

Abstract
In Sindbis virus, initiation of nucleocapsid core assembly begins with recognition of the encapsidation signal of the viral RNA genome by capsid protein. This nucleation event drives the recruitment of additional capsid proteins to fully encapsidate the genome, generating an icosahedral nucleocapsid core. The encapsidation signal of the Sindbis virus genomic RNA has previously been localized to a 132-nucleotide region of the genome within the coding region of the nsP1 protein, and the RNA-binding activity of the capsid was previously mapped to a central region of the capsid protein. It is unknown how capsid protein binding to encapsidation signal leads to ordered oligomerization of capsid protein and nucleocapsid core assembly. To address this question, we have developed a mobility shift assay to study this interaction. We have characterized a 32 amino acid peptide capable of recognizing the Sindbis virus encapsidation signal RNA. Using this peptide, we were able to observe a conformational change in the RNA induced by capsid protein binding. Binding is tight (Kdapp = 12 nM), and results in dimerization of the capsid peptide. Mutational analysis reveals that although almost every predicted secondary structure within the encapsidation signal is required for efficient protein binding, the identities of the bases within the helices and hairpin turns of the RNA do not need to be maintained. In contrast, two purine-rich loops are essential for binding. From these data, we have developed a model in which the encapsidation signal RNA adopts a highly folded structure and this folding process directs early events in nucleocapsid assembly.
Keywords: alphavirus assembly, RNA–protein interactions, viral packaging signal
INTRODUCTION
Many viruses have developed mechanisms by which capsid proteins accurately recognize genomic RNAs for packaging and assembly into mature virions. The genomes of some of these viruses contain encapsidation signals that possess the sequence or structure necessary for recognition by capsid proteins, and thereby target the RNA genome for packaging into virus particles. Encapsidation signal sequences within genomic RNAs have been identified in multiple animal virus families including Coronaviridae (Cologna and Hogue 2000; Narayanan and Makino 2001), Togaviridae (Geigenmuller-Gnirke et al. 1993), Bunyaviridae (Severson et al. 2001; Xu et al. 2002), Orthomyxoviridae (Tchatalbachev et al. 2001), and Retroviridae (McBride and Panganiban 1996; Banks and Linial 2000; Griffen et al. 2001; Beasley and Hu 2002). In many of these examples, secondary structural elements, including stem–loops and bulges, have been identified as the major structural determinants of packaging. Structures have been solved for the interaction of the viral RNA encapsidation signal sequence and its respective packaging protein for the bacteriophage MS2 and the human immunodeficiency virus type-1 (Valegard et al. 1994; DeGuzman et al. 1998). These structures have laid a foundation for understanding the mechanism of this important step of the virus life cycle.
Members of the genus alphavirus within the family Togaviridae are enveloped, positive-strand RNA viruses exhibiting an extremely wide host range with transmission occurring through an arthropod host (Strauss and Strauss 1994). Many alphaviruses are human and animal pathogens known to cause symptoms such as encephalitis, fever, arthritis, and rash. Because they are arthropod-borne, with mosquitoes being the usual vector, alphaviruses possess the potential for rapid spread of infection (Strauss and Strauss 1994). Because of the combinations or severity of these symptoms, the alphaviruses represent a serious threat to human health in multiple regions of the world and they can be costly to agricultural endeavors. Viral assembly is a potential target for therapeutics that can be used to block viral replication.
Alphavirus virions possess an icosahedral nucleocapsid, enveloped in a tight-fitting membrane whose glycoprotein components are also present in an icosahedral lattice (Mukhopadhyay et al. 2002; Zhang et al. 2002). The nucleocapsid core consists of the single-stranded, plus-sense RNA genome of ~12 kb surrounded by 240 copies of a single species of a 30-kD capsid protein (Strauss and Strauss 1994). Nucleocapsids are readily formed in the cytoplasm and then associate with viral glycoproteins at membrane surfaces. At the plasma membrane, this association drives budding of a newly assembled virus particle (Tellinghuisen et al. 2001a).
The capsid protein of Sindbis virus, the prototype of the alphavirus genus, is composed of 264 amino acids and has multiple functional domains, as described in Figure 1 ▶ (Tellinghuisen et al. 2001a). Sindbis virus has been a useful system for studying the process of nucleocapsid assembly because infectious RNA transcripts of the virus can be generated from cDNA clones, thus permitting the construction and rescue of defined mutants, and nucleocapsid cores can be reconstituted in vitro (Tellinghuisen et al. 1999). This has allowed extensive analyses of the role of the nucleocapsid in viral replication and assembly (Tellinghuisen and Kuhn 2000). Sindbis virus genomic RNA is preferentially packaged into nucleocapsid cores (Owen and Kuhn 1996), suggesting that this virus has an accurate mechanism of recognition of its genomic RNA. Initiation of this packaging event is thought to begin with binding of a single capsid protein to the encapsidation signal of the viral RNA genome. Following nucleation, additional capsid proteins are recruited to complete the packaging of the genomic RNA within the nucleocapsid shell. Under most conditions, nucleic acid is essential to formation of the nucleocapsid core (Tellinghuisen et al. 1999), and electrostatic interactions between the positively charged N terminus of the capsid protein and the negatively charged RNA backbone are likely involved in this process. It is of particular interest that a protein that has a high sequence-independent RNA binding activity is able to recognize a specific RNA sequence and target only that sequence for packaging.
FIGURE 1.

Sindbis virus capsid protein constructs. Full-length capsid protein consists of 264 amino acid residues (top). The N-terminal region (amino acids 1–113) is involved in RNA binding and nucleocapsid assembly, whereas the C-terminal domain (114–264) adopts a protease fold and forms the capsomeres visible on the outer shell of the nucleocapsid core. Within the N-terminal domain are regions that have been implicated in nonspecific RNA binding (1–80), nucleocapsid oligomerization (81–113), and coiled-coil formation (38–55). The hatched region is the minimum domain previously shown to retain specific binding to the encapsidation signal. The 81–264 fragment has been shown to be competent to assemble into core-like particles in vitro. The GST fusion 76–132 fragment (GST76–132) has been shown to retain specific binding using a mobility shift assay. Also synthesized was a GST fusion to capsid protein residues 81–132 (GST81–132). CP81–112 is the capsid protein fragment spanning residues 81–112 of the Sindbis capsid protein, and expressed with the addition of a single serine at the N terminus.
For Sindbis virus, regions of the viral RNA genome and the capsid protein essential for initiation of assembly have been identified (Fig. 1 ▶; Geigenmuller-Gnirke et al. 1993; Weiss et al. 1994). The encapsidation signal within the genomic RNA (Fig. 2 ▶) has been localized to a 132-nt fragment spanning nt 945–1076 within the coding region for the nsP1 protein, a viral methyltransferase. Two positive-strand RNA species are generated during replication: 49S full-length genomic RNA, and a subgenomic 26S RNA, which spans the coding region for the structural proteins and is generated by transcription from an internal promotor. The location of the encapsidation signal within the coding region for nsP1 ensures that the genomic RNA, and not the subgenomic transcript, is packaged. The region of the capsid protein presumed to be involved in recognition of genomic RNA spans amino acids 76–132 (Fig. 1 ▶). Further characterization of the interaction has been hampered by lack of a robust in vitro assay. Additionally, it is unknown how a single binding event leads to the ordered oligomerization of capsid protein and nucleocapsid assembly. Binding of capsid protein to RNA may produce a scaffold for the addition of adjacent capsid protein or it may alter the structure of the capsid protein in a way that favors binding to other capsid proteins (Tellinghuisen et al. 2001a). There is a size discrepancy between the partners in this protein–RNA interaction. The region of capsid protein believed to bind the encapsidation signal is ~5 kD, whereas the encapsidation signal RNA is ~40 kD. This suggests that either the protein must recognize a small fragment of the encapsidation signal, or it must recognize a surface on the face of the RNA that is only formed on folding of the RNA into a defined tertiary structure.
FIGURE 2.

Predicted fold of the encapsidation signal RNA. The secondary structure of the encapsidation signal RNA was predicted by mfold (Zuker 2003). Base-paired regions of the RNA are named with a P, hairpin loops are named with an L, and “single-stranded” connecting regions are named with a J. The GGA at the 5′-terminus of the molecule is a nonviral sequence included to increase efficiency of transcription by T7 RNA polymerase. Mutations produced to characterize the RNA–protein interaction are drawn, and bases that differ from the wild-type sequence are indicated in boldface.
We have continued to investigate the mechanics of this interaction by developing a model system that contains the minimum regions of the capsid protein necessary for recognition of the encapsidation signal RNA. Using a mobility shift assay, we have identified a 32 amino acid peptide that retains the ability to bind the encapsidation signal RNA with formation of a single, discrete complex containing two molecules of the protein. This complex has a compact tertiary structure as assayed by native gel mobility and as probed by mutational analysis. These data suggest a model in which tertiary structure of the encapsidation signal RNA plays a significant role in initiating viral assembly in Sindbis virus.
RESULTS
The goal of this study was to determine how Sindbis virus RNA is initially recognized for packaging by its capsid protein. This could not be accomplished with full-length protein and RNA because rapid nucleocapsid core assembly follows the initial RNA-binding event (Tellinghuisen et al. 1999). To study the initial RNA recognition event in nucleocapsid assembly, we developed a simple system that contained short fragments of the Sindbis capsid protein and the genomic RNA. If these fragments are designed correctly, they should form a protein–RNA complex that possesses many of the same structural contacts as those formed by the interaction of full-length RNA and capsid protein during initiation of assembly.
Deletion fragment of Sindbis capsid protein, residues 81–132 fused to glutathione-S-transferase (GST), specifically binds the encapsidation signal RNA
Prior work by Weiss et al. (1994) demonstrated that a fragment formed by residues 76–132 of the Sindbis capsid, when expressed as a GST fusion, is able to specifically bind encapsidation signal RNA (Fig. 1 ▶). This GST fusion protein was chosen as a starting point to identify the binding domain of the capsid protein. The binding of two fusion proteins, GST76–132 and GST81–132, was examined by native gel mobility shift analysis (Fig. 3 ▶). These fusion proteins had identical properties. Therefore, analysis was continued only with the GST81–132 protein. The fusion demonstrated the ability to bind and form discrete complexes with the encapsidation signal RNA. The mobility of these complexes is slower than that of the free RNA (Fig. 3 ▶). When the concentration of fusion protein in the binding reaction was increased beyond 500 nM, the RNA was observed to form up to four discrete complexes of increasingly reduced mobility (data not shown).
FIGURE 3.

Native gel mobility shift assay comparing the ability of GST81–132 capsid fusion protein to bind to RNA containing either the encapsidation signal sequence or to an unrelated RNA molecule. For each sample, 1 nM of 32P-labeled RNA was mixed with variable amounts of protein in the binding buffer: 50 mM Hepes (pH 7.4), 150 mM potassium acetate, 2 mM magnesium acetate, 0.05 μg ssDNA fragments, and 10% glycerol. Samples were loaded onto a nondenaturing 6% polyacrylamide gel to allow separation of protein bound complexes from unbound RNA. hBST-177 is a 177-nt fragment of Bacillus stearothermophilus RNase P RNA.
As a control, binding of GST81–132 to RNAs with unrelated sequences was tested. Surprisingly, these RNAs demonstrated the ability to be bound with similar affinity to the GST fusion protein. Differences in binding affinities between the encapsidation signal and unrelated RNAs were observable only on the addition of sheared salmon sperm DNA competitor (Fig. 3 ▶). In the presence of ssDNA, the dissociation constants for capsid protein binding to the encapsidation signal RNA and to hBst-177, an unrelated 177 nucleotides RNA derived from Bacillus stearothermophilus RNase P RNA, were roughly estimated to be 180 nM and 540 nM, respectively. These numbers were estimated based on the concentration of protein required to shift half the RNA into bound species.
Competition assays were used to further characterize fusion protein binding to the encapsidation signal and to unrelated RNAs. These equilibrium assays contained trace radiolabeled RNA and sufficient protein to form multiple protein–RNA complexes. Unlabeled RNA was included to assess the ability to compete with the radiolabeled sample. Encapsidation signal RNA was able to disrupt the preformed protein–RNA complexes significantly more effectively than unrelated RNA (Fig. 4 ▶). These experiments demonstrate that, although the estimated affinities for specific and nonspecific RNAs are not dramatically different, the fusion protein does possess some specificity for the encapsidation signal RNA.
FIGURE 4.

Competition assays revealed discrimination between viral RNA and unrelated sequences. For each sample, 10 nM 32P-labeled RNA was mixed with variable amounts of unlabeled competitor RNA and 100 nM protein in the binding buffer: 50 mM Hepes (pH 7.4), 150 mM potassium acetate, 2 mM magnesium acetate, and 10% glycerol. Samples were loaded onto a nondenaturing 6% polyacrylamide gel to allow separation of protein bound complexes from unbound RNA. HBst-177 is a 177-nt RNA derived from B. stearothermophilus RNase P.
Identification of the stoichiometric ratio of RNA to protein in each of these complexes was made difficult because the GST fusion protein forms nonnative dimers under conditions favorable for RNA–protein binding, as confirmed by ultracentrifugation (data not shown). Each of the two monomeric fusion proteins of the GST dimer would then possess the specific RNA binding domain, giving each dimer at least two equivalent RNA binding sites. Thus the second, slower complex observed in the mobility shift assay could contain either one RNA molecule and two protein dimers or two RNA molecules and one protein dimer. Both of the RNA binding domains of one dimer could potentially bind simultaneously to the same RNA molecule, complicating the ability to measure the affinity of that protein for the RNA. We therefore turned to capsid protein fragments that could be obtained free of the fusion with GST.
Deletion fragment 81–112 represents a minimum sequence that maintains specific binding for encapsidation signal sequence
The capsid protein fragment 81–112 was synthesized as a fusion with Smt3 protein (Mossessova and Lima 2000). Fusion protein was cleaved with Ulp1 protease, enabling purification of the capsid protein fragment 81–112. This 32-residue fragment, named CP81–112, was expressed with the addition of a single serine residue at the N terminus. A native gel mobility shift revealed that the peptide retained the ability to bind and form a single discrete complex with the encapsidation signal RNA (Fig. 5 ▶). As observed for the GST fusion proteins, it was expected that this complex would have a slower mobility. In this case, however, the presence of CP81–112 capsid protein fragment resulted in an increased mobility of the encapsidation signal RNA (Fig. 5 ▶). This was surprising because protein is highly basic. Binding of the protein to the RNA should both increase the mass and reduce the negative charge of the complex relative to the free RNA. The altered mobility of the RNA–protein complex on the native gel could be caused by folding of the RNA into a more compact structure or by cleavage of the RNA. The latter was ruled out by size analysis of the RNA using denaturing gel electrophoresis (data not shown). This suggested that the RNA undergoes a conformational change on protein binding that produces a more compact form that can move more rapidly through the gel matrix.
FIGURE 5.

Native gel mobility shift assay comparing the ability of CP81–112 to bind either the encapsidation signal RNA or its reverse complement. Increasing amounts of protein were mixed with 0.2 nM of 5′-end labeled RNA, as indicated. The mobilities of the free RNAs are indicated with an asterisk. Binding of CP81–112 to encapsidation signal sequence RNA results in formation of a single discrete complex, whereas interaction with the complement strand of RNA results in nonspecific shifting to multiple complexes.
It is possible that this peptide is acting as a chaperone, altering the conformation of the RNA, but not remaining bound. The mobility of the complex, however, is dependent on the length of the peptide. Additionally, the preformed complex is sensitive to proteinase K digestion (data not shown). Thus, the faster moving species appears to contain both peptide and RNA.
Trace RNA was mixed with varying concentrations of protein before loading onto a native gel. After electrophoresis, the amounts of bound and unbound RNA were quantitated and the fraction of RNA bound was plotted. This reaction was repeated, extending the incubation time from 10 min to 60 min (data not shown). No differences in the binding reaction could be detected, suggesting that these measurements are being made at equilibrium. Binding of CP81–112 resulted in the formation of only one shifted complex; however, Hill plots of protein binding to RNA revealed a Hill coefficient of 2 ± 0.2 (Fig. 6 ▶). Additionally, the saturation curve does not fit well to a model in which one molecule of capsid peptide binds to one molecule of RNA (Fig. 6 ▶). It is nonhyperbolic and suggestive of positive cooperativity. These data suggest that two molecules of peptide are binding to the RNA to generate the high-mobility complex observed on the native gel. We looked for intermediates in assembly of the trimolecular complex, but no bands of intermediate mobility corresponding to a single peptide bound to the encapsidation signal were observed. The binding interaction was modeled such that the RNA possesses two capsid-protein binding sites that display complete positive cooperativity. A semiempirical measure of the dissociation constant (Kdapp) was obtained by fitting the data to the Hill equation as follows:
FIGURE 6.

Plots of CP81–112 binding to encapsidation signal RNA analyzed by native gel mobility shift assay. (A) Plot of saturation binding used to determine the apparent dissociation constant. The dashed line indicates the best fit to a simple binding isotherm in which one RNA molecule would bind to one protein molecule. The solid line represents the best fit to a model in which two peptide molecules bind to one RNA molecule with complete positive cooperativity. The estimated Kdapp from this plot was 12 nM. (B) Corresponding Hill plot for CP81–112 binding to encapsidation signal RNA. This Hill plot has a slope (n = 2), suggesting that there are two interacting sites for protein on the encapsidation signal sequence. The error bars indicate the standard deviations observed from three independent measurements.
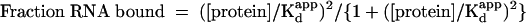 |
The protein binds wild-type encapsidation signal RNA with an estimated dissociation constant Kdapp = 12 nM (summarized in Table 1 ▶). This value is not a true dissociation constant, but it is related to Kd1 and Kd2, the dissociation constants for the binding of the first and second protomer, respectively. If the binding is essentially perfectly cooperative, Kdapp = (Kd1*Kd2)1/2.
TABLE 1.
Binding affinity of CP81–112 to nucleic acid
| Nucleic acid | Average Kdapp (nM) |
| Encapsidation signal (135 nt) | ~ 12 |
| Complement of the encapsidation signal (135 nt) | ~100 |
| hBst-177 RNA (177 nt) | ~100 |
| Pseudoknot RNA (125 nt) | N.B. |
| dsRNA (135 base pairs) | N.B. |
| ssDNA (48 nt) | N.B. |
(N.B.) No binding (protein concentration up to 5 μM).
Unrelated RNAs were also capable of binding CP81–112. The reverse complement of the encapsidation signal sequence was chosen for comparison because it is the same length, and secondary structure predictions suggest that it has the potential for formation of the same number of stem–loops. The complement strand was observed to bind CP81–112 (Fig. 5 ▶). Protein–RNA complexes formed by the interaction of CP81–112 and nonspecific RNAs are faster in mobility than the naked RNA, but multiple complexes of different mobilities are observed, and higher concentrations of protein are required for binding (Fig. 5 ▶). The Kd ~100 nM is a lower boundary for the dissociation constant; it is estimated based on the concentration of protein required to shift half the RNA. This binding never saturates with increasing protein concentration up to 5 μM, suggesting nonspecific binding. The same characteristics of binding were also observed for hBst-177. This RNA, like the complement strand, possesses regions of single-stranded RNA that may allow the peptide to bind nonspecifically.
The capsid peptide does not have high affinity for double-stranded RNA. Duplex RNA formed by annealing the encapsidation signal to its complement strand was not bound by CP81–112 (Table 1 ▶). Also tested were a 125-nt RNA possessing a pseudoknot structure, and a single-stranded DNA of 48 nucleotides that has been shown to be competent for assembly of nucleocapsid cores in vitro (Tellinghuisen et al. 1999). Neither of these nucleic acids was found to be capable of binding CP81–112 (Table 1 ▶).
Mutational analysis of the Sindbis encapsidation signal
To characterize the regions of RNA that are crucial for protein recognition and RNA folding, we performed a mutational analysis of the RNA. Previous studies had suggested that the packaging signal spanned the entire 132-nt RNA sequence from position 945–1076 in the viral genome. There is a great difference in size between the two binding partners, suggesting that the peptide is either recognizing a small fragment of the RNA, or else it binds to a surface of folded RNA.
Defining the 5′- and 3′-ends of the encapsidation signal
The initial mutations produced were targeted at minimizing the total length of the RNA sequence. Five nucleotides at a time were removed from each end of the RNA molecule to identify the minimal required length of the encapsidation signal sequence. All of the RNAs were transcribed with a GGA trinucleotide at the 5′-terminus to facilitate transcription by T7 RNA polymerase. Measuring the binding affinity of CP81–112 for RNAs with deletions from the 5′-end revealed that the first five nucleotides could be removed, but not 10 or more (data not shown). Deletion analysis at the 3′-end demonstrated that up to 10 nucleotides could be deleted without disrupting function (data not shown). However, a deletion of 15 nucleotides from the 3′-end interfered with RNA folding and protein binding.
Stem–loop modifications
Many viral encapsidation signals involve protein binding to nucleotides within hairpin turns of the RNA. Therefore, the RNA secondary structures important for RNA folding and protein interaction were investigated by modifying each of the stem–loops P3, P5, P6, and P7 in turn. To accomplish this, each stem–loop was truncated so that it now possessed a stem of 4 bp in length with a GAAA tetraloop for stabilization (Fig. 2 ▶). The mFold server (http://www.bioinfo.rpi.edu/~zukerm/) was used to verify that the calculated secondary structure would not be altered by the mutation (Zuker 2003). The mutated RNAs were analyzed on native gels to verify that their electrophoretic mobilities were similar to that of the wild-type encapsidation signal. This suggested that the structure of the RNA in the absence of protein was not altered by the mutation. The average affinity of each of the RNAs for CP81–112 was determined by native gel mobility shift analysis and the estimated Kdapp’s are listed in Table 2 ▶. Modification of P3, P5, and P7 did not affect protein binding. However, when P6 was shortened, no mobility shift in the presence of protein could be detected.
TABLE 2.
Estimated binding affinity of CP81–112 for encapsidation signal RNA
| RNA | Average Kdapp (nM) |
| WT (135 nt) | 12 ± 2 |
| mP3 (128 nt) | 17 ± 2 |
| mP5 (126 nt) | 13 ± 1 |
| mP6 (132 nt) | >100 |
| mP7 (125 nt) | 13 ± 1 |
| mP6a (135 nt) | 15 ± 1 |
| mP6b (135 nt) | 20 ± 2 |
| delP3 (118 nt) | 11 ± 1 |
| delP5 (116 nt) | N.B. |
| delP6 (121 nt) | N.B. |
| delP7 (115 nt) | N.B. |
| mJ2/3 (135 nt) | >100 |
| mJ5/6 (135 nt) | >100 |
| mSwitchJ (135 nt) | 15 ± 2 |
| mP4 (73 nt) | >100 |
| mP4ext (147 nt) | ~100 |
(N.B.) No binding (protein concentration up to 5 μM); (WT) wild-type RNA; m- and del- denote mutant RNAs; see text and Figure 2 ▶ for additional details.
Because shortening of the P6 stem–loop altered the ability of CP81–112 to produce a band shift, additional mutations were introduced to characterize the role of P6 in RNA binding. The role of the nucleotides in the paired region of P6 were investigated by making a variant (mP6a) in which the base-pairing potential was conserved, but the identities of the bases were changed (Fig. 2 ▶). This variant was able to bind protein in a manner similar to that of the wild-type encapsidation signal. To investigate the role of the nucleotides within the hairpin turn, we mutated the loop from the wild-type sequence ACA to UUU (Fig. 2 ▶). This variant (mP6b) is also able to bind the encapsidation signal. This suggests that although the P6 stem–loop structure is required for efficient protein binding, there is no sequence recognition within the P6 helix. Lack of observed binding with the truncation mutant mP6 may be the result of destabilization of the RNA structure, allowing formation of an undetected alternate RNA fold. Alternatively, the mobility of the protein–RNA complex may not be significantly different than that of the free RNA when P6 is shortened.
Because many of the stem–loop mutations were found to have no effect on the ability of the RNA to bind CP81–112, the process was then taken a step further and each helix was completely deleted to determine if the helices were important for stabilization of surrounding structures (delP3, delP5, delP6, and delP7 in Table 2 ▶). Of the four helices tested (P3, P5, P6, and P7) only the P3 stem–loop deletion was still capable of binding to CP81–112. Thus, although most of the secondary structures within this RNA are required for CP81–112 binding, bases in the hairpin turns do not appear to be involved in protein recognition.
Junction modifications
We then investigated the role of the single-stranded junction regions in protein binding. The nucleotides that make up the major junctions (J2/3 and J5/6) were all mutated to either C or U, depending on what was necessary at each position to avoid alternative secondary structure folding of the molecule. Of these two mutants (mJ2/3 and mJ5/6), both were found to have lost the ability to bind CP81–112. It is not clear if these mutations directly interfere with binding, or if they interfere indirectly by disrupting an RNA tertiary structure required for protein binding. A mutant in which the purine-rich sequences in J2/3 and J5/6 were exchanged (mSwitchJ) was able to bind protein with a calculated dissociation constant similar to that of the wild-type RNA.
P4 mutations
In addition to the peripheral stem–loop truncations, we also synthesized a truncation and an insertion at helix P4. The first variant was a large mutation that involved deleting the molecule at the P4 stem by replacing 62 nucleotides of the molecule with a GAAA tetraloop (Fig. 2 ▶). This deletion mutant was found to possess only weak ability to bind CP81–112. The second variant extended the helix P4 by 6 bp, corresponding to about half of a turn of a helix. This molecule could bind to the capsid protein; however, multiple complexes were formed and these were slower in mobility than that formed with the wild-type RNA. The estimated dissociation constant for peptide binding to this molecule was ~100 nM.
DISCUSSION
In this study, the initiation of Sindbis virus nucleocapsid core assembly was investigated. The nucleocapsid core is composed of 240 copies of a single capsid protein species that encases a single molecule of genomic RNA (Strauss and Strauss 1994). High-resolution structural data have been obtained by X-ray crystallography of the capsid protein, revealing the structure of the C-terminal 151 amino acids. In several crystal structures, the N terminus of the protein is disordered (Choi et al. 1991; Tong et al. 1993; Choi et al. 1996). Placement of the atomic resolution crystal structure of the capsid protein into an 11-Å resolution cryoelectron microscopy map of Sindbis virus has provided detailed information about the exterior of the core and the arrangement of the C-terminal half of the capsid protein in the mature virus particle (Zhang et al. 2002). There are only minimal protein–protein contacts between the C-terminal domain of capsid proteins in the nucleocapsid core, suggesting that much of the driving force for nucleocapsid assembly comes from protein–RNA interactions and protein–protein interactions involving the N terminus of the capsid protein. This is also in agreement with the observations that nucleocapsid assembly is nucleic acid dependent. Unfortunately, averaging of the density in the 11-Å map corresponding to capsid protein–RNA interaction results in little structural data for interpretation of this region of the capsid protein (Zhang et al. 2002). In terms of value to understanding assembly, this static view of the fully assembled core may not provide information about the protein–nucleic acid interactions that take place early and promote the assembly process. Despite the various structural studies on Sindbis virus, little is known about the interior of the core or how the interaction of nucleic acid and protein is able to drive assembly.
To understand how nucleocapsid assembly is initiated and to understand early RNA–protein recognition events, we have sought to identify stable assembly intermediates for study. This is nontrivial because following early RNA recognition events, rapid oligomerization of capsid proteins to form intact nucleocapsid cores occurs. An assembly-competent dimer of capsid protein has been identified by chemical cross-linking (Tellinghuisen et al. 2001b). Formation of the cross-linked dimer is dependent on the presence of nucleic acids. It is not, however, dependent on formation of intact cores, as a mutation that prevents nucleocapsid assembly can be cross-linked in the presence of RNA (Tellinghuisen and Kuhn 2000). It has been postulated that dimeric capsid protein, formed in the presence of nucleic acid, is a transient intermediate in the assembly process.
Residues 81–112 of Sindbis capsid protein are sufficient for encapsidation signal recognition
A fragment of the Sindbis capsid protein, spanning residues 81–112, was identified and found to bind to the encapsidation signal sequence. This 32-residue fragment was of interest because of its unique binding properties. First, it could bind with formation of a single, discrete complex. This distinction from larger capsid protein fragments may be the result of either reduced nonspecific RNA-binding affinity or the loss of domains important for protein–protein interactions that enable higher levels of oligomerization. The RNA-binding properties of this fragment correspond with the observations of Geigenmuller-Gnirke et al. (1993). In addition to these investigators finding a 68-residue fragment that possessed near wild-type binding, they also identified a protein fragment, residues 76–116, that retained specific binding but at only 35% the intensity of wild type. Their assay was performed by radiolabeling protein fragments and quantitating the amount of protein that could bind to the encapsidation signal RNA and migrate with it in an agarose gel (Geigenmuller-Gnirke et al. 1993). The design of this assay does not control for the oligomeric state of the capsid proteins that are binding to RNA or for a stronger nonspecific RNA binding potential. It is possible that the reason for differences in the observed affinities is simply due to differences in the oligomerization potential for each fragment.
The second observation from CP81–112 binding was that it could bind the encapsidation signal sequence with formation of a complex of faster mobility, implying that the RNA was becoming more compact on protein binding (Kjems et al. 1992; Baer et al. 1994; Batey and Williamson 1996). Compaction of the RNA implies a change in its tertiary structure induced or stabilized by protein binding. This phenomenon has previously been observed on binding of ribosomal protein S15 to a fragment of ribosomal RNA corresponding to a high-affinity protein-binding site. Binding of ribosomal protein S15 organizes a three-helix junction into a defined structure that travels with faster mobility through an acrylamide gel (Batey and Williamson 1996). The encapsidation signal RNA contains a three-helix junction and a four-helix junction. The increased mobility of the RNA on protein binding may result from a reorganization of the RNA’s tertiary structure in the presence of protein.
The Hill coefficient of two for CP81–112 binding to RNA suggests that the encapsidation signal possesses two protein-binding sites. There are two possible mechanisms that would lead to two protein molecules bound to the RNA. The protein may bind to the RNA as a preformed dimer, or the two protein molecules may bind to the RNA in a stepwise fashion. If the protein existed as a preformed dimer and bound to give a single complex, the data would have a Hill coefficient of one. Consistent with this, analysis of the capsid protein by ultracentrifugation reveals only monomers, suggesting the concentration of dimeric protein in solution is undetectably small (T. Tellinghuisen, J. Burgner, and R. J. Kuhn, unpubl.). There was no observable shifted RNA species corresponding to an RNA bound to a single molecule of protein. This species was either unstable or unobservable under our assay conditions.
All RNAs of unrelated sequence that demonstrated any interaction with CP81–112, formed multiple bands with significant smearing when analyzed by native gel mobility shift, suggesting the lack of a specific interaction. The observed binding was analogous to that observed when spermidine is added to each of the RNAs, suggesting that the interaction allows RNAs with folding potential to condense and increase in their mobility on the gel, but not with any kind of specific interaction between the protein and RNA. This compaction activity may aid the nucleocapsid protein in packaging the remainder of the 12-kb genomic RNA.
Mutational analysis suggests that the minimal effective packaging signal RNA is only slightly smaller than that originally identified by Weiss et al. (1994). Of the modifications to stem–loops and junction domains, only the junction domains demonstrated a loss of binding that could not be explained by gross alterations in the fold of the RNA. This suggested that the nucleotides present in each of the two major junction regions were directly or indirectly important to capsid protein binding.
A model for an intermediate in viral RNA packaging
How does a relatively small protein recognize a very large sequence within the viral RNA? The encapsidation signal of Sindbis virus is significantly different from the packaging signals that have previously been structurally characterized. Those RNAs can be described as relatively simple stem–loop structures with recognition determinants embedded within them. The Sindbis virus encapsidation signal is a 132-nt RNA with a secondary structure that can be characterized as containing a three-way helical junction and a four-way helical junction. Binding to individual stem–loops within this structure is not likely because all of these helices can be altered without detrimental effects on protein binding. The sequence of the single-stranded junctions J2/3 and J5/6 are important for protein binding. This may result from direct recognition of these sequences, but purine-rich “single-stranded” junctions often adopt stable tertiary structures that stabilize helical packing (Pley et al. 1994; Scott et al. 1995; Cate et al. 1996; Klein et al. 2001). Because the encapsidation signal appears to fold into a compact structure in the absence of the protein, it is likely that these single-stranded, purine-rich junctions are involved in protein binding because they stabilize the tertiary structure of the RNA. The recognition element for the protein is likely to be a face of the RNA generated by formation of RNA tertiary structure.
Inspection of the predicted secondary structure of the encapsidation signal suggests a repeated secondary structure motif within the RNA. This motif consists of a double-helical stem, followed by a purine-rich “single-stranded” region, a second double helical stem, a single adenosine, and a third double helical stem. Motif 1 spans P2, J2/3, P3, and P4, whereas P5, J5/6, P6, and P7 make up motif 2. It is possible that these motifs are half-binding sites for the dimer. We tested this hypothesis with several mutated sequences. First, the purine-rich sequences in J2/3 and J5/6 were swapped to determine if they could substitute for each other. This mutation was tolerated, and the protein-binding properties of this RNA are very similar to that of wild type. Second, the P4 helix was lengthened by six nucleotides. This should both alter the distance between the two motifs, and, because this corresponds to about half of a turn of an A-form helix, it should change their relative orientation. This molecule failed to bind to the capsid protein with affinity similar to that of the wild type. Furthermore, binding of protein to this extended RNA results in the formation of multiple complexes that are less compact than the wild-type complex. These data support a model in which helix P4 separates two highly-cooperative protein-binding sites. Finally, we truncated the encapsidation signal at P4 to create a molecule containing a single motif. This molecule exhibits a markedly reduced affinity to bind protein, consistent with the requirement for the protein dimer for efficient RNA binding.
How does a protein with significant nonspecific RNA-binding activity promote specific recognition of viral RNA within the cellular RNA pools? Binding of amino acid residues 81–112 to each of the motifs within the encapsidation signal RNA may stabilize a conformation of the capsid protein that promotes the formation of a stable dimer. Protein–protein contacts previously identified include the putative coiled-coil region in the N-terminal domain, and the C-terminal domain contacts that are observed in the mature virion (Perera et al. 2001; Zhang et al. 2002). This dimer could then serve as a nucleation point for the formation of the icosahedral core. Nonspecific binding appears to occur with relatively high affinity and a dissociation constant that is only ~10-fold higher than that for encapsidation signal binding. Such binding, however, may not be able to promote efficient dimerization of the capsid protein under physiological conditions.
The three-dimensional structure of the encapsidation signal RNA does not appear to be a simple protein binding site, or packaging signal. Rather, it has the potential to act as a scaffold on which the capsid protein can dimerize and thereby serve as a nucleus for further assembly of the nucleocapsid core. Thus, the encapsidation signal RNA appears to take an active role in nucleocapsid assembly, and folding of this RNA may initiate packaging of the Sindbis virus RNA.
MATERIALS AND METHODS
Sindbis virus capsid protein fragment cloning, expression, and purification
Amino acid residues 76–132 and 81–132 of the Sindbis capsid protein were expressed as GST fusion proteins using the pGEX-KT vector (Pharmacia). The nucleotide sequences encoding the desired peptides were amplified by PCR using the full-length capsid protein from a full-length genomic clone (pTOTO64) as a template (Owen and Kuhn 1996). These PCR products were ligated into vector pGEX-KT at a unique BamH I site in the polylinker region. The resulting plasmid encodes the desired peptide with a GST domain fused to its C terminus by a linker region containing a thrombin cleavage site. Ligation products were then transformed into Escherichia coli strain XL-1 Blue. Clones were sequenced to verify that the fragments were inserted correctly (Purdue Genomics Core Facility). The plasmids were transformed into E. coli strain BL21(DE3) for protein expression. Fusion proteins were isolated using a glutathione-agarose affinity column and their purity examined by SDS PAGE. All protein concentrations reported were determined by measuring absorption at 280 nm (ɛ280 = 52,000 M−1cm−1).
Amino acid residues 81–112 of the Sindbis capsid protein were also expressed as an Smt3 fusion protein in a modified pET28b vector (Mossessova and Lima 2000). The nucleotide sequence encoding the desired peptide was amplified by PCR using the full-length capsid protein gene from a genomic clone (Owen and Kuhn 1996) as the template. This PCR product was ligated into vector pET28b between unique BamH I and HinD III sites in the polylinker region. The ligation product was transformed into E. coli strain XL-1 Blue. Clones were sequenced to verify that the fragment was inserted correctly, and the plasmid was transformed into E. coli strain BL21(DE3) for protein expression. Fusion protein was isolated using an Ni-agarose affinity column, and cleaved at 4°C for 6 h by Ulp1 protease using a 1000:1 ratio by mass of fusion protein to protease. Ulp1 was expressed and purified as previously described (Mossessova and Lima 2000). The cleaved capsid protein fragment was loaded onto an HiTrap SP HP column (Amersham), equilibrated with 25 mM Tris-HCl (pH 7.4), 100 mM NaCl, and eluted with a linear salt gradient from 100 mM to 1 M NaCl in 25 mM Tris-HCl (pH 7.4). Fractions containing capsid protein fragment were concentrated with Centricon YM-3 centrifugal filter devices at 4°C and exchanged into 25 mM Hepes (pH 7.4), 150 mM potassium acetate. Protein concentrations were determined by acid hydrolysis and amino acid analysis.
In vitro transcription
A plasmid encoding the encapsidation signal sequence of Sindbis virus was kindly provided by Sondra Schlessinger (Weiss et al. 1994). The nt 945–1076 were amplified by PCR and ligated into pUC19 between the unique HinD III and Xba I restriction sites. The RNA sequences were amplified by PCR using oligomers containing the T7 promoter and an Ear I recognition site for generation of the desired 3′-end by run-off transcription. The trinucleotide GGA was inserted between the T7 promotor and the native gene sequence to facilitate transcription by T7 RNA polymerase. Mutations in the native encapsidation signal sequence were generated by PCR mutagenesis. Ligation products were transformed into E. coli strain XL-1 Blue. Clones were sequenced to verify that the fragment was inserted correctly. Plasmid purification was performed using a QIAGEN plasmid purification kit. Plasmids were linearized by Ear I digestion and phenol:chloroform extracted prior to use in transcription reactions. RNAs were synthesized using T7 RNA polymerase as previously described (Milligan et al. 1987).
RNAs were purified from the transcription reactions by gel electrophoresis on a 6% polyacrylamide gel containing TBE, 7 M urea, 19.5:1 acrylamide:bis(acrylamide); TBE is 0.1 M Tris base, 0.83 M boric acid, and 1 mM EDTA. RNA products were visualized by UV-shadowing. Bands containing the desired RNA were excised and crushed, and the RNA was eluted into TEN (10 mM Tris-HCl at pH 7.5, 1 mM EDTA, and 250 mM NaCl). After removal of the gel fragments by centrifugation, the RNAs were ethanol precipitated and dissolved in a desired amount of H2O. RNAs for use in mobility shift assays were radiolabeled with 32P either at the 5′-end using γ–32P–ATP and polynucleotide kinase (Promega) or by incorporation of α–32P–ATP during in vitro transcription.
Native gel mobility shift assays
Prior to use in gel mobility assays, RNA was prewarmed to 90°C for 2 min and then allowed to cool to room temperature for 3 min before addition to the protein-buffer mixture. For mobility shift assays with the GST fusion protein, 20 μL samples contained 1 nM 32P-labeled RNA, 50 mM Hepes (pH 7.4), 150 mM potassium acetate, 2 mM magnesium acetate, 0.05 mg/mL DNA fragments, and 10% glycerol, plus variable amounts of protein. After addition of protein, the mixtures were vortexed for 2 sec and then centrifuged briefly to remove aggregates. Samples were incubated for 10 min and then loaded onto a 6% polyacrylamide gel containing TBE. The gel was prerun for 30 min at 400 V in a TBE running buffer in a cold room at 4°C. Gels were dried and exposed to a phosphor storage screen for imaging and quantitation with a Typhoon phosphoimager and Imagequant software (Amersham Biosciences).
For competition binding experiments, samples were 20 μL in volume and contained 10 nM 32P-labeled RNA, 100 nM GST-81–132 protein fragment, 50 mM Hepes (pH 7.4), 150 mM potassium acetate, 2 mM magnesium acetate, 10% glycerol, plus variable amounts of unlabeled RNA competitor. RNA was prewarmed to 90°C for 2 min and then allowed to cool to room temperature for 3 min. Radiolabeled and unlabeled RNA were then mixed in binding buffer. Sample preparation, electrophoresis, and image analysis were performed as described earlier.
For band-shift assays with capsid protein fragments 76–112 and 81–112, samples were 10 μL in volume and contained 0.2 nM 32P-labeled RNA, 25 mM Hepes (pH 7.4), 150 mM potassium acetate, 5 mM magnesium acetate, and 6% glycerol, in addition to a variable concentration of protein for affinity binding studies. RNA was prewarmed to 90°C for 2 min and then allowed to cool to room temperature for 3 min before addition to the protein-buffer mixture. After addition of protein, the mixtures were vortexed for 2 sec and then centrifuged briefly to remove aggregates. The reaction mixtures were incubated for 5 min and then loaded onto a 6% polyacrylamide gel containing TBE. The gel was prerun for 50 min at 500 V in TBE running buffer. Electrophoresis was then carried out at 500 V for 55 min with the temperature regulated by a water bath to maintain 25°C. Gels were dried and image analysis was performed as described earlier.
Acknowledgments
We thank Christopher Lima for providing the plasmids pSUMO and pSMT3, and Ranjit Warrier and Chanakha Navaratnarahah for critical discussions. This work was supported by a Shawalter Award to B.L.G. and a Public Health Service grant (GMS6279) to R.J.K.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.5127104.
REFERENCES
- Baer, M.L., Houser, F., Loesch-Fries, L.S., and Gehrke, L. 1994. Specific RNA binding by amino-terminal peptides of alfalfa mosaic virus coat protein. EMBO J. 13: 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks, J.D. and Linial, M.L. 2000. Secondary structure analysis of a minimal avian leucosissarcoma virus packaging signal. J. Virol. 74: 456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey, R.T. and Williamson, J.R. 1996. Interaction of the Bacillus stearothermophilus ribosomal protein S15 with 16S rRNA: Defining the minimal RNA site. J. Mol. Biol. 261: 536–549. [DOI] [PubMed] [Google Scholar]
- Beasley, B.E. and Hu, W. 2002. Cis-acting elements important for retroviral RNA packaging specificity. J. Virol. 76: 4950–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cate, J.H., Gooding, A.R., Podell, E., Zhou, K., Golden, B.L., Kundrot, C.E., and Cech, T.R. 1996. Crystal structure of a group I ribozyme domain: Principles of RNA packing. Science 273: 1678–1685. [DOI] [PubMed] [Google Scholar]
- Choi, H.K., Tong, L., Minor, W., Dumas, P., Boege, U., Rossmann, M.G., and Wengler, G. 1991. Structure of Sindbis virus core protein reveals a chymotrypsin-like serine proteinase and the organization of the virion. Nature 354: 37–43. [DOI] [PubMed] [Google Scholar]
- Choi, H., Lee, S., Zhang, Y., McKinney, B., Wengler, G., Rossmann, M.G., and Kuhn, R.J. 1996. Structural analysis of Sindbis virus capsid mutants involving assembly and catalysis. J. Mol. Biol. 262: 151–167. [DOI] [PubMed] [Google Scholar]
- Cologna, R. and Hogue, B.G. 2000. Identification of bovine coronavirus packaging signal. J. Virol. 74: 580–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGuzman, R.N., Wu, Z.R., Stallings, C.C., Pappalardo, L., Borer, P.N., and Summers, M.F. 1998. Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element. Science 279: 384–388. [DOI] [PubMed] [Google Scholar]
- Geigenmuller-Gnirke, U., Nitschko, H., and Schlesinger, S. 1993. Deletion analysis of the capsid protein of Sindbis virus: Identification of the RNA binding region. J. Virol. 67: 1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffen, S.D.C., Allen, J.F., and Lever, A.M.L. 2001. The major human immunodeficiency virus type 2 (HIV-2) packaging signal is present on all HIV-2 RNA species: Cotranslational RNA encapsidation and limitation of Gag protein confer specificity. J. Virol. 75: 12058–12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjems, J., Calnan, B., Frankel, A.D., and Sharp, P.A. 1992. Specific binding of a basic peptide from HIV-1 Rev. EMBO J. 11: 1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, D.J., Schmeing, T.M., Moore, P.B., and Steitz, T.A. 2001. The kink-turn: A new RNA secondary structure motif. EMBO J. 20: 4214–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride, M.S. and Panganiban, A.T. 1996. The human immunodeficiency virus type 1 encapsidation site is a multipartite RNA element composed of functional hairpin structures. J. Virol. 70: 2963–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan, J.F., Groebe, D.R., Witherell, G.W., and Uhlenbeck, O.C. 1987. Oligoribonucleotide synthesis using T7 RNA-polymerase and synthetic DNA templates. Nucleic Acids Res. 15 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossessova, E. and Lima, C.D. 2000. Ulp1-SUMO crystal structure and genetic analysis revealed conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell 5: 865–876. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, S., Chipman, P.R., Hong, E.M., Kuhn, R.J., and Rossmann, M.G. 2002. In vitro-assembled alphavirus core-like particles maintain a structure similar to that of nucleocapsid cores in mature virus. J. Virol. 76: 11128–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan, K. and Makino, S. 2001. Cooperation of an RNA packaging signal and a viral envelope protein in coronavirus RNA packaging. J. Virol. 75: 9056–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen, K.E. and Kuhn, R.J. 1996. Identification of a region in the Sindbis virus nucleocapsid protein that is involved in specificity of RNA encapsidation. J. Virol. 70: 2757–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera, R., Owen, K.E., Tellinghuisen, T.L., Gorbalenya, A.E., and Kuhn, R.J. 2001. Alphavirus nucleocapsid protein contains a putative coiled coil α-helix important for core assembly. J. Virol. 75: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pley, H.W., Flahery, K.M., and McKay, D.B. 1994. Three-dimensional structure of a hammerhead ribozyme. Nature 372: 68–74. [DOI] [PubMed] [Google Scholar]
- Scott, W.G., Finch, J.T., and Klug, A. 1995. The crystal structure of an all-RNA hammerhead ribozyme: A proposed mechanism for RNA catalytic cleavage. Cell 81: 991–1002. [DOI] [PubMed] [Google Scholar]
- Severson, W.E., Xu, X., and Jonsson, C.B. 2001. Cis-acting signals in encapsidation of Hantaan virus S-segment viral genomic RNA by its N protein. J. Virol. 75: 2646–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss, J.H. and Strauss, E.G. 1994. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 58: 491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchatalbachev, S., Flick, R., and Hobom, G. 2001. The packaging signal of influenza viral RNA molecules. RNA 7: 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellinghuisen, T.L. and Kuhn, R.J. 2000. Nucleic-acid dependent cross-linking of the nucleocapsid protein of Sindbis virus. J. Virol. 74: 4302–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellinghuisen, T.L., Hamburger, A.E., Fisher, B.R., Ostendorf, R., and Kuhn, R.J. 1999. In vitro assembly of alphavirus cores by using nucleocapsid protein expressed in Escherichia coli. J. Virol. 73: 5309–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellinghuisen, T.L., Perera, R., and Kuhn, R.J. 2001a. Genetic and biochemical studies of the assembly of an enveloped virus. Genetic Engineering (N Y) 23: 83–112. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. In vitro assembly of Sindbis virus core-like particles from cross-linked dimers of truncated and mutant capsid proteins. J. Virol. 75: 2810–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, L., Wengler, G., and Rossmann, M.G. 1993. Refined structure of Sindbis virus core protein and comparison with other chymotrypsin-like serine proteinase structures. J. Mol. Biol. 230: 228–247. [DOI] [PubMed] [Google Scholar]
- Valegard, K., Murray, J.B., Stockley, P.G., Stonehouse, N.J., and Liljas, L. 1994. Crystal structure of an RNA bacteriophage coat protein-operator complex. Nature 371: 623–626. [DOI] [PubMed] [Google Scholar]
- Weiss, B., Geigenmuller-Gnirke, U., and Schlesinger, S. 1994. Interactions between Sindbis virus RNAs and a 68 amino acid derivative of the viral capsid protein further defines the capsid binding site. Nucleic Acids Res. 22: 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X., Severson, W.E., Villegas, N., Schmaljohn, C.S., and Jonsson, C.B. 2002. The RNA binding domain of the Hantaan virus N protein maps to a central, conserved region. J. Virol. 76: 3301–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W., Mukhopadhyay, S., Pletnev, S.V., Baker, T.S., Kuhn, R.J., and Rossmann, M.G. 2002. Placement of the structural proteins in Sindbis virus. J. Virol. 76: 11645–11658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]