

Abstract
We describe a strategy for the ultra-sensitive detection of nucleic acids using “half” ribozymes that are devoid of catalytic activity unless completed by a trans-acting target nucleic acid. The half-ribozyme concept was initially demonstrated using a construct derived from a multiple turnover Class I ligase. Iterative RNA selection was carried out to evolve this half-ribozyme into one activated by a conserved sequence present in the hepatitis C virus (HCV) genome. Following sequence optimization of substrate RNAs, this HCV-activated half-ribozyme displayed a maximal turnover rate of 69 min−1 (pH 8.3) and was induced in rate by approximately 2.6 × 109-fold by the HCV target. It detected the HCV target oligonucleotide in the zeptomole range (6700 molecules), a sensitivity of detection roughly 2.6 × 106-fold greater than that previously demonstrated by oligonucleotide-activated ribozymes, and one that is sufficient for molecular diagnostic applications.
Keywords: Ribozyme, diagnostic, nucleic acid, hepatitis C virus, riboswitch
INTRODUCTION
Many reports have described ribozymes whose activity is modulated by molecular recognition of an oligonucleotide (Lorsch and Szostak 1995; Porta and Lizardi 1995; Komatsu et al. 1999; Robertson and Ellington 1999; Kuwabara et al. 2000; Stojanovic et al. 2001; Wang and Sen 2001; Burke et al. 2002; Iyo et al. 2002; Komatsu et al. 2002), a protein (Robertson and Ellington 2001; Hartig et al. 2002; Vaish et al. 2002; Wang and Sen 2002), or a small molecule (Tang and Breaker 1997; Araki et al. 1998; Koizumi et al. 1999; Soukup and Breaker 1999; Robertson and Ellington 2000; Wang et al. 2002a). In these systems, molecular recognition is directly converted into catalytic events that can be monitored in homogeneous solution phase assays (Hartig et al. 2002; Vaish et al. 2002; Mei et al. 2003), ELISA-like assays (Seetharaman et al. 2001), array-based assays (Hesselberth et al. 2003), or by RT-PCR (Robertson and Ellington 1999). Consequently, such reagents have been proposed for a variety of biosensor applications (Hoffman et al. 2001; Breaker 2002; Rajendran and Ellington 2002).
Nucleic acid-activated ribozymes are most commonly produced through strategies in which a target oligonucleotide induces a conformational rearrangement of the ribozyme to properly orient active site residues for catalysis. In this allosteric strategy, the rate enhancement provided by a saturating amount of an oligonucleotide target (a maximum of 10,000-fold [Robertson and Ellington 1999]) reflects the change in the equilibrium distribution of the ribozyme between active and inactive conformer(s) induced by target. In a second “expansive” strategy, Watson-Crick association of a substrate RNA with a ribozyme (or deoxyribozyme) is enhanced by a target oligonucleotide, thereby increasing the observed rate at concentrations of substrate that are not saturating in the absence of target (Wang and Sen 2001; Wang et al. 2002b).
The allosteric and “expanded” ribozymes reported to date promote substantial catalysis in the absence of target (Lorsch and Szostak 1995; Porta and Lizardi 1995; Komatsu et al. 1999; Robertson and Ellington 1999; Kuwabara et al. 2000; Stojanovic et al. 2001; Wang and Sen 2001; Burke et al. 2002; Iyo et al. 2002; Komatsu et al. 2002). Such target-independent catalysis limits the ability of these ribozymes to detect nucleic acid targets present in substoichiometric amounts because low levels of a target result in only a marginal increase in observed rate.
As an example of the limitations on substoichiometric detection imposed by target-independent catalysis, consider a target-bound ribozyme induced 250-fold in rate by a nucleic acid target. More than one of every 250 ribozymes must be activated by target to increase the observed rate twofold over that in the absence of target. Signal in the presence versus the absence of target would not appreciably change when less than one of every 500 ribozymes was activated. Thus, assuming a concentration of 1 μM ribozyme is required to rapidly and quantitatively bind the target, the limit of detection (LOD) using this ribozyme would be 2 nM (or 1.2 × 1010 target molecules in 10 μL).
We describe the development of a new class of target-activated ribozymes that are completely devoid of catalytic activity in the absence of an oligonucleotide target. These ribozymes are distinct from other nucleic acid-activated ribozymes because the LOD they afford is dictated by the substantial rate difference between catalysis promoted by the target-bound ribozyme and the corresponding uncatalyzed reaction, rather than the rate of target-independent ribozyme catalysis. We show that such ribozymes can be used to detect a clinically relevant oligonucleotide target present in the zeptomole range.
RESULTS
Half-ribozymes
To produce a ribozyme that was essentially devoid of catalytic activity in the absence of an oligonucleotide target, we developed a “half-ribozyme” approach in which a target nucleic acid served to complete a partial ribozyme that lacked residues absolutely essential for catalysis. Although in principle this approach is general to large ribozymes that use extensive Watson-Crick base pairs, we developed it using the extensively characterized Class I ligase motif of Bartel and colleagues (Ekland and Bartel 1995; Ekland et al. 1995; Bergman et al. 2000; Glasner et al. 2000, 2002).
To derive a half-ribozyme from a multiple turnover version of a Class I ligase, the 207t construct (Bergman et al. 2000) was 3′ truncated in the loop that terminates the P7 helix (Fig. 1A ▶). The sequence 3′ to the truncation point serves as a trans-acting oligonucleotide target. It was anticipated that such a severe truncation would completely debilitate the half-ribozyme. In addition, this system has the advantage that the bimolecular substrate RNA complex should not appreciably interact with the half-ribozyme in the absence of target because it interacts (through Watson-Crick base pairs) only with target sequence in the target-half-ribozyme complex (Fig. 1A ▶). Thus, the half-ribozyme design scheme incorporates elements of both allosteric and “expansive” approaches.
FIGURE 1.

A half-ribozyme derived from the Class I ligase. (A) A half-ribozyme based on the sequence of the 207t Class I ligase (black) was derived from the constitutive ligase by introducing a nick (dashed line) so that a trans-acting target nucleic acid (green) will associate and allow ligation (red arrow) of a substrate RNA with a 3′ cis-diol (SOH, red) to a substrate RNA with a 5′-triphosphate (pppS, blue). Note that pppS and SOH form a bimolecular substrate RNA complex that interacts with the half-ribozyme-target complex. Nomenclature for paired regions (P1–P7) is analogous to that used for the constitutive ligase. (B) Observed rate of multiple turnover ligation promoted by the 207t half-ribozyme in the presence (closed red circle) versus the absence (open red circle) of bound target relative to that observed without half-ribozyme and target (blue square). Reaction conditions were as follows: buffer (30 mM Tris-HCl at pH 7.5; 60 mM MgCl2), 1.0 μM half-ribozyme, 0.1 μM target, 25 μM each SOH and pppS.
Kinetic analysis of the nicked 207t construct demonstrated that half-ribozymes are significantly activated by an oligonucleotide target (Fig. 1B ▶). Target-bound half-ribozyme displayed a multiple turnover rate of 0.2 min−1 (Fig. 1B ▶). In the absence of target, longer time courses demonstrated that the 207t half-ribozyme did not stimulate substrate RNA ligation above the rate of ligation attributed to the intrinsic chemical reactivity of the substrate RNA complex (kobs = 5.0 × 10−8 min−1 versus 5.4 × 10−8 min−1 in the presence or absence of ribozyme, respectively). Thus, this half-ribozyme was devoid of activity in the absence of target and displayed an observed rate increase of approximately 4 × 106-fold when bound to target.
Half-ribozymes activated by hepatitis C virus (HCV) sequences
To determine whether clinically relevant oligonucleotide sequences could activate half-ribozyme catalysis, we designed a half-ribozyme that could interact with a sequence in the genome of HCV. To develop a half-ribozyme that recognized the broadest range of HCV genotypes possible, a highly conserved sequence of 41 nucleotides within the 5′-UTR was chosen as a half-ribozyme target (nucleotide positions −177 to −137 by conventional numbering; Fig. 2A ▶).
FIGURE 2.

Half-ribozyme that interacts with HCV sequences. (A) Conserved sequence of HCV 5′-UTR used as target oligonucleotide (green) and minor sequence variants (red) as a sequence logo (Schneider and Stephens 1990). The height of each letter represents its absolute number of occurrences in ~1500 GenBank entries. (B) Half-ribozyme (black) was engineered to associate with target nucleic acid (green) and to direct the ligation of a substrate RNA with a 3′ cis-diol (SOH, red) to a substrate RNA with a 5′-triphosphate (pppS, blue).
The 207t half-ribozyme was redesigned in order to recognize this HCV target by changing the sequence of paired regions P4, P7, and the sequence of the 3′ substrate (pppS) that interacts with the HCV target to form P2 (Fig. 2B ▶). Because many of the positions that were changed are highly conserved in the Class I ligase motif (Ekland and Bartel 1995), it was not surprising that the rate of the HCV-activated mutant half-ribozyme (kobs < 1 × 10−6 min−1, data not shown) was greatly diminished relative to target-activated 207t half-ribozyme (kobs = 0.2 min−1; Fig. 1B ▶).
Iterative RNA selection
The observed rate of the HCV-activated half-ribozyme was increased through an iterative RNA selection scheme similar to that previously described (Ekland and Bartel 1995), but one that required the HCV target to activate catalysis. The half-ribozyme library used in this iterative RNA selection carried sequence variation in a total of 62 nucleotide positions of the half-ribozyme that were not required to form Watson-Crick base pairs with substrate RNA or the HCV target (Fig. 3A ▶). Each of the ~1010 half-ribozyme sequence variants in this library carried an average of 18 changes relative to the parental HCV-half-ribozyme.
FIGURE 3.

HCV-activated half-ribozyme from initial selection. (A) Half-ribozyme sequence library (black and yellow) that performs autoligation for use in iterative RNA selection carried non-parental sequence at 62 positions (yellow) and can interact with the HCV target (green) to direct ligation of an extended SOH substrate RNA that allows PCR (extension not shown). Helix P7 was extended by 17 nucleotides relative to the 207t half-ribozyme (Fig. 1A ▶) and the initial HCV half-ribozyme (Fig. 2B ▶) to provide a nonrandomized sequence of sufficient length to allow reverse transcription. (B) Sequence of the clone 8/7 half-ribozyme isolated from iterative selection. Input sequence (panel A) is black and sequence changes relative to it are indicated (yellow). Three different multiple turnover versions of the clone 8/7 half-ribozyme were constructed by 5′ truncation (numbered blue arrows). Two guanosine residues were added to the 5′ end of each multiple turnover half-ribozyme to allow efficient transcription by T7 RNA polymerase (not shown). (C) Observed rate of autoligation promoted by the clone 8/7 half-ribozyme as a function of pH. Reaction conditions: buffer (30 mM MES at pH 6.0 and 6.5 or Tris-HCl at pH 7.0–8.3, 60 mM MgCl2), 1 μM half-ribozyme, 1.1 μM target, trace SOH. (D) Observed rate of autoligation as a function of the concentration of MgCl2 of the clone 8/7 half-ribozyme (red circles) and the final half-ribozyme sequence library obtained from a second iterative RNA selection (blue squares; see text and Fig. 4 ▶). Reaction conditions: buffer (30 mM Tris-HCl at pH 7.5; MgCl2 as indicated), 1 μM half-ribozyme, 1.1 μM target, trace SOH.
In successive cycles of selection, HCV-activated half-ribozyme complexes with increased catalytic rates were selected by decreasing the time allowed for ligation of the half-ribozyme library to substrate RNA. Iterative RNA selection was terminated when the library of half-ribozyme complexes displayed an autoligation rate of 0.03 min−1 (data not shown), a rate similar to that displayed by the final library of the original iterative RNA selection from which the Class I ligase was produced (Bartel and Szostak 1993).
The kinetic properties and sequences of 33 half-ribozymes isolated from this iterative RNA selection were characterized. All selected half-ribozymes comprised one sequence family (Fig. 3B ▶) and promoted autoligation at ~1.5 to 2 min−1 in the presence of a stoichiometric amount of the HCV target (Fig. 3C ▶; data not shown). The optimal half-ribozyme obtained from iterative selection, clone 8/7, contained 10 nucleotide changes relative to the input sequence and one nucleotide deletion (Fig. 3B ▶, yellow residues). The most highly conserved nucleotide change in this sequence family is a G-to-U transversion in J3/4 that changes the identity of a nucleotide that is universally conserved in the Class I ligase and all of its functional variants (Ekland and Bartel 1995, 1996; Wright and Joyce 1997; Johnston et al. 2001; McGinness et al. 2002). Although all members of the clone 8/7 half-ribozyme family carried sequence changes in P6, a helix that is highly conserved in the Class I motif (Ekland and Bartel 1995), these changes were not conserved among family members. All family members also carried distinct changes in the portion of J1/3 that is not conserved in the Class I motif.
To examine whether a chemical or nonchemical step determined the observed rate of the clone 8/7 half-ribozyme, we measured kobs as a function of pH because the chemical step of ligation promoted by the Class I ligase ribozyme is log-linear with pH (Bergman et al. 2000; Glasner et al. 2002). The rate of this half-ribozyme displayed a log-linear increase up to pH 7.0 and then reached at plateau at ~2 min−1 (Fig. 3C ▶). Other members of this half-ribozyme sequence family displayed similar profiles (data not shown). Such a plateau indicates that the rate of autoligation of this half-ribozyme is limited by a nonchemical step proceeding at ~2 min−1.
Divalent metal ions control the folding and stability of the Class I motif and are essential for catalysis (Glasner et al. 2002). Because magnesium was the only metal ion used in the iterative RNA selection from which the clone 8/7 half-ribozyme was developed, the dependence of kobs on magnesium ion concentration was examined. Indeed, the autoligation rate of the HCV target-bound clone 8/7 half-ribozyme increased as a function of magnesium at all concentrations tested (Fig. 3D ▶). Given that a nonchemical step appears to limit kobs at this pH (Fig. 3C ▶), we hypothesized that the catalytically competent structure of the clone 8/7 half-ribozyme was not fully formed or that a catalytically essential magnesium ion binding site was not fully saturated at even the highest concentration tested.
We predicted that any difficulty in forming the catalytic conformation of the autoligation construct would be exacerbated when it was converted to a multiple turnover catalyst. Nevertheless, the clone 8/7 half-ribozyme was 5′ truncated at various positions to produce several different half-ribozymes for multiple turnover reactions (blue arrows in Fig. 3B ▶). The sequence 5′ of this position was supplied as a trans-acting pppS substrate RNA. As expected, the observed rate of all of these constructs was greatly reduced relative to the rate of autoligation in both single turnover and multiple turnover regimes (kobs < 10−4 min−1, data not shown). We hypothesized that these low turnover rates were due to a combination of factors including weak magnesium ion binding (Fig. 3D ▶) and weak binding or alternate folding of the various RNA components. Therefore, a second iterative RNA selection was undertaken to optimize the folding of this half-ribozyme and further increase its catalytic activity.
Four new libraries of sequence variants based on the clone 8/7 half-ribozyme were produced for this second iterative RNA selection (Fig. 4A ▶). The first library contained completely randomized sequence at all single-stranded positions except those whose identity changed in the selection of the 8/7 half-ribozyme. The other three pools carried an additional domain of random sequence inserted into the 8/7 HCV-half-ribozyme at one of three distinct locations. The first library contained 1010 different sequences, all possible sequence combinations, whereas the latter three each consisted of ~1 × 1015 sequence variants but sampled only a fraction of sequence space.
FIGURE 4.

Efficient HCV sequence-activated multiple turnover half-ribozymes. (A) Four sequence libraries (libraries 1–4) based on the autoligation version of the clone 8/7 half-ribozyme (black) maintained the nucleotide changes in the clone 8/7 half-ribozyme sequence relative to the 207t half-ribozyme (yellow). Library 1 carried completely randomized sequence in all other single-stranded positions (blue highlight). Libraries 2–4 carried seven positions that were randomized at a 6% frequency (boxed) and an inserted random sequence domain (blue boxes). The four libraries were 3′ truncated relative to the clone 8/7 half-ribozyme and were used with an appropriately truncated HCV target sequence (green) because a reverse transcription primer could adequately anneal to a defined sequence internal to that used in the initial iterative RNA selection. SOH was extended at its 5′ end to allow for PCR amplification (not shown). (B) The clone 21 half-ribozyme isolated from iterative selection maintained all of the sequence changes produced in the clone 8/7 half-ribozyme isolated from the initial iterative RNA selection (yellow) and carried an inserted region that, on examining all members of this sequence family, had a conserved portion (blue) and a variable portion (purple). Two conserved changes were present in J3/4 (blue). An alternate P3 helix allowed by the inserted region is shown and was tested using mutations (pink) in the 3′ side of P3 (M1) and sequence, allowing either the alternate (M2) or original (M3) P3 base-pairing arrangement. Three different multiple turnover versions of the clone 21 half-ribozyme were constructed by 5′ truncation (numbered blue arrows). Two guanosine residues were added to the 5′ end of each multiple turnover half-ribozyme to allow efficient transcription by T7 RNA polymerase (not shown). Joining regions J1/3 and J3/4 are indicated (gray). (C) Time course of autoligation of the clone 21 half-ribozyme (red circles) or variants either carrying mutations M1 and M2 (blue squares) or mutations M1 and M3 (green triangles). Reaction conditions: buffer (30 mM Tris-HCl at pH 7.5, 10 mM MgCl2), 1 μM half-ribozyme, 1.1 μM target, trace SOH. (D) Observed rate of multiple turnover ligation promoted by the clone 21 half-ribozyme in the presence (solid red circle) or absence (open red circle) of bound target relative to that observed without half-ribozyme or target (blue square). Reaction conditions: buffer (30 mM Tris-HCl at pH 7.5, 60 mM MgCl2), 1 μM half-ribozyme, 0.01 μM target, 10 μM each SOH and pppS.
The four random sequence libraries were used separately in an iterative RNA selection scheme similar to our original selection, but one that required catalysis at progressively lower concentrations of MgCl2 (20 mM to 1 mM). Interestingly, the library with randomized single-stranded regions, but containing no additional sequence, failed to show activity greater than the clone 8/7 half-ribozyme as the magnesium ion concentration was decreased. In contrast, the activity of the other three pools increased with each round (data not shown). To obtain the best performing half-ribozymes from these latter three sequence libraries, we mixed all three together after six cycles of selection and subjected them to two additional cycles. Iterative RNA selection was terminated after the second pooled selection cycle. At that point, the pooled half-ribozyme libraries displayed a single turnover rate of 0.02 min−1 at 1 mM MgCl2 (data not shown), a rate greatly enhanced from the starting sequence pool at this magnesium concentration (<10−5 min−1, data not shown).
The rate of autoligation of the final pool of sequences was determined as a function of MgCl2 concentration and compared with similar data obtained for the clone 8/7 half-ribozyme (Fig. 3D ▶). At all MgCl2 concentrations tested, the rates of ligation promoted by the pooled half-ribozyme libraries obtained from the second iterative selection were dramatically greater than those displayed by the clone 8/7 half-ribozyme. Indeed, observed rates of the pooled half-ribozyme libraries were too fast to measure at Mg2+ concentrations above 30 mM (>12 min−1). Thus, iterative RNA selection using libraries containing an additional sequence “domain” relative to the 207t half-ribozyme yielded catalysts with greatly increased rates at reduced MgCl2 concentrations, whereas the library without additional sequence did not.
The rates of autoligation promoted by 80 individual half-ribozymes derived from the second iterative RNA selection were characterized under suboptimal conditions so that rates were slowed enough to permit quantification. Twenty-one of these individuals displayed a rate slightly greater than the pooled half-ribozyme libraries and comprised one family of related sequences derived from library 3 (Fig. 4B ▶; data not shown). Like the pooled libraries, the optimal half-ribozyme from this sequence family, clone 21, displayed an observed autoligation rate too fast to measure (>12 min−1) and a plateau level of ligation of 58% at pH 7.5, 60 mM MgCl2 (data not shown).
The conserved sequence in the inserted region of the clone 21 half-ribozyme family allows an alternate P3 pairing relative to the clone 8/7 and the 207t half-ribozymes (Fig. 4B ▶). This alternate pairing shifts a portion of the inserted region 3′ to P3, dramatically lengthening J3/4 and changing its position relative to J1/3. Because both of these joining regions are highly conserved in the Class I ribozyme (Ekland and Bartel 1995), this alternate P3 pairing may change the architecture of the clone 21 half-ribozyme relative to the constitutive Class I ribozyme.
To directly test whether the alternate P3 pairing interaction was required for catalysis, we made three nucleotide changes in the 3′ side of P3 (M1, Fig. 4B ▶) and combined them either with mutations that restore the alternate pairing (M2, Fig. 4B ▶) or with the original pairing (M3, Fig. 4B ▶). At the magnesium ion concentration used in this experiment (10 mM), the original clone 21 half-ribozyme displayed biphasic kinetics (Fig. 4C ▶). The first phase of the reaction occurs with a rate essentially identical to the constitutive Class I ligase when assayed at an identical magnesium ion concentration and pH (3.6 min−1 versus 3.9 min−1, respectively (Ekland and Bartel 1995). A variant of the clone 21 half-ribozyme that allows the alternate pairing but not the original (M1/M2, Fig. 4B ▶) displayed a first phase rate of 2.8 min−1 and a plateau slightly lower than the original clone 21 half-ribozyme. In contrast, a variant allowing the original P3 helix (M1/M3, Fig. 4B ▶) was devoid of activity (kobs < 10−4 min−1). Thus, the catalytic conformation of the clone 21 half-ribozyme relies on this alternate P3 base-pair configuration and is only modestly sensitive to its sequence composition.
In addition to the inserted region, most members of the clone 21 sequence family carried A-to-U and A-to-C transversions in two highly conserved J3/4 positions (Fig. 4B ▶; Ekland and Bartel 1995). Thus, our iterative selections ultimately yielded a half-ribozyme carrying changes in three of the four nucleotides in J3/4 that were previously universally conserved among Class I ligase variants isolated from several independent iterative RNA selections (Ekland and Bartel 1995, 1996; Wright and Joyce 1997; Johnston et al. 2001; McGinness et al. 2002).
Several multiple turnover configurations of the clone 21 half-ribozyme were developed by 5′ truncation at various positions in a fashion identical to that used in the production of multiple turnover variants of the clone 8/7 half-ribozyme (blue arrows in Figs. 3B ▶ and 4B ▶, respectively). Again, the sequence 5′ to the indicated position was supplied as a trans-acting pppS substrate RNA. All multiple turnover configurations of the clone 21 half-ribozyme displayed turnover rates of 0.7 min−1 at a saturating concentration of substrate RNA and an equimolar concentration of target (Fig. 4D ▶; data not shown), greater than a 7000-fold improvement over the multiple turnover rate of the clone 8/7 half-ribozyme. Like the 207t half-ribozyme, the observed rate of substrate RNA ligation in the absence of its target oligonucleotide was essentially equivalent to the rate of uncatalyzed ligation of the bimolecular substrate RNA complex (3.5 × 10−8 min−1 and 4.4 × 10−8 min−1 in the presence versus absence of half-ribozyme, respectively; Fig. 4D ▶), indicating that the clone 21 half-ribozyme was completely devoid of catalytic activity in the absence of its target. Thus, the multiple turnover rate of the clone 21 half-ribozyme is stimulated ~2 × 107-fold by the HCV target.
Optimization of the substrate RNAs improves turnover rate
Multiple turnover rates of HCV target-bound clone 21 half-ribozyme (Fig. 4D ▶) were more than 17-fold lower than its observed rate of autoligation (Fig. 4C ▶). We investigated which step limited the turnover rate of this half-ribozyme and whether the rate of that step could be improved. To that end, the observed rate of ligation promoted by target-bound clone 21 half-ribozyme was determined in a single turnover regime to define whether the rate-limiting step occurred before or after the first catalytic cycle (Fig. 5A ▶). The first catalytic cycle was too fast to accurately measure, and indistinguishable from the rate of autoligation, when the two substrates were preannealed in water. Unexpectedly, the rate of the first catalytic cycle decreased to 1.1 min−1 if the substrate RNAs were preequilibrated in reaction buffer. [Note: This rate is equivalent to the turnover rate when adjusted for the fraction of active ribozyme (58%; Fig. 5A ▶).] Further studies identified MgCl2 as the buffer component responsible for this phenomenon (data not shown). Thus, the reduced rate of multiple turnover catalysis relative to autoligation could result from a MgCl2-dependent phenomenon involving the substrate RNA complex—not from issues relating to the functional characteristics of the clone 21 half-ribozyme itself.
FIGURE 5.

Characterization and optimization of clone 21 in multiple turnover configuration 3. (A) The rate of the initial catalytic cycle of multiple turnover configuration 3 (blue) was compared with the rate of autoligation (red) when their respective substrate RNAs were preincubated as indicated. Reaction conditions: buffer (30 mM Tris-HCl at pH 7.5, 60 mM MgCl2). Autoligation reactions contained 1 μM half-ribozyme, 1.1 μM target, and trace SOH; reactions using the multiple turnover half-ribozyme contained 10 μM half-ribozyme, 10 μM and 1 μM each SOH and pppS. (B) Turnover rates of configuration 3 afforded by mutant SOH and pppS substrate RNAs expressed relative to the initial substrate RNA pair (red and blue). P2 interaction with target nucleic acid (black line) and potential wobble base pairs (green highlight) are indicated. Circles indicate relative rates of mutations further characterized. Reaction conditions: buffer (30 mM Tris-HCl at pH 7.5, 100 mM KCl, 60 mM MgCl2), 1 μM half-ribozyme, 0.01 μM target, 10 μM each SOH and pppS. (C) Turnover rate of configuration 3 afforded by original (red circles), C8U/flip-13 (blue squares), or C8U/flip-13/A5G (green diamonds) substrate RNA pairs as a function of substrate RNA concentration. Reaction conditions were the same as panel B, except the concentration of SOH and pppS varied, as indicated. (D) Turnover rate of configuration 3 using the C8U/flip-13/A5G substrate RNA pair as a function of MgCl2 and KCl concentrations. Reaction conditions: buffer (30 mM Tris-HCl at pH 7.5, MgCl2, and KCl concentration varied as indicated), 1 μM half-ribozyme, 0.001 μM target, 1 μM each SOH and pppS. (E) Maximum turnover rate of multiple turnover configuration 3 using the C8U/flip-13/A5G substrate RNA pair as a function of pH inferred from Lineweaver-Burk analysis (closed red circles) compared with direct measurement of rate in the absence of target nucleic acid at identical pH values (open red circles).
Following this hypothesis, we attempted to abrogate the effect of buffer using sequence variants of the two substrate RNAs. The turnover rates of 46 mutant substrates were determined (Fig. 5B ▶). Only two afforded turnover rates greater than the original substrate pair (each an increase of ~1.5-fold). The favorable changes were C8U in P2 and a base pair “flip” in P1 that exchanges the identity of the 3′-most nucleotide of SOH and its pairing partner in pppS (flip-13). Notably, a SOH and pppS substrate RNA pair that contained both C8U and flip-13 mutations afforded a turnover rate that was slightly greater than the sum of the two individual mutants, resulting in a turnover rate that was 3.4-fold greater than the initial substrate pair (Fig. 5B ▶).
Because the C8U/flip-13 substrate pair introduced a wobble base pair in P2 that could affect its affinity for the target-half-ribozyme complex, its activity as a function of substrate RNA concentration was compared with the original substrate RNA pair (Fig. 5C ▶). Significantly, the multiple turnover rate of the clone 21 half-ribozyme bound to target did not appreciably change as the concentration of the original substrate pair was varied between 100 nM to 20 μM. In contrast, the turnover rate of target-bound clone 21 half-ribozyme increased as the concentration of the C8U/flip-13 substrate pair was increased, displaying a turnover rate of 3.4 min−1 at 20 μM substrate RNA. A triple mutant (C8U/flip-13/A5G) that was predicted by an RNA folding program (Xia et al. 1998) to have an increased affinity (0.5 kcal/mole) for the target relative to the C8U/flip-13 substrate RNA pair showed an increase in activity relative to the double mutant at all substrate RNA concentrations examined. Thus, further efforts to optimize turnover rate were focused on the C8U/flip-13/A5G substrate RNA pair.
To identify reaction conditions that increased the affinity of the C8U/flip-13/A5G substrate RNA pair and therefore increased clone 21 half-ribozyme activity at lower concentrations of substrate RNA (i.e., increasing kmax/ KM), we examined the kinetic performance of the half-ribozyme at various concentrations of monovalent and divalent metal ions (Fig. 5D ▶). These results indicated that turnover rate increased as ionic strength increased (compare kobs at increasing KCl concentrations; Fig. 5D ▶). Optimal MgCl2 concentration was ~150 mM. We interpreted these data to reflect both a magnesium ion-specific effect and an ionic strength effect on turnover rate through a modulation of substrate RNA affinity. At optimal salt concentrations (0.9 M KCl, 150 mM MgCl2), the turnover rate of the ribozyme increased to 2.6 min−1 at 1 μM substrate complex, representing an eightfold increase in rate relative to the original substrate in the original buffer condition (Fig. 5C ▶).
Using the C8U/flip-13/A5G substrate RNA pair and the optimized metal ion concentrations, kobs was determined as a function of substrate RNA concentration and pH. Lineweaver-Burk analysis of these data was used to generate KM and kmax at each pH (data not shown). KM of the substrate complex varied little from pH 6.0 to pH 8.3 (from 5 μM to 16 μM, data not shown). When adjusted for the active fraction of the target-bound clone 21 half-ribozyme (84% from single turnover kinetics, data not shown), kmax is predicted to be 8.1 min−1 at the lowest pH tested and 69.0 min−1 at the highest pH tested (Fig. 5E ▶). Maximal rate did not display a log-linear relationship with pH, but instead increased roughly eightfold over slightly more than two pH units (Fig. 5E ▶). Thus, in contrast to the constitutive Class I ligase (Bergman et al. 2000; Glasner et al. 2002), the multiple turnover rate of ligation afforded by the C8U/flip-13/A5G substrate RNA pair is not limited solely by a hydroxide ion-dependent chemical step.
The uncatalyzed rate of ligation of the C8U/flip-13/A5G substrate RNA pair in the absence of target was also determined as a function of pH (Fig. 5E ▶). As expected, the rate of the uncatalyzed reaction displayed a log-linear increase with pH. The difference in the rate of substrate RNA ligation in the presence versus absence of target, therefore, ranged from 2.6 × 109-fold at pH 6.0 to 1.7 × 108-fold at pH 8.3. These HCV-target induced rate differentials are comparable to the rate enhancement provided by the constitutive Class I ligase (Bergman et al. 2000; Glasner et al. 2002).
Limit of detection for HCV target sequence
Because half-ribozymes are devoid of catalytic activity in the absence of their targets (Figs. 1B ▶, 4B ▶), the LOD they afford is determined by the difference in the rate of product formation as a result of target-activated half-ribozyme catalysis and the rate of product formation through uncatalyzed ligation. The concentration of the bimolecular substrate RNA complex affects both of these rates, but not proportionally. Therefore, the LOD will vary with the concentration of the substrate RNA complex. To determine the substrate RNA concentration that afforded the lowest possible LOD, we used the kinetic analysis of the clone 21 half-ribozyme described earlier (Fig. 5E ▶) to develop a mathematical model to predict how the LOD would vary with substrate RNA concentration and pH.
Both catalyzed and uncatalyzed rates can be derived from rate equations; the former is given by:
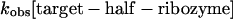 |
(1) |
and the latter is given by:
 |
(2) |
If the concentration of target-half-ribozyme is taken to equal the total concentration of target (and will be if the target is quantitatively captured), then target increases the amount of ligation product twofold above that seen in its absence when:
 |
(3) |
kobs at any substrate RNA concentration is defined by kmax and KM parameters using the Michaelis-Menten relationship:
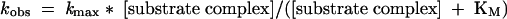 |
(4) |
Substitution of Equation 4 into Equation 3 and rearranging allows solution for the concentration of target-half-ribozyme (which is taken as the concentration of target), where the signal due to catalyzed and uncatalyzed ligation are equal:
 |
(5) |
Equation 5 allows the LOD to be determined as a function of substrate RNA concentration from known kuncat, kmax, and KM values. Solution of Equation 5 gives the target concentration yielding a twofold increase in product formation compared with that observed in the absence of target. We define the LOD as half this number of target molecules, that is, the number of target molecules that produce an amount of ligation product that is indistinguishable from the amount produced in its absence. Equation 5 was used to calculate LOD (as number of HCV molecules) at various pH values and substrate RNA concentrations (Fig. 6A ▶) after substrate RNA concentration was converted to number of molecules in a reaction of a specific volume (5 μL in this case). Importantly, the kmax value used to solve Equation 5 reflected the total concentration of target-activated half-ribozyme and not the fraction of target-bound half ribozyme that was catalytically competent.
FIGURE 6.

Limit of detection of HCV sequence. (A) Calculated LOD was determined by solving Equation 5 (text) after substrate RNA concentration was converted to number of molecules using kinetic parameters established for clone 21 half-ribozyme in configuration 3 and substrate RNA pair C8U/flip-13/A5G. Calculated LOD is expressed as a function of substrate RNA concentration and pH. The intersection of lines on the surface of the graph indicates specific substrate RNA concentrations and pH values. The surface corresponding to 104 and 105 target molecules is indicated (dashed white line). The condition used to experimentally determine LOD is indicated (blue circle). (B) Ligation product from duplicate reactions, examining product formation as a function of HCV target copy number. The minor species migrating more rapidly than the major species observed in some lanes is derived from N−1 pppS substrate RNA generated from in vitro transcription and was not used in quantification. The large amount of unligated SOH carrying radiolabel was well separated from ligated product (not shown). Reaction conditions: buffer (30 mM MES at pH 6.0, 150 mM MgCl2, and 0.9 M KCl), 1 μM half-ribozyme, target as indicated, 0.1 μM each SOH and pppS. (C) Quantification of product formation as a function of HCV target copy number from four half-ribozyme reactions: two reactions each from two independent serial dilutions. Dashed red line with arrowhead indicates LOD extrapolated from a power function fit to signal from 107 to 104 HCV copies to signal observed in the absence of HCV target. Standard deviation from four separate trials amounted to <10% of the average half-ribozyme activity in the presence of 107 to 104 molecules and <15% in reactions containing 0, 102, and 103 target molecules. Power function fit to signal observed with 107 to 104 HCV molecules with R2 = 0.99946.
As expected, the calculated LOD improves as pH is lowered because the uncatalyzed reaction displays a logarithmic increase with pH but the catalyzed reaction does not (Fig. 5E ▶). Interestingly, the calculated LOD increases (becomes less sensitive) as substrate RNA concentration approaches and exceeds KM because the amount of product formed through uncatalyzed ligation increases to a greater extent than the increase in product formation provided by the enhanced rate of half-ribozyme catalysis. As substrate RNA concentration is lowered, the calculated LOD asymptotically approaches 6900 target molecules. Thus, the calculated LOD is maximized at concentrations of substrate RNA that actually attenuate half-ribozyme catalysis. However, because the maximal LOD is asymptotically approached as substrate RNA concentration is decreased, a substrate RNA concentration can be identified that supports significant ribozyme activity but does not appreciably compromise LOD. For example, target-bound clone 21 half-ribozyme is predicted to display a turnover rate of 0.13 min−1 and an LOD of 7050 HCV molecules at 100 nM substrate RNA in a 5-μL reaction buffered at pH 6.0.
To test the validity of this method of calculating the LOD, we determined the amount of ligation product produced by the clone 21 HCV-half-ribozyme as a function of HCV target copy number in 5-μL reactions carried out at pH 6.0, 100 nM substrate RNA (Fig. 6B ▶). Ligation product above background was clearly evident with as few as 104 copies of the HCV target (3.3 fM; Fig. 6B ▶). Half-ribozyme catalysis quantitatively detected target amounts ranging from 104 copies to the highest amount tested (107 molecules; Fig. 6C ▶). Extrapolated turnover rates in each of these reactions averaged to 0.16 ± 0.01 min−1. These values are in concordance with the calculated rate obtained from the LOD model (0.13 min−1) and therefore serve to validate the model. As expected, the amount of ligation product decreases with a log–log relationship to target copy number.
The LOD was defined as the intercept of a power (xy) function fit to data from 107 to 104 molecules with the average background signal observed in the absence of the HCV target. This method of defining the LOD is in direct analogy to that used in the model to calculate the theoretical LOD. The resultant value of 6700 HCV molecules (~11 zeptomoles, ~2.2 fM) is in excellent agreement with the LOD calculated in the model (7050 molecules, Fig. 6A ▶). As expected from the theoretical model (Fig. 6A ▶), signal observed in the presence of 1000 and 100 HCV molecules was indistinguishable from the signal observed in the absence of HCV target oligonucleotide (Fig. 6B,C ▶). Thus, half-ribozymes afford detection of oligonucleotide targets present in the zeptomole range, and this detection sensitivity can be accurately predicted from their kinetic properties.
DISCUSSION
Half-ribozymes afford billion-fold rate enhancements
We developed half-ribozymes by 3′-truncation of the Class I ligase (Figs. 1B ▶, 4B ▶). A trans-acting nucleic acid target completes half-ribozymes and thereby allows ribozyme catalysis. Because interaction with target is absolutely essential for catalysis (Figs. 1B ▶, 3D ▶), half-ribozymes are both qualitatively and quantitatively different from other oligonucleotide-activated ribozymes.
Half-ribozymes are qualitatively different because the rate change afforded by target reflects the rate differential between target-induced catalysis and the uncatalyzed reaction (not the target-dependent and target-independent rates of the ribozyme). Thus, unlike previous approaches, the intrinsic chemical reactivity of the substrates is the only source of product in the absence of target. Consequently, to optimize performance, it is important to choose a ribozyme for this approach that not only displays a fast absolute rate, but also uses substrates with low intrinsic chemical reactivity.
The magnitude of the rate difference in the presence versus absence of target makes this approach quantitatively different from other efforts. Previously, the maximum rate difference in the presence versus absence of an oligonucleotide target was 104-fold (Robertson and Ellington 1999). The initial half-ribozyme based on the 207t ligase displayed a multiple turnover rate ~4 × 106-fold greater than the rate of uncatalyzed ligation (Fig. 1B ▶), and the target-bound clone 21 half-ribozyme enhanced the rate of product formation ~2.6 × 109-fold after optimization of the sequence of its substrate RNAs (Fig. 5 ▶). Thus, the half-ribozyme approach affords target-induced rate enhancements that are 2.6 × 105-fold greater than the previously reported maximum (Robertson and Ellington 1999) and roughly 5 × 106-fold greater than more typical rate enhancements (Burke et al. 2002; Komatsu et al. 2002; Wang et al. 2002b).
Although the half-ribozyme approach is most easily applied to ribozymes activated by oligonucleotides, a similar mechanism of activation might be available for protein-activated ribozymes. Robertson and Ellington (2001) reported a protein-activated ligase whose autoligation rate was increased 7.5 × 104-fold by a saturating amount of its protein target, representing the largest induction of ribozyme activity by a target prior to the development of half-ribozymes. The rate of target-independent catalysis was very close to the uncatalyzed rate of reaction of similar substrates assayed under similar conditions [compare 3.3 × 10−7 min−1 at pH 7.7 (Robertson and Ellington 2001) to Fig. 3 ▶ in Rohatgi et al. 1996]. This protein-activated ribozyme was developed through an iterative RNA selection scheme that used a ribozyme sequence library in which the catalytic core was replaced with random sequence that directed protein binding. It was suggested to use functional groups from the protein target to produce a “nucleoprotein” catalyst (Robertson and Ellington 2001). Thus, target-assisted catalysis may be a strategy available to ribozymes activated by both nucleic acids (this work) and proteins (Robertson and Ellington 2001). Such systems are analogous to protein enzymes engineered so that a substrate RNA is required to supply functional groups to complete an enzyme’s active site (Carter and Wells 1987).
Remodeling of the Class I ligase motif
Using iterative RNA selection techniques and biochemical optimization, an HCV sequence-responsive half-ribozyme was developed that displayed an autoligation rate indistinguishable from the parent ligase (to the limits of kinetic measurements by manual pipeting; Fig. 3D ▶ and Glasner et al. 2002). The multiple turnover rate of a version of this half-ribozyme was also equivalent to, or greater than, the multiple turnover rate of the constitutive Class I ligase (Fig. 5E ▶; Bergman et al. 2000; Glasner et al. 2002). Thus, iterative RNA selection was effectively used to develop a half-ribozyme that was activated by an HCV oligonucleotide sequence, indicating that half-ribozymes activated by an arbitrarily chosen RNA target sequence can be produced.
Although the double pseudoknotted structure of the constitutive Class I motif is evident in the clone 21 half-ribozyme, it is likely that dramatic remodeling was required to confer the capacity to respond to the HCV target. Most interestingly, insertion of an additional sequence “domain” was required to produce HCV-activated half-ribozymes active in low concentrations of magnesium ions (Fig. 3D ▶). Because higher concentrations of magnesium ions permit the folding and association of otherwise unstable RNA structures (Reich et al. 1988), it is reasonable to assume that the additional sequence in the clone 21 family promoted folding. The hammerhead ribozyme has recently been shown to display enhanced function at reduced magnesium ion concentrations when adjacent viroid genomic sequences are added to it (Fedor and Westhof 2002). Thus, the iterative RNA selection that yielded these HCV-activated half-ribozymes may have accessed a strategy used in nature to stabilize otherwise loosely ordered ribozymes, namely, the use of nonessential domains to enhance the proper ordering of active site residues.
In further contrast to the constitutive Class I ligase, highly conserved positions are changed in the clone 21 half-ribozyme. For example, universally conserved nucleotides 5′-AGAG (positions 44 to 47) in the constitutive motif (Ekland and Bartel 1995) are changed to 5′-UGCU (Fig. 4B ▶). However, the nearly invariant decanucleotide sequence 5′-AAAAAGACAAAU-3′ that immediately precedes P3 is maintained in sequence and position in the clone 21 half-ribozyme. In effect, selective pressure “pushed” the inserted domain 3′ of the P3 helix. Selective pressure also resulted in changes in conserved Watson-Crick pairing identities in P6 and potentially lengthened P3.
Kinetic analysis of target-bound clone 21 half-ribozyme activity with the variant substrate RNA pairs indicates that substrate RNA complex recognition and utilization is not identical to that reported for the constitutively active Class I ligase (Ekland and Bartel 1995; Ekland et al. 1995; Bergman et al. 2000; Glasner et al. 2000, 2002). Most strikingly, mutation of A3 only slightly decreased (A3G and A3C) or increased (A3U) activity, although this position is highly conserved in the Class I ligase. Similarly, the most severe decrease in activity displayed by mutation of G2 was a 0.3-fold reduction, although it, too, is highly conserved in the constitutive motif. The P2 region tolerated G-U wobble base pairs only in a subset of P2 locations, indicating that, unlike the Class I ligase, it cannot be altered in sequence. Thus, not only is it likely that the HCV-activated half-ribozyme has an architecture that differs from the constitutive motif, but it also appears to recognize its substrate RNAs differently.
HCV sequence detection
The HCV-activated half-ribozyme described here affords the greatest sensitivity of detection reported to date for an oligonucleotide-activated ribozyme. Its LOD for the HCV target is 6700 molecules (Fig. 6C ▶), representing an improvement of ~2.6 × 106-fold in LOD (Robertson and Ellington 1999). The HCV-activated half-ribozyme LOD was accurately predicted from the fundamental kinetic parameters of target-mediated (kcat and KM) and uncatalyzed ligation (kuncat; Fig. 6A ▶). Importantly, the mathematical model we constructed to predict LOD (Equation 5) allows exploration of half-ribozyme LOD under a variety of conditions that alter its kinetic performance (such as substrate RNA concentration, pH, and magnesium ion concentration), as long as its kinetic characteristics under these conditions are known. Thus, we provide a theoretical framework to examine detection of substoichiometric targets by half-ribozymes.
This model emphasizes that the LOD is controlled by the ratio of target-induced and background ligation rates; data confirm that the LOD is limited by background ligation that obscures the effector-dependent signal at extremely low effector concentrations (Fig. 6B,C ▶). The lack of target-independent ribozyme catalysis allows the target-bound HCV-activated half-ribozyme to attain a 2.6 × 109-fold rate enhancement; a dramatic improvement relative to the previously maximum rate enhancement of 10,000-fold (Robertson and Ellington 1999). Thus, the significant improvement of LOD afforded by half-ribozymes relative to previously reported target-activated ribozymes is due to the low level of target-independent product accumulation relative to that observed in other systems.
The model we have developed can be applied easily to a ribozyme that promotes catalysis in the absence of target, provided that a rate equation describing target-independent ribozyme catalysis replaces the rate equation for uncatalyzed reaction (see Equation 3). Therefore, this model is generally applicable to calculate the LOD of any target-activated ribozyme, provided that its kinetic characteristics in the absence of target are known. In direct analogy to the half-ribozyme system, sensitive detection using such a ribozyme is still predicted to require a fast target-mediated rate and a low rate in the absence of target.
The close agreement between experimentally determined LODs and those predicted by our mathematical model indicates that the sensitivity of the HCV-activated half-ribozyme can be improved by modulating the kinetic parameters of the ribozyme. Most important, we note that the affinity of the substrate complex for the ribozyme is low (ranging from 4 μM at pH 6.0 to 16 μM at pH 8.3, data not shown), requiring a high concentration of substrate RNA for maximal catalytic activity. As predicted by our mathematical model, an increased kmax/ KM would improve the LOD because lower substrate RNA concentrations would support more target-bound half-ribozyme activity. At the same time, the ability to work at lower substrate concentrations would lower background ligation. An increase in the KM of the substrate RNA complex may be possible through the incorporation of modified nucleotides in pppS that stabilize P2. Substitution of A residues in P2 with 2,6-diamino-A, U with 2-thio U, or inclusion of 2′-O-CH3 nucleotides all represent viable routes to this end because such modifications stabilize nucleic acid duplexes (Lebedev et al. 1996; Kumar and Davis 1997; Verma and Eckstein 1998). If such substitutions allow the half-ribozyme to function at its maximal rate at pH 6.0 with 100 nM substrate (kmax = 6.8 min−1 when not corrected for the fraction of active target-half-ribozyme), rather than at 0.6 min−1, then the LOD is predicted to improve by a proportional amount (from 6700 molecules [Fig. 6C ▶] to 100 molecules).
Improvement in LOD is also possible in different assay formats. Most significantly, we note that the amount of product formed from the uncatalyzed reaction is proportional to assay volume, whereas the amount of product formed from half-ribozyme catalysis is not. Consequently, microfluidic devices that decrease the reaction volume should improve the LOD. Thus, although the half-ribozyme system we report here already affords an LOD that greatly exceeds previously reported oligonucleotide-activated ribozymes (for example, see Robertson and Ellington 1999), avenues to even greater sensitivity of detection using this system should be available.
Applications
Applications ranging from basic research to molecular diagnostics require ultrasensitive detection of nucleic acids. Several technologies are currently used for these purposes, most notably polymerase chain reaction (PCR), transcription-mediated amplification (TMA), and rolling circle amplification. Although PCR, TMA, and rolling circle amplification offer excellent sensitivity, their use in clinical diagnostic applications is problematic because they are time-consuming assays that require technically skilled operators and are difficult to run in high throughput (Conry-Cantilena 1997). Consequently, a major goal in molecular diagnostics is to develop methods that afford sensitive detection in a simple assay that can be run in high throughput.
The half-ribozyme approach described here permits sensitive detection of nucleic acid targets in a simple isothermal assay. Because the two substrate RNAs used by half-ribozymes can be made by solid-phase oligonucleotide synthesis, each can be independently and site specifically labeled, allowing the half-ribozyme system to be integrated with numerous assay formats. For example, ELISA-like detection is possible if one substrate RNA is labeled with a hapten and the second is labeled with a capture agent such as biotin. In this format, half-ribozyme assays could be run in high-throughput mode in commercially available autosamplers currently used for immunoassays. Such a format has the benefit of incorporating an additional signal amplification step through hapten detection.
Half-ribozymes are well suited for applications in which many samples are screened for only a limited number of targets, applications such as the screening of blood products for HIV, HCV, and other viral contaminants. The need for such a technology is clear because the immunodiagnostic methods currently used for this purpose offer limited sensitivity (Majid and Gretch 2002). Given that half-ribozymes currently display a sensitivity of detection approaching validated PCR assays (Dufour 2002) and can be formatted in ELISA-like assays compatible with established immunodiagnostic platforms, this technology holds promise for such applications.
MATERIALS AND METHODS
RNA preparation
SOH substrate RNAs were produced through standard oligoribonucleotide synthesis procedures. pppS substrate RNAs were made by in vitro T7 RNA polymerase transcription of the annealed oligonucleotides DNA (5′-GCTAATACGACTCACTATAGGAAATCCAAACGACTGGTACAA-3′ and 5′-TTGTACCAGTCGTTTGGATTTCCTATAGTGAGTCGTATTAGC-3′). pppS sequence variants were produced from versions of these oligonucleotides that carried the appropriate sequence changes. Half-ribozymes were produced by in vitro T7 RNA polymerase transcription of two overlapping antiparallel DNA oligonucleotides that were first extended to completion with Taq polymerase. Their sequences were 5′-GCTAATACGACTCACTATAGGAAAAGACAAATGTGCCCTCAGAGC-3′ and 5′-AAAGCCACCGGGGGGTGCCTCCCCTGGATCCGAAGATCTTCTCAAGCTCTGAGGGCACATTTGTCTT-3′ (207t); 5′-GCTAATACGACTCACTATAGGAAATCCAAACGACTGGTACAAAAAAGACAAATGTGCCCTCAGATCAA GGAC-3′ and 5′-CCAGGACGACTGCAGGGTGCCACCTGTAGATCCGAAGATCGGTCCTTGATCTGAGGGCAC-3′ (autoligation version of clone 8/7). The autoligation version of clone 21 was prepared by colony PCR using oligonucleotides 5′-GCTAATACGACTCACTATAGGAAATCCAAACGACTGGTAC-3′ and 5′-CCAGGACGACTGCAGGGTGCC-3′ as primers. Other bacterial clones obtained following iterative RNA selection were produced by PCR using these same primers. The underlined sequence indicates T7 RNA polymerase promoter. Multiple turnover versions of clone 8/7 and clone 21 half-ribozymes were produced using oligonucleotides 5′-GCTAATACGACTCACTATAGGTACAAAAAAAGACAAATGTGC-3′, 5′-GCTAATACGACTCACTATAGGAAAAAAAGACAAATGTGC-3′, and 5′-GCTAATACGACTCACTATAGGAAAAAGACAAATGTGC-3′ (configuration 1, 2, and 3, respectively) and 3′ primer identical to that used to produce autoligation constructs.
Kinetic assays
Kinetic assays were performed at 23°C in 30 mM MES (pH 6.0, 6.5) or Tris-HCl (pH 7.0, 7.5, 8.0, 8.3) as indicated, which included MgCl2 and KCl as indicated. Half-ribozyme and target were heated at 80°C for 2 min., 5X buffer was added, and the reaction was allowed to cool to 23°C over 5 min. Reactions were initiated by addition of substrate RNAs. Native gel analysis was used to confirm that 100% of added target was bound.
Autoligation
A trace concentration of 5′-32P-labeled SOH (<5 nM) was incubated with 1 μM of the appropriate half-ribozyme and 1.1 μM target nucleic acid. Aliquots were quenched with gel loading dye at different times depending on half-ribozyme rate. Observed single turnover rates were determined by fitting the quantified data either to a linear equation (fraction ligated versus time) or to the single exponential equation: fraction ligated = Fa(1−e−kt), where t equals time, k equals rate of catalysis, and Fa equals the fraction of ligated SOH at completion. A double exponential fit was used in cases where biphasic kinetics were evident: fraction ligated = Fa(1−e−k1t) + Fb(1−e−k2t), where k1 and k2 represent the rate constants for the first and second phase of the reaction, respectively. Data were fit using Kalidagraph (Synergy Software).
Multiple turnover constructs in single turnover regime
Reactions (10–20 μL) included 1 μM each SOH and pppS (plus trace 5′-radiolabeled SOH), 10 μM half-ribozyme, and 10 μM target nucleic acid. Aliquots were quenched with gel loading dye at different times depending on half-ribozyme rate. Observed single turnover rates were determined by fitting data as for autoligation.
Multiple turnover constructs in multiple turnover regime
Reactions (10–20 μL) included SOH and pppS (trace 5′-radiolabeled SOH), half-ribozyme, and target nucleic acid as indicated. Aliquots were quenched with gel loading dye at different times depending on half-ribozyme rate. Observed turnover rates were calculated from the initial phase of the reaction (<20% substrate converted to ligated product) and fit to [ligated product]/([target-half-ribozyme]*time). Michaelis-Menten parameters were established by varying concentration of SOH/pppS substrate RNA complex and data fit to v = [E][S]kmax/(KM+[S]), where v equals rate at each [S], S represents SOH/pppS substrate RNA complex concentration, KM equals apparent binding constant for half-maximal activation, and kmax equals the maximal catalytic rate.
Iterative RNA selections
Initial iterative RNA selection
The sequence library was derived from the 207 Class I ligase (Bergman et al. 2000; Glasner et al. 2002). The library contained a central region of 62 positions mutagenized to 30% that was flanked on both sides by a constant sequence region (5′-ACACCG GAATTGCCAGGACGACCGgggggtgcctcccctggatccgaagatcggtcctt gctctgagggcacatttgtcttttacgGTACCAGTCGTTTGGATTTCC-3′; lowercase indicates mutagenized positions). The library was amplified in a 5-mL PCR reaction with primers used to amplify the clone 8/7 half-ribozyme (see earlier). Library complexity was 1 × 1010. One nanomole of the DNA library was transcribed in 2 mL and RNA purified on a 10% polyacrylamide gel. The initial round of selection was carried out with 2 × 1015 molecules (4 nmoles) of half-ribozyme library RNA and 1.1 equivalents of the HCV target RNA (5′-ACACCGGAAUUGCCAGGACGACCGGGUCCUUUCUUG GAUAA-3′). Reactions were assembled by heating the library and target to 80°C in water for 3 min and cooling to room temperature for 10 min. 2× selection buffer was added (final buffer conditions: 30 mM Tris at pH 7.5, 200 mM KCl, 0.6 mM EDTA, and 60 mM MgCl2), along with 2.2 equivalents of a radiolabeled and 5′ extended SOH variant that allowed visualization of ligation product and product-specific PCR (5′-AAGCAUCUAAGCAUCUCAAG CAAACCAGUC-3′). After a defined period of incubation, reactions were stopped by addition of gel loading dye, and half-ribozymes ligated to SOH were purified on a 10% polyacrylamide/7 M urea gel. Purified RNA was amplified by RT-PCR with primers specific for the 5′ and the 3′ constant sequence, and the T7 promoter was restored through nested PCR. A total of eight rounds of selection were performed. At each round, selection stringency was increased by progressively decreasing the ligation time from 16 h to 15 sec. A negative selection step entailing 20 h incubation in the absence of target was introduced in the forth round to prevent the amplification of target-independent half-ribozymes.
Secondary iterative RNA selection
Single-stranded DNAs representing four distinct half-ribozyme sequence libraries were synthesized based on the clone 8/7 half-ribozyme. Library 1 was made from a single oligonucleotide (5′-CAGGACGACTGCAGGGTGCCACCTGNAGATCCGAAGATCGGTCCTTNA-N6-GGCAC-N11-TTGTACCAGTCGTTTGGATTTCC-3′). Libraries 2–4 were made by extension of overlapping oligonucleotides (library 2: 5′-GGAAATCCAAACGACTGGTACAA-N26-AAAAGACAAATGTGCCCTCAGATCAAGGACCGATCTTCGGATCTACAGG-3′ and 5′-CCAGGACGACTGCAGGGTGCCACCTGTAGATCCGAAGATCGGTCC-3′, library 3: 5′-GGAAATCCAAACGACTGGTACAAAAAAGACAAAT-N26-GTGCCCTCAGATCAAGGACCGATCTTCGGATCTACAGG-3′ and 5′-CCAGGACGACTGCAGGGTGCCACCTGTAGATCCGAAGATCGGTCC-3′, library 4: 5′-CCAGGACGACTGCAGGGTGCCACCTGTAGATC-N30-GATCGGTCCTTGATCTGAGGGCACATTTGTCTTTTTTG-3′ and 5′-GCTAATACGACTCACTATAGGAAATCCAAACGACTGGTACAAAAAAGACAAATGTGCC-3′). All four libraries were amplified in a 5-mL PCR reaction using a 5′ primer that added the T7 RNA polymerase promoter (5′-GCTAATACGACTCACTATAGGAAATCCAAACGACTGGTAC-3′) and a 3′ primer (5′-CCAGGACGACTGCAGGGTGCC-3′). Final library complexity was 2.7 × 1011 (library 1) and ~3 × 1014 (libraries 2–4). Approximately 0.6 nmole of each library was transcribed separately in 1 mL and RNA purified independently on a 10% polyacrylamide gel. Four parallel selections were carried out; each started with ~1.6 nmole of half-ribozyme library. Because manual pipetting may have limited our ability to select for half-ribozymes with the fastest catalytic rates represented in the libraries, selection was carried out at pH 6.0, with the hypothesis that half-ribozymes selected at pH 6.0 might display higher rates at a higher pH. Reactions were assembled by heating each of the four half-ribozyme libraries and 1.1 equivalents of truncated HCV target RNA (5′-CCAGGACGACCGGGUCCUUUCUUGGAUAA-3′) in water to 80°C for 3 min. Following addition of 4× buffer (final buffer conditions: 30 mM MES at pH 6.0, 0.6 mM EDTA and 0.1% NP40, variable MgCl2), reactions were initiated by addition of 2.2 equivalents of radiolabeled SOH. Reactions were further processed as described in initial iterative RNA selection. MgCl2 concentration (20 mM in round 1 to 3 mM in round 8) and incubation time (10 min in round 1 to 5 sec in round 8) were progressively decreased to increase selective pressure. A negative selection step entailing a 20-h incubation step was introduced in rounds 5, 7, and 8 to prevent the amplification of target-independent half-ribozymes.
Cloning and sequencing
DNA from the final round of each selection was cloned into Escherichia coli using TOPO TA cloning kit (Invitrogen). Cloned DNA from individual colonies was amplified by the colony PCR method using M13 forward and M13 reverse primers. Both strands of selected clones were PCR sequenced using dideoxy-terminated sequencing and fluorescent dyes (Applied Biosystems) and sequence alignments were performed using DS Gene software (Accelrys).
Limit of detection (LOD) determinations
LOD determinations were carried out at 23°C in 30 mM MES at pH 6.0, 0.9 M KCl, and 150 mM MgCl2. Reactions were assembled as described earlier except that they contained either no or 102 to 107 truncated HCV target molecules as indicated. Target was prepared by serial dilution into 100 ng/μL yeast tRNA from a concentrated stock. Concentration of target in the HCV RNA stock solution was determined by OD260 using the extinction coefficient of the HCV target oligonucleotide (ɛ = 2.8324 × 105 M−1 cm−1). Two independent serial dilutions using different pipets were prepared and each used for duplicate half-ribozyme reactions (total n = 4).
Acknowledgments
We thank Olke Uhlenbeck, Lawrence Blatt, Tatsuki Matsuno, and members of Sirna Therapeutics for helpful discussion during the course of this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
REFERENCES
- Araki, M., Okuno, Y., Hara, Y, and Sugiura, Y. 1998. Allosteric regulation of a ribozyme activity through ligand-induced conformational change. Nucleic Acids Res. 26: 3379–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel, D.P. and Szostak, J.W. 1993. Isolation of new ribozymes from a large pool of random sequences. Science 261: 1411–1418. [DOI] [PubMed] [Google Scholar]
- Bergman, N.H., Johnston, W.K., and Bartel, D.P. 2000. Kinetic framework for ligation by an efficient RNA ligase ribozyme. Biochemistry 39: 3115–3123. [DOI] [PubMed] [Google Scholar]
- Breaker, R.R. 2002. Engineered allosteric ribozymes as biosensor components. Curr. Opin. Biotechnol. 13: 31–39. [DOI] [PubMed] [Google Scholar]
- Burke, D.H., Ozerova, N.D., and Nilsen-Hamilton, M. 2002. Allosteric hammerhead ribozyme TRAPs. Biochemistry 41: 6588–6594. [DOI] [PubMed] [Google Scholar]
- Carter, P. and Wells, J.A. 1987. Engineering enzyme specificity by “substrate-assisted catalysis.” Science 237: 394–399. [DOI] [PubMed] [Google Scholar]
- Conry-Cantilena, C. 1997. Hepatitis C virus diagnostics: Technology, clinical applications and impacts. Trends Biotechnol. 15: 71–76. [DOI] [PubMed] [Google Scholar]
- Dufour, D.R. 2002. Hepatitis C: Laboratory tests for diagnosis and monitoring of infection. Clinical Lab. News 10,12,14.
- Ekland, E.H. and Bartel, D.P. 1995. The secondary structure and sequence optimization of an RNA ligase ribozyme. Nucleic Acids Res. 23: 3231–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1996. RNA-catalysed RNA polymerization using nucleoside triphosphates. Nature 382: 373–376. [DOI] [PubMed] [Google Scholar]
- Ekland, E.H., Szostak, J.W., and Bartel, D.P. 1995. Structurally complex and highly active RNA ligases derived from random RNA sequences. Science 269: 364–370. [DOI] [PubMed] [Google Scholar]
- Fedor, M.J. and Westhof, E. 2002. Ribozymes: The first 20 years. Mol. Cell 10: 703–704. [DOI] [PubMed] [Google Scholar]
- Glasner, M.E., Yen, C.C., Ekland, E.H., and Bartel, D.P. 2000. Recognition of nucleoside triphosphates during RNA-catalyzed primer extension. Biochemistry 39: 15556–15562. [DOI] [PubMed] [Google Scholar]
- Glasner, M.E., Bergman, N.H., and Bartel, D.P. 2002. Metal ion requirements for structure and catalysis of an RNA ligase ribozyme. Biochemistry 41: 8103–8112. [DOI] [PubMed] [Google Scholar]
- Hartig, J.S., Najafi-Shoushtari, S.H., Grune, I., Yan, A., Ellington, A.D., and Famulok, M. 2002. Protein-dependent ribozymes report molecular interactions in real time. Nat. Biotechnol. 20: 717–722. [DOI] [PubMed] [Google Scholar]
- Hesselberth, J.R., Robertson, M.P., Knudsen, S.M., and Ellington, A.D. 2003. Simultaneous detection of diverse analytes with an aptazyme ligase array. Anal. Biochem. 312: 106–112. [DOI] [PubMed] [Google Scholar]
- Hoffman, D., Hesselberth, J., and Ellington, A.D. 2001. Switching nucleic acids for antibodies. Nat. Biotechnol. 19: 313–314. [DOI] [PubMed] [Google Scholar]
- Iyo, M., Kawasaki, H., and Taira, K. 2002. Allosterically controllable maxizymes for molecular gene therapy. Curr. Opin. Mol. Ther. 4: 154–165. [PubMed] [Google Scholar]
- Johnston, W.K., Unrau, P.J., Lawrence, M.S., Glasner, M.E., and Bartel, D.P. 2001. RNA-catalyzed RNA polymerization: Accurate and general RNA-templated primer extension. Science 292: 1319–1325. [DOI] [PubMed] [Google Scholar]
- Koizumi, M., Kerr, J.N., Soukup, G.A., and Breaker, R.R. 1999. Allosteric ribozymes sensitive to the second messengers cAMP and cGMP. Nucleic Acids Symp. Ser. 42: 275–276. [DOI] [PubMed] [Google Scholar]
- Komatsu, Y., Yamashita, S., Kazama, N., and Ohtsuka, E. 1999. Induction of ribozyme activity by anti-ribozyme oligonucleotides. Nucleic Acids Symp. Ser. 42: 279–280. [DOI] [PubMed] [Google Scholar]
- Komatsu, Y., Nobuoka, K., Karino-Abe, N., Matsuda, A., and Ohtsuka, E. 2002. In vitro selection of hairpin ribozymes activated with short oligonucleotides. Biochemistry 41: 9090–9098. [DOI] [PubMed] [Google Scholar]
- Kumar, R.K. and Davis, D.R. 1997. Synthesis and studies on the effect of 2-thiouridine and 4-thiouridine on sugar conformation and RNA duplex stability. Nucleic Acids Res. 25: 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara, T., Warashina, M., and Taira, K. 2000. Allosterically controllable maxizymes cleave mRNA with high efficiency and specificity. Trends Biotechnol. 18: 462–468. [DOI] [PubMed] [Google Scholar]
- Lebedev, Y., Akopyants, N., Azhikina, T., Shevchenko, Y., Potapov, V., Stecenko, D., Berg, D., and Sverdlov, E. 1996. Oligonucleotides containing 2-aminoadenine and 5-methylcytosine are more effective as primers for PCR amplification than their nonmodified counterparts. Genet. Anal. 13: 15–21. [DOI] [PubMed] [Google Scholar]
- Lorsch, J.R. and Szostak, J.W. 1995. Kinetic and thermodynamic characterization of the reaction catalyzed by a polynucleotide kinase ribozyme. Biochemistry 34: 15315–15327. [DOI] [PubMed] [Google Scholar]
- Majid, A.M. and Gretch, D.R. 2002. Current and future hepatitis C virus diagnostic testing: Problems and advancements. Microbes Infect. 4: 1227–1236. [DOI] [PubMed] [Google Scholar]
- McGinness, K.E., Wright, M.C., and Joyce, G.F. 2002. Continuous in vitro evolution of a ribozyme that catalyzes three successive nucleotidyl addition reactions. Chem. Biol. 9: 585–596. [DOI] [PubMed] [Google Scholar]
- Mei, S.H., Liu, Z., Brennan, J.D., and Li, Y. 2003. An efficient RNA-cleaving DNA enzyme that synchronizes catalysis with fluorescence signaling. J. Am. Chem. Soc. 125: 412–420. [DOI] [PubMed] [Google Scholar]
- Porta, H. and Lizardi, P.M. 1995. An allosteric hammerhead ribozyme. Biotechnology (N Y) 13: 161–164. [DOI] [PubMed] [Google Scholar]
- Rajendran, M. and Ellington, A.D. 2002. Selecting nucleic acids for biosensor applications. Comb. Chem. High Throughput Screen. 5: 263–270. [DOI] [PubMed] [Google Scholar]
- Reich, C., Olsen, G.J., Pace, B., and Pace, N.R. 1988. Role of the protein moiety of ribonuclease P, a ribonucleoprotein enzyme. Science 239: 178–181. [DOI] [PubMed] [Google Scholar]
- Robertson, M.P. and Ellington, A.D. 1999. In vitro selection of an allosteric ribozyme that transduces analytes to amplicons. Nat. Biotechnol. 17: 62–66. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Design and optimization of effector-activated ribozyme ligases. Nucleic Acids Res. 28: 1751–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2001. In vitro selection of nucleoprotein enzymes. Nat. Biotechnol. 19: 650–655. [DOI] [PubMed] [Google Scholar]
- Rohatgi, R., Bartel, D.P., and Szostak, J.W. 1996. Kinetic and mechanistic analysis of nonenzymatic, template-directed oligoribonucleotide ligation. J. Am. Chem. Soc. 118: 3332–3339. [DOI] [PubMed] [Google Scholar]
- Schneider, T.D. and Stephens, R.M. 1990. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 18: 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seetharaman, S., Zivarts, M., Sudarsan, N., and Breaker, R.R. 2001. Immobilized RNA switches for the analysis of complex chemical and biological mixtures. Nat. Biotechnol. 19: 336–341. [DOI] [PubMed] [Google Scholar]
- Soukup, G.A. and Breaker, R.R. 1999. Design of allosteric hammerhead ribozymes activated by ligand-induced structure stabilization. Structure Fold. Des. 7: 783–791. [DOI] [PubMed] [Google Scholar]
- Stojanovic, M.N., de Prada, P., and Landry, D.W. 2001. Catalytic molecular beacons. Chembiochem 2: 411–415. [DOI] [PubMed] [Google Scholar]
- Tang, J. and Breaker, R.R. 1997. Rational design of allosteric ribozymes. Chem. Biol. 4: 453–459. [DOI] [PubMed] [Google Scholar]
- Vaish, N.K., Dong, F., Andrews, L., Schweppe, R.E., Ahn, N.G., Blatt, L., and Seiwert, S.D. 2002. Monitoring post-translational modification of proteins with allosteric ribozymes. Nat. Biotechnol. 20: 810–815. [DOI] [PubMed] [Google Scholar]
- Verma, S. and Eckstein, F. 1998. Modified oligonucleotides: Synthesis and strategy for users. Annu. Rev. Biochem. 67: 99–134. [DOI] [PubMed] [Google Scholar]
- Wang, D.Y. and Sen, D. 2001. A novel mode of regulation of an RNA-cleaving DNAzyme by effectors that bind to both enzyme and substrate. J. Mol. Biol. 310: 723–734. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Rationally designed allosteric variants of hammerhead ribozymes responsive to the HIV-1 Tat protein. Comb. Chem. High Throughput Screen. 5: 301–312. [DOI] [PubMed] [Google Scholar]
- Wang, D.Y., Lai, B.H., and Sen, D. 2002a. A general strategy for effector-mediated control of RNA-cleaving ribozymes and DNA enzymes. J. Mol. Biol. 318: 33–43. [DOI] [PubMed] [Google Scholar]
- Wang, D.Y., Lai, B.H., Feldman, A.R., and Sen, D. 2002b. A general approach for the use of oligonucleotide effectors to regulate the catalysis of RNA-cleaving ribozymes and DNAzymes. Nucleic Acids Res. 30: 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, M.C. and Joyce, G.F. 1997. Continuous in vitro evolution of catalytic function. Science 276: 614–617. [DOI] [PubMed] [Google Scholar]
- Xia, T., SantaLucia Jr. J., Burkard, M.E., Kierzek, R., Schroeder, S.J., Jiao, X., Cox, C., and Turner, D.H. 1998. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 37: 14719–14735. [DOI] [PubMed] [Google Scholar]