairs are not reversible in either the correct cleavage or miscleavage assays. Because both of these Watson-Crick pairs are geometrically reversible, this implies other constraints beyond base pair geometry. The presence of additional constraints on this interaction is consistent with nucleotide analog interference mapping data that indicate functional groups on the Hoogsteen face of A248 are important for substrate binding (Siew et al. 1999).
As noted above, miscleavage by A248 mutants requires the deletion of the 2′-OH at N(-1). This result can best be explained if the deletion of this 2′-OH selectively destabilizes complexes with the correct phosphodiester bond docked into the active site. As illustrated for miscleavage and rescue for the A248U mutant in Figure 8B ▶, we propose that interactions with both N(-1) and the 2′-OH stabilize the correct cleavage complex (Fig. 8B ▶, column 1). A complex with the incorrect N(-2) base docked into the active site is likely to be intrinsically destabilized relative to the correct complex, and will lack the N(-1) base interaction. Thus, the correct complex has the lowest free energy and only cleavage at the correct site is observed. It is apparent from our data that simple mutation of A248 is not sufficient to significantly destabilize the correct cleavage site relative to the miscleavage site, even if an interaction with N(-2) is possible (Fig. 8B ▶, column 2). However, when the A248U mutant is combined with an N(-1) deoxy substrate (Fig. 8B ▶, column 3), the deletion of the 2′-OH destabilizes the correct cleavage complex relative to the miscleavage complex, leading to predominant miscleavage. Finally, in the presence of the dA(-1) substrate (Fig. 8B ▶, column 4), restoration of a contact between N(-1) and the A248U mutant is restored in the correct complex. This restored interaction causes the correct cleavage complex to once again have a lower free energy, explaining the observed miscleavage rescue.
Importantly, the interaction proposed here is consistent with previous results obtained with other substrates and RNase P RNAs from other species. Several studies have established that mutations at N(-1) affect RNase P function, and that a U at N(-1) is optimal (Kufel and Kirsebom 1996; Crary et al. 1998; Loria and Pan 1998; Loria et al. 1998; Zuleeg et al. 2001; Brannvall et al. 2002). Also, Kirsebom and coworkers have shown that in the context of an RNase P RNA with weak RCCA motif recognition, mutation of N(-2) can lead to miscleavage in a manner consistent with the proposed A248-N(-1) interaction (Brannvall et al. 1998). Finally, Pan and coworkers, examining the cleavage of a yeast tRNAPHE substrate by the RNase P from B. subtilis, showed that a substrate with an AU-rich leader and a U at N(-1), is cleaved at a faster rate than one with a GC-rich leader and a C at N(-1) (Loria and Pan 1998). Thus, the interaction identified here explains a number of observations in the literature regarding the importance of N(-1) and N(-2) for RNase P function.
While our results indicate the presence of an interaction between A248 and N(-1), it is clear that this interaction cannot be considered in isolation. The complex set of interactions that constitute the enzyme’s active site clearly impinge on the role of the A248-N(-1) interaction. For example, the observation that N(-1) is important for the correct processing of several substrates and RNase P species and the conserved preference for U at N(-1) argue that this interaction is a general feature of RNase P substrate recognition. However, the preference for U at N(-1) is not universal, and even in species with a strong preference for U at N(-1) not all pre-tRNAs exhibit this feature. Thus, as shown experimentally, correct processing is not absolutely dependent on the recognition of N(-1). This is consistent with the general observation that RNase P overdefines its cleavage site. That is, although RNase P is capable of recognizing a number of substrate features proximal to the cleavage site, many in vivo substrates possess only a subset of these features. In species such as B. subtilis none of the important features proximal to the cleavage site are found in all pre-tRNA molecules. Thus, it seems likely that only a subset of the possible interactions, such as the A248-N(-1) interaction, need be present for correct processing by RNase P.
The interaction described here is notable in that it takes place immediately adjacent to the scissile phosphate, and therefore defines part of the RNase P active site. In addition, the findings presented here are important in that they indicate that the RNase P RNA subunit, like the protein subunit, directly contacts the 5′ leader sequence. In a series of studies, Fierke and colleagues found that the B. subtilis RNase P protein directly contacts the 5′ leaders of pre-tRNAs (Crary et al. 1998; Kurz et al. 1998; Loria et al. 1998; Niranjanakumari et al. 1998). In addition, Fierke and colleagues noted that a 5′ leader, consisting of a single nucleotide, decreases substrate binding affinity relative to the tRNA product (Crary et al. 1998). They propose that this destabilization is caused by unfavorable ground state interactions that are offset by favorable interactions involving other nucleotides in the 5′ leader, in a manner similar to the case of the group I intron (Narlikar et al. 1995; Crary et al. 1998). Given the direct involvement of N(-1), the source of the observed destabilization, in the interaction described here, it seems likely that the A248–N(-1) interaction plays a role in this process. Taken together, these observations highlight the intimate relationship between RNA and protein subunits at this critical interface in the enzyme–substrate complex.
MATERIALS AND METHODS
General methods
Reagents were obtained commercially unless otherwise specified. RNA oligonucleotides were purchased from Dharmacon, Inc. RNase P RNAs were synthesized using run-off transcription according to standard protocols (Kaye et al. 2002). Pre-tRNA substrates were generated by ligating a 5′ end-labeled oligonucleotide encoding substrate tRNAASP nucleotides −5 to +9 to an oligonucleotide encoding nucleotides +10 to +77 according to standard protocols (Moore and Sharp 1992; Loria and Pan 1997). RNAs were renatured by incubating at 50°C for 50 min, followed by 30 min at 37°C before use. Unless otherwise noted, reactions were carried out in 1 M ammonium acetate, 25 mM MgCl2, and 20 mM PIPES at pH 6.0 at 37°C.
Analysis of ribozyme binding and kinetics
Single turnover kinetic analyses were performed as described (Siew et al. 1999). Rates were measured for two concentrations of enzyme, generally 1 and 5 μM, although higher concentrations were used as necessary. Enzyme concentrations were considered to be saturating if the two observed rates differed by less than the uncertainty in the measurements. Binding affinities were measured using gel mobility shift, as described previously (Kaye et al. 2002). In the case of the native enzyme and some mutants, tight binding made it difficult to maintain the substrate concentration significantly lower than the lowest enzyme concentration. In these cases, experiments were performed with a final substrate concentration of 2 nM, and the resulting data fit to Equation 1,
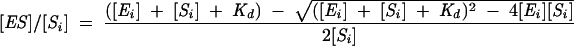 |
(1) |
where [ES]/[Si] is the fraction of the substrate that is shifted, [Ei] is the total enzyme concentration, [Si] is the total substrate concentration, and Kd,obs is the observed dissociation constant (Pleiss and Uhlenbeck 2001).
Analysis of ribozyme cleavage specificity
Reactions were performed under standard conditions, using either ligated substrates or annealed substrates. Annealed substrate reactions took advantage of the ability of the oligonucleotides for pre-tRNAASP positions −5 to +9 and +10 to +77 to form a potential substrate through base-pairing interactions without being ligated. These two oligonucleotides are capable of forming seven intermolecular base pairs in the acceptor stem and it has been shown previously that RNase P efficiently recognizes such bipartite substrates (Hansen et al. 2001).
Ribozyme miscleavage reactions contained 2 μM RNase P RNA. For ligated substrates, the final substrate concentration was 50 nM. Annealed substrate reactions contained 50 nM of the −5 to +9 oligonucleotide and 100 nM of +10 to +77 oligonucleotide, which were renatured together. Reactions were allowed to proceed overnight and products were separated by 22.5% denaturing PAGE.
For holoenzyme reactions, full-length E. coli C5 protein was purified using the IMPACT T7 protein purification system from New England Biolabs, and stored in buffer containing 50% glycerol, 10 mM Tris at pH 8.0, 0.1 mM EDTA, 30 mM DTT, 0.1% Triton X-100, and 0.5 M NaCl. Enzyme and substrate RNAs were renatured separately in 100 mM NaCl, 17.5 mM MgCl2, and 50 mM Tris at pH 8.0 at 37°C. The C5 protein was added to RNase P RNAs 10 min before adding substrates. Reactions were terminated after 10 min. Final reaction conditions were 5% glycerol, 50 mM Tris at pH 8.0, 0.01 mM EDTA, 3 mM DTT, 0.01% Triton X-100, 100 mM NaCl, 17.5 mM MgCl2, 20 nM RNase P RNA, 25 nM RNase P protein, and 25 nM pre-tRNAASP. Reactions with annealed substrates contained 25 nM of the labeled 5′ oligonucleotide and 50 nM of the 3′ tRNA oligonucleotide.
Analysis of pre-tRNA leader sequences in Bacteria and Archaea
Sequences upstream of tRNA genes in Bacteria and Archaea were identified using information from The Genomic tRNA Database (Lowe and Eddy 1997; http://rna.wustl.edu/GtRDB/) and genome sequences available from GenBank (Benson et al. 2003). A series of scripts using gawk (http://www.gnu.org/software/gawk) located the start site of each tRNA in the genome, confirmed the location of the tRNA and reported the sequence of eight nucleotides, N(-8) through N(-1), upstream of each tRNA. Copies of scripts are available upon request. Equation 2 was used to calculate the probability, p, of finding the given number of uridine residues at N(-1), r, given the frequency of uridine in N(-1) through N(-8), |gP, and the number of tRNA genes in the species, n (Rosner 2000).
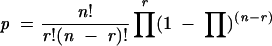 |
(2) |
Acknowledgments
We thank the members of the Harris laboratory for support and critical reading of the manuscript. This work was supported by National Institutes of Health (NIH) grant GM56742 to MEH. NHZ was supported by NIH training grant GM008056.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.5220703.
REFERENCES
- Benson, D.A., Karsch-Mizrachi, I., Lipman, D.J., Ostell, J., and Wheeler, D.L. 2003. GenBank. Nucleic Acids Res. 31: 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannvall, M., Mattsson, J.G., Svard, S.G., and Kirsebom, L.A. 1998. RNase P RNA structure and cleavage reflect the primary structure of tRNA genes. J. Mol. Biol. 283: 771–783. [DOI] [PubMed] [Google Scholar]
- Brannvall, M., Fredrik Pettersson, B.M., and Kirsebom, L.A. 2002. The residue immediately upstream of the RNase P cleavage site is a positive determinant. Biochimie 84: 693–703. [DOI] [PubMed] [Google Scholar]
- Brown, J.W. 1999. The ribonuclease P database. Nucleic Acids Res. 27: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgin, A.B. and Pace, N.R. 1990. Mapping the active site of ribonuclease P RNA using a substrate containing a photoaffinity agent. EMBO J. 9: 4111–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian, E.L. and Harris, M.E. 1999. The track of the pre-tRNA 5′ leader in the ribonuclease P ribozyme–substrate complex. Biochemistry 38: 12629–12638. [DOI] [PubMed] [Google Scholar]
- Christian, E.L., McPheeters, D.S., and Harris, M.E. 1998. Identification of individual nucleotides in the bacterial ribonuclease P ribozyme adjacent to the pre-tRNA cleavage site by short-range photo-cross-linking. Biochemistry 37: 17618–17628. [DOI] [PubMed] [Google Scholar]
- Christian, E.L., Zahler, N.H., Kaye, N.M., and Harris, M.E. 2002. Analysis of substrate recognition by the ribonucleoprotein endonuclease RNase P. Methods 28: 307–322. [DOI] [PubMed] [Google Scholar]
- Crary, S.M., Niranjanakumari, S., and Fierke, C.A. 1998. The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5′ leader sequence of pre-tRNAAsp. Biochemistry 37: 9409–9416. [DOI] [PubMed] [Google Scholar]
- Frank, D.N. and Pace, N.R. 1998. Ribonuclease P: Unity and diversity in a tRNA processing ribozyme. Annu Rev Biochem 67: 153–180. [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada, C. and Altman, S. 1984. Catalytic activity of an RNA molecule prepared by transcription in vitro. Science 223: 285–286. [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada, C., Gardiner, K., Marsh, T., Pace, N., and Altman, S. 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857. [DOI] [PubMed] [Google Scholar]
- Hansen, A., Pfeiffer, T., Zuleeg, T., Limmer, S., Ciesiolka, J., Feltens, R., and Hartmann, R.K. 2001. Exploring the minimal substrate requirements for trans-cleavage by RNase P holoenzymes from Escherichia coli and Bacillus subtilis. Mol. Microbiol. 41: 131–143. [DOI] [PubMed] [Google Scholar]
- Hardt, W.D., Erdmann, V.A., and Hartmann, R.K. 1996. Rp-deoxy-phosphorothioate modification interference experiments identify 2′-OH groups in RNase P RNA that are crucial to tRNA binding. RNA 2: 1189–1198. [PMC free article] [PubMed] [Google Scholar]
- Heide, C., Feltens, R., and Hartmann, R.K. 2001. Purine N7 groups that are crucial to the interaction of Escherichia coli RNase P RNA with tRNA. RNA 7: 958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye, N.M., Christian, E.L., and Harris, M.E. 2002. NAIM and site-specific functional group modification analysis of RNase P RNA: Magnesium dependent structure within the conserved P1–P4 multi-helix junction contributes to catalysis. Biochemistry 41: 4533–4545. [DOI] [PubMed] [Google Scholar]
- Kirsebom, L.A. and Svard, S.G. 1994. Base pairing between Escherichia coli RNase P RNA and its substrate. EMBO J. 13: 4870–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knitt, D.S., Narlikar, G.J., and Herschlag, D. 1994. Dissection of the role of the conserved G.U pair in group I RNA self-splicing. Biochemistry 33: 13864–13879. [DOI] [PubMed] [Google Scholar]
- Kufel, J. and Kirsebom, L.A. 1996. Different cleavage sites are aligned differently in the active site of M1 RNA, the catalytic subunit of Escherichia coli RNase P. Proc. Natl. Acad. Sci. 93: 6085–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz, J.C. and Fierke, C.A. 2000. Ribonuclease P: A ribonucleoprotein enzyme. Curr. Opin. Chem. Biol. 4: 553–558. [DOI] [PubMed] [Google Scholar]
- Kurz, J.C., Niranjanakumari, S., and Fierke, C.A. 1998. Protein component of Bacillus subtilis RNase P specifically enhances the affinity for precursor-tRNAAsp. Biochemistry 37: 2393–2400. [DOI] [PubMed] [Google Scholar]
- Leontis, N.B., Stombaugh, J., and Westhof, E. 2002. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 30: 3497–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria, A. and Pan, T. 1997. Recognition of the T stem-loop of a pre-tRNA substrate by the ribozyme from Bacillus subtilis ribonuclease P. Biochemistry 36: 6317–6325. [DOI] [PubMed] [Google Scholar]
- ———. 1998. Recognition of the 5′ leader and the acceptor stem of a pre-tRNA substrate by the ribozyme from Bacillus subtilis RNase P. Biochemistry 37: 10126–10133. [DOI] [PubMed] [Google Scholar]
- ———. 1999. The cleavage step of ribonuclease P catalysis is determined by ribozyme–substrate interactions both distal and proximal to the cleavage site. Biochemistry 38: 8612–8620. [DOI] [PubMed] [Google Scholar]
- Loria, A., Niranjanakumari, S., Fierke, C.A., and Pan, T. 1998. Recognition of a pre-tRNA substrate by the Bacillus subtilis RNase P holoenzyme. Biochemistry 37: 15466–15473. [DOI] [PubMed] [Google Scholar]
- Lowe, T.M. and Eddy, S.R. 1997. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25: 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, M.J. and Sharp, P.A. 1992. Site-specific modification of pre-mRNA: The 2′-hydroxyl groups at the splice sites. Science 256: 992–997. [DOI] [PubMed] [Google Scholar]
- Narlikar, G.J. and Herschlag, D. 1997. Mechanistic aspects of enzymatic catalysis: Lessons from comparison of RNA and protein enzymes. Annu. Rev. Biochem. 66: 19–59. [DOI] [PubMed] [Google Scholar]
- ———. 1998. Direct demonstration of the catalytic role of binding interactions in an enzymatic reaction. Biochemistry 37: 9902–9911. [DOI] [PubMed] [Google Scholar]
- Narlikar, G.J., Gopalakrishnan, V., McConnell, T.S., Usman, N., and Herschlag, D. 1995. Use of binding energy by an RNA enzyme for catalysis by positioning and substrate destabilization. Proc. Natl. Acad. Sci. 92: 3668–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjanakumari, S., Stams, T., Crary, S.M., Christianson, D.W., and Fierke, C.A. 1998. Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc. Natl. Acad. Sci. 95: 15212–15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, B.K. and Pace, N.R. 1994. Interaction of the 3′-end of tRNA with ribonuclease P RNA. Nucleic Acids Res. 22: 4087–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, N.R. and Brown, J.W. 1995. Evolutionary perspective on the structure and function of ribonuclease P, a ribozyme. J. Bacteriol. 177: 1919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, T., Loria, A., and Zhong, K. 1995. Probing of tertiary interactions in RNA: 2′-hydroxyl-base contacts between the RNase P RNA and pre-tRNA. Proc. Natl. Acad. Sci. 92: 12510–12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss, J.A. and Uhlenbeck, O.C. 2001. Identification of thermodynamically relevant interactions between EF-Tu and backbone elements of tRNA. J. Mol. Biol. 308: 895–905. [DOI] [PubMed] [Google Scholar]
- Pyle, A.M., Moran, S., Strobel, S.A., Chapman, T., Turner, D.H., and Cech, T.R. 1994. Replacement of the conserved G.U with a G-C pair at the cleavage site of the Tetrahymena ribozyme decreases binding, reactivity, and fidelity. Biochemistry 33: 13856–13863. [DOI] [PubMed] [Google Scholar]
- Rosner, B. 2000. Fundamentals of biostatistics. Duxbury, Pacific Grove, CA.
- Siew, D., Zahler, N.H., Cassano, A.G., Strobel, S.A., and Harris, M.E. 1999. Identification of adenosine functional groups involved in substrate binding by the ribonuclease P ribozyme. Biochemistry 38: 1873–1883. [DOI] [PubMed] [Google Scholar]
- Smith, D. and Pace, N.R. 1993. Multiple magnesium ions in the ribonuclease P reaction mechanism. Biochemistry 32: 5273–5281. [DOI] [PubMed] [Google Scholar]
- Su, L.J., Qin, P.Z., Michels, W.J., and Pyle, A.M. 2001. Guiding ribozyme cleavage through motif recognition: The mechanism of cleavage site selection by a group ii intron ribozyme. J. Mol. Biol. 306: 655–668. [DOI] [PubMed] [Google Scholar]
- Tallsjo, A., Kufel, J., and Kirsebom, L.A. 1996. Interaction between Escherichia coli RNase P RNA and the discriminator base results in slow product release. RNA 2: 299–307. [PMC free article] [PubMed] [Google Scholar]
- Willkomm, D.K., Feltens, R., and Hartmann, R.K. 2002. tRNA maturation in Aquifex aeolicus. Biochimie 84: 713–722. [DOI] [PubMed] [Google Scholar]
- Zuleeg, T., Hansen, A., Pfeiffer, T., Schubel, H., Kreutzer, R., Hartmann, R.K., and Limmer, S. 2001. Correlation between processing efficiency for ribonuclease P minimal substrates and conformation of the nucleotide-1 at the cleavage position. Biochemistry 40: 3363–3369. [DOI] [PubMed] [Google Scholar]