

Abstract
The kinetic pathway of a trans-acting delta ribozyme includes an essential structural rearrangement involving the P1 stem, a stem that is formed between the substrate and the ribozyme. We performed cross-linking experiments to determine the substrate position within the catalytic center of an antigenomic, trans-acting, delta ribozyme. Substrates that included a 4-thiouridine either in position −1, +4, or +8 (i.e., adjacent to the cleavage site, or located either in the middle of or at the 3′-end of the P1 stem, respectively) were synthesized and shown to be efficiently cleaved. Examination of the cross-linking conditions, the use of various mutated ribozymes, as well as the probing and characterization of the resulting ribozyme–substrate complexes, revealed several new features of the molecular mechanism: (1) the close proximity of several bases between nucleotides of the substrate and ribozyme; (2) the active ribozyme–substrate complex folds in a manner that docks the middle of the P1 stem on the P3 stem, while concomitantly the scissile phosphate is in close proximity to the catalytic cytosine; and, (3) some complexes appear to be compatible with being active intermediates along the folding pathway, while others seem to correspond to misfolded structures. To provide a model representation of these data, a three-dimensional structure of the delta ribozyme was developped using several RNA bioinformatic software packages.
Keywords: ribozyme, RNA structure–function, UV cross-linking, folding pathway, Hepatitis delta virus
INTRODUCTION
Both the genomic and antigenomic hepatitis delta virus (HDV) RNA strands include a self-cleaving RNA motif (for reviews, see Shih and Been 2002; Bergeron et al. 2003). According to the pseudoknot model secondary structure, which is well supported by experimental data, this ribozyme is composed of one stem (P1 stem), one pseudoknot (P2 stem), two stem–loops (P3–L3 and P4–L4), and three single-stranded junctions (J1/2, J1/4, and J4/2; see Fig. 1 ▶). Both the J1/4 junction and the L3 loop are single-stranded in the initial stages of folding, but are subsequently involved in the formation of a second pseudoknot that consists of two Watson-Crick base pairs (Ferré D’Amaré et al. 1998; Wadkins et al. 1999; Deschênes et al. 2003). The X-ray diffraction and the NMR spectrums have provided high-resolution definition of the ternary structure of the delta ribozyme (Ferré D’Amaré et al. 1998; Tanaka et al. 2002a). Overall, these approaches have shown that the catalytic core includes two coaxial helices formed by the stacking of the P1–P1.1–P4 stems and of the P2–P3 stems.
FIGURE 1.

Secondary structure of the antigenomic trans-acting delta ribozyme. This trans-acting delta ribozyme system has been reported previously (e.g., see Mercure et al. 1998; Deschênes et al. 2003). The substrate and ribozyme are represented by S and Rz, respectively. The numbering system is according to Shih and Been (2002). The Δ in the L4 loop indicates the P4 deletion (compared to the natural variants). The pseudoknot P1.1 and the G28–U23 base pair are indicated by dotted lines. The homopurine base pair at the top of the P4 stem is represented by two large dots (G••G), while the Wobble base pair is represented by a single large dot (G•U). The arrow indicates the cleavage site.
The cis-acting self-cleaving sequence has been separated into two molecules to develop trans-acting systems in which one molecule, identified as a ribozyme (Rz), possesses the catalytic properties required to successively cleave several molecules of substrate (S). The trans-acting delta ribozymes possesses a complex kinetic pathway. Initially, the ribozyme binds its substrate through formation of the P1 stem, yielding an RzS complex. Next, upon the addition of at least one magnesium ion, a structural transition, that has been shown to be essential for the formation of an active RzS′ complex, occurs (Mercure et al. 1998; Ananvoranich and Perreault 2000). One of the consequences of this conformational transition is to bring the P1 stem within the catalytic core. It is only after this structural rearrangement that the chemical step takes place. It has been demonstrated that the highly conserved cytosine residue at position 76 is involved in an acid-base catalysis (for a review, see Bevilacqua et al. 2003).
The involvement of the P1 stem is an essential structural transition that was initially suggested based on data from directed mutagenesis of the residue in the middle of this helix. Specifically, the introduction of mismatches in either position G+3–C37 or U+4–A36 resulted in a complete lack of cleavage activity Fig. 1 ▶; Ananvoranich et al. 1999). More-over, when the original U+4–A36 base pair was replaced by an U+4–G36, a C+4–G36 or an A+4–U36 base pair, cleavage was observed, although at different levels. In contrast, the presence of a G+4–C36 base pair was detrimental to the cleavage. These nucleotides have to be important not only in substrate recognition, but also in the subsequent steps (e.g., a tertiary interaction that leads to a conformation change that produces a transition complex). The details of these interactions, however, remain unknown.
To determine the position of the substrate within the catalytic center of a delta ribozyme, cross-linking experiments in the presence of a 4-thiouridine (s4U) photoactivable nucleotide appear to be the best approach (Fig. 2A ▶). Upon UV irradiation (>300 nm), the s4U is activated and can form a tertiary interaction via a covalent bond (cross-link) to a stacked, neighboring nucleotide (~3 Å). This approach has proved to be successful in the extensive mapping of several ribozymes (e.g., Pinard et al. 1999; Blount and Uhlenbeck 2002; Hiley et al. 2002). In the case of the delta ribozyme, the results of cross-linking experiments using a 4-thiouridine-2′-deoxynucleotide in position −2 from the cleavage site (see Fig. 1 ▶) have been reported previously (Bravo et al. 1996). The s4U-analog was shown to cross-link with nucleotides C24, G28, and G41. When introduced into a cleavable substrate, the s4U-analog was shown to cross-link with residue C76. Here, using cross-linking experiments between 4-thiouridine modified substrates and a trans-acting delta ribozyme, we suggest a dynamic interaction in the middle of the P1 stem. These results, combined with those of previous reports, led us to propose a more complete folding pathway for the ribozyme-catalyzed reaction.
FIGURE 2.

Structure and cleavage activity of the substrates harboring a 4-thiouridine. (A) Structure of 4-thiouridine. (B) Sequences of the different substrates. The 4-thiouridines are indicated by s4U. The arrows indicate the cleavage sites. (C) Graphical representation of time courses for the cleavage reactions of Swt (opened squares), SC−1U (opened circles), SC−1s4U (closed circles), S+4s4U (closed triangles), and S+8s4U (closed diamonds). The inset shows a typical autoradiogram of the denaturing PAGE gel for the analysis of the cleavage reaction of Swt. The positions of the bromophenol blue (BPB), the 11-nt substrate (S), and the 4-nt product (P) are indicated.
RESULTS
Synthesis and characterization of a collection of substrates
To determine the position of the substrate within the catalytic core of a delta ribozyme, we used the trans-acting version of delta ribozyme for which the kinetic behavior has been characterized under both single- and multiple-turnover conditions (Mercure et al. 1998; Ananvoranich et al. 1999). This version, which is derived from the HDV anti-genomic strands, is composed of 57 nt and cleaves a model substrate (11 nt) into products of 4 and 7 nt (see Fig. 1 ▶).
Initially, a collection of substrates either with, or without, a ribonucleotide 4-thiouridine (s4U) was synthesized (Fig. 2B ▶). Our objective was to introduce an s4U in the substrate either in the 5′, the middle, or 3′ region of the P1 stem. The original substrate possesses a cytosine in position −1 (i.e., adjacent to the scissile phosphate [Swt]). In this fashion, a substrate with an s4U in position −1 (SC−1s4U) and the corresponding unmodified substrate (SC−1U), were thus synthesized. To introduce a photoactivable residue into the middle of the stem, a substrate including an s4U in position +4 was synthesized (S+4s4U). This substrate was synthesized by in vitro transcription because it only contained a single uridine. Finally, a substrate with an s4U in position +8, which is 3′ adjacent to the P1 stem, was synthesized (S+8s4U). The latter substrate and the SC−1s4U include two uridine residues; consequently, they were chemically synthesized to incorporate an unmodified uridine in position +4 and the substitution in either position −1 or +8. An additional guanosine residue was added at the 3′-end for S+8s4U because s4U linked to a matrix is not commercially available for chemical synthesis (i.e., given a 13-nt substrate).
To ensure that the introduction of s4U into the molecules did not induce misfolding of the ribozyme–substrate complex or alter the kinetic pathway, the cleavage activities of all substrates was determined under single-turnover conditions. Trace amounts of 32P5′-end-labeled substrate (<1 nM) were incubated in the presence of an excess of ribozyme (100 nM) and the cleavage activity was monitored. A graphical representation of the cleavage activity is shown in Figure 2C ▶, and a typical autoradiogram is shown in the inset. Briefly, the cleavage activity appears to be relatively similar, regardless of the composition of the substrate. To more accurately reveal the differences between these substrates, kinetic analyses were performed using 5 to 400 nM of ribozyme, and the pseudo first-order (k2 and KM) and second-order (k2/KM) cleavage constants were determined (see Table 1 ▶). The original substrate had k2 and KM values of 0.34 min−1 and 7.7 nM, respectively, which are similar to those reported for this ribozyme in independent experiments (0.23 min−1 and 9.3 nM, Mercure et al. 1998; 0.29 min−1 and 16 nM; Ananvoranich and Perreault 2000). The two substrates possessing the modification in position −1 were cleaved with second-order rate constants similar to those of the original substrate. The k2/KM values of the substrate SC−1U and SC−1s4U substrate were 5.9 × 107 min−1M−1 and 4.8 × 107 min−1M−1, respectively, compared to 4.4 × 107 min−1M−1 for the Swt substrate (Table 1 ▶). More specifically, the k2 and the KM values of the SC−1s4U substrate were virtually identical to those of the original one, while for SC−1U both values were approximately threefold smaller. Finally, the two other substrates (i.e., S+4s4U and S+8s4U) had k2, KM, and k2/KM values that were virtually identical to those of the original substrate (see Table 1 ▶). Thus, the modifications performed to the various substrates did not significantly affect the cleavage rate of the ribozyme. These results suggest that all substrates adopt the same tertiary conformation in the overall substrate–ribozyme folding pathway, and therefore, those including an s4U have the potential to probe the active catalytic center.
TABLE 1.
Kinetic parameters of various substrates
| Substrates | k2(min−1) | KM (nM) | k2/KM (min−1M−1) |
| Swt | 0.34 ± 0.03 | 7.7 ± 2.9 | 4.4 × 107 |
| SC−1U | 0.14 ± 0.01 | 2.3 ± 0.8 | 5.9 × 107 |
| SC−1s4U | 0.38 ± 0.03 | 7.9 ± 3.2 | 4.8 × 107 |
| S+4s4U | 0.25 ± 0.02 | 7.6 ± 3.5 | 3.3 × 107 |
| S+8s4U | 0.35 ± 0.02 | 5.7 ± 1.6 | 6.2 × 107 |
Cross-linking of RzS complexes
To obtain structural information on the active RzS complex, a cross-linking procedure that reflected the conditions of the cleavage reaction catalyzed by the ribozyme was adopted. For example, the experiments were performed in a solution containing 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2 and an excess of ribozyme (compared to the 5′-end-labeled substrate that was present in trace amounts) to maintain single turnover conditions. Once the ribozyme and substrate were mixed together, but prior to the addition of magnesium, they were subjected to heat denaturation, snap-cooling, and preincubation at 37°C with Tris, a treatment that favors the formation of the RzS complexes. To increase the probability of cross-linking a rapid folding event, all manipulation after these preliminary incubations all manipulations (i.e., the addition of MgCl2 and UV irradiations) were performed on ice to reduce the cleavage of the substrate by the ribozyme and to prevent the evaporation of the solution exposed to the UV irradiation. However, because the annealing protocols were identical for both procedures, they should result in identical folding. This procedure was optimized and characterized with respect to several aspects (e.g., time of irradiation and UV wavelength).
After UV irradiation, the mixtures were fractionated by denaturing 20% polyacrylamide gel electrophoresis (PAGE). Cross-linked RNA species are known to have an aberrant electrophoretic mobility in denaturing PAGE gels due to the formation of either a lariat or a three-branched structure (Saenger et al. 1976). A typical autoradiogram is shown in Figure 3A ▶. When the unmodified substrate was submitted to the cross-linking procedure, the level of cleavage was not altered by the irradiation (compare lanes 1 and 2). This observation shows that the cross-linking procedure supports the cleavage activity of the ribozyme. The next lanes in this gel represent various reaction conditions examined in the presence of the S+4s4U substrate. For example, in the absence of either ribozyme or MgCl2, no cross-linked complex was observed. However, overexposition revealed a faint band for the UV-irradiated mixture containing the ribozyme in the absence of magnesium. When both the ribozyme and MgCl2 were added to the mixture, but the irradiation was omitted, a band corresponding to the cleavage product (4 nt) was observed. When UV irradiation was performed in addition to the previous conditions, a novel band with the characteristic slow mobility of cross-linked complex was observed. When the electrophoresis was pursued for an extended period, overexposition of the autoradiogram allowed the detection of one dark and three faint bands (see Fig. 3A ▶, inset). The four different bands corresponded to different cross-linked complexes (identified ×1 to ×4).
FIGURE 3.

Typical autoradiograms of cross-linking experiments. (A) and (C) are the experiments performed using the S+4s4U and SC−1s4U substrates, respectively. The various conditions tested are indicated at the top of the gels. The positions of the origin of migration (ori), xylene cyanol (XC), and bromophenol blue (BPB) are indicated on the left of the gels. The positions of the 11-nt substrates (S), the 4-nt product (P), and the cross-linked complexes (RzS) are indicated on the right of the gels. The inset shows a higher resolution migration that reveals several bands of slower migration. (B) NaOH ladder of the purified RzS complexes where the ribozyme was labeled at its 3′-end with 32PCp. The first lane is a partial hydrolysis with RNase T1 to identify the guanosines, and the second lane is the partial NaOH ladder of the noncross-linked ribozyme. The third and fourth lanes are the partial NaOH ladders of the cross-linked ×1 and ×2 complexes, respectively. The positions of the stops are indicated on the right.
To identify the cross-linked residues, the ribozyme were either 5′ or 3′-end-labeled (i.e., 32P or 32PCp), and larger amounts of RzS complexes used in the cross-linking experiments. A corresponding banding pattern including one dark (fastest migration), and either two or three faint bands, was observed under these conditions (data not shown). To properly reveal the cross-linked sites present in the RzS complexes, alkaline hydrolysis was performed on the gel extracted bands. At the site of cross-linking, a tri-branched structure is formed; consequently, alkaline hydrolysis of the phosphodiester bonds releases a branched RNA species that possesses a slower electrophoretic mobility in denaturing PAGE gels (i.e., causes a shift of all bands corresponding to the hydrolysis products that are produced over the cross-linked sites). For example, complex ×1 produces a ladder of bands from the labeled 3′-end up to nucleotide U17. After this position the shifts were observed (Fig. 3B ▶). In the case of complex ×2, the ladder of bands goes up to nucleotide C22, although a trace amount of ladder also reached position U23 (Fig. 3B ▶). In the latter case, this observation was due to a contamination of the species by the upper bands on the previous purification gel as ×3 corresponds to U23. The cross-linking at these two positions has been confirmed by separate experiments. Finally, no readable ladder was obtained with complex ×4, which was only detected in trace amounts. In conclusion, the S+4s4U substrate cross-linked complexes were observed in the presence of all components essential for ribozyme cleavage (Rz, S, and Mg2+), and it cross-linked with the nucleotides U17 (in the most abundant species), C22 and U23.
Using the 5′ labeled SC−1s4U substrate the results were almost identical to those of the S+4s4U substrate (Fig. 3C ▶). It was noticed that trace amounts of the complexes were detected in the absence of magnesium. For this substrate a long migration revealed up to five bands (one predominant (>80%) and four minor). The identification of the cross-linked nucleotides was performed as described above using a 5′-end-labeled ribozyme. The predominant species cross-linked with residue U77, while the four minor bands corresponded to shifts from positions U26, U68, A79, and G81. Similar results were obtained when the experiment was performed using a 3′-end-labeled ribozyme. The cross-link with U77 is in good agreement with a previous report showing a cross-link between a 4-thiouridine located in position −2 of a cleavable substrate and C76 (Bravo et al. 1996). Finally, similar experiments were performed using the S+8s4U substrate (data not shown). The predominant species cross-linked with G31 of the P3 stem.
Considering the secondary structure of the delta ribozyme depicted in Figure 1 ▶, this cross-linking data allowed us to propose that the ribozyme is folded in such a manner that it docks the middle of P1 stem onto the P3 stem, while the scissile phosphate is found in close proximity to C76. This is in agreement with the observation that the predominant complex with the s4U at position −1 gave a link with U77 that is adjacent to the essential cytosine. At the other extremity of the P1 stem, the s4U in position +8 cross-links with G31 of the P3 stem. The cross-links with C22 and U23, at the junction of the P3 stem and L3 loop observed for s4U in the middle of the P1 stem (i.e., +4), appeared to be in agreement with this hypothesis. However, the predominant cross-linked species found using the S+4s4U substrate was that with the U17 residue of the P2 stem of the ribozyme, which at first sight seems inconsistent with the previous results. Clearly, a better understanding of the cross-linking reactions during folding, and more specifically for the S+4s4U substrate, would be instructive.
Behavior of the cross-links versus the cleavage reaction
Several experiments were performed to characterize the cross-linking reactions, specifically that with the S+4s4U substrate. To verify whether or not the cross-linked complexes correspond to structures that include an RzS conformation compatible with being an active intermediate in the reaction pathway, each one was isolated and incubated under optimal conditions for self-cleavage (i.e., 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 60 min at 37°C). With the exception of the cross-linked complexes of S+4s4U that give the most intense band (×1; inset Fig. 3A ▶), the two other complexes exhibited self-cleavage activity, although at different levels (Fig. 4A ▶, complexes ×2 and ×3). These results suggest that the species including the cross-link with the residues C22 and U23 correspond to productive ternary complexes. However, the species showing the cross-link with U17 exhibited no detectable self-cleavage under the conditions tested, suggesting that the ribozyme most likely had been cross-linked in a nonproductive complex. Conversely, the predominant complexes cross-linked in the presence of the SC−1s4U and S+8s4U substrates (U77 and G31, respectively) exhibited self-cleavage, suggesting that they were also compatible with being active intermediates in the reaction pathway (data not shown).
FIGURE 4.

Behavior of the cross-link. (A) Self-cleavage assay of three cross-linked complexes obtained from the high yield resolution purification of a single cross-linking experiment with 5′-end-labeled S+4s4U and the ribozyme. Shown in the inset is the corresponding autoradiogram where the first lane is the full-length 11-nt substrate and the last lane is the cleaved 4-nt product. (B) Cross-linking experiments of 32PCp-labeled S+4s4U and the ribozyme in the presence of a chaotropic agent. Various concentrations of urea were added before the addition of the magnesium and UV irradiation. Gray and white bars indicate the percentages of cleavage and cross-linking, respectively. The corresponding autoradiogram is shown in the inset. (C) Percentage of cross-linking of 32PCp-labeled S+4s4U onto the ribozyme when the incubation at 37°C is performed in the presence of magnesium prior to the UV irradiation. The corresponding autoradiogram is shown in the inset.
The hypothesis of a misfolded species being the predominant species cross-linked in the presence of the S+4s4U substrate receives physical support from the demonstration that the addition of urea (up to a concentration of 6 M) prior to UV irradiation does not modify the banding pattern (Fig. 4B ▶). More importantly, increasing amounts of urea rapidly decreased the cross-linking at U17, while the cleavage remained almost unaffected. Because the moderate presence of a chemical denaturant can partially unfold mis-folded structures (Pan et al. 1997), we would expect that the addition of urea prior to the cross-linking step might allow unfolding, at least partially, of the nonproductive structure. As a result, this treatment significantly decreases the percentage of cross-linked complexes, while the level of cleavage increases slightly up to 2.0 M urea before decreasing. The absence of a significant increase in the cleavage level may be explained by several factors that contribute to preventing the proper rearrangement of the RzS complex (e.g., mixture at 4°C, presence of urea, etc.). Together, these results led us to suggest that this misfolded RzS complex may be a kinetic trap. However, this does not exclude the hypothesis that the presence of the cross-link bond prevents the subsequent conformational rearrangment essential for the chemical reaction from taking place.
Subsequent experiments were performed to determine whether the cross-linking reaction occurred before or after the chemical cleavage. The ribozyme and the 3′-end-labeled substrate were incubated together in the presence of magnesium at 37°C for various times prior to cooling the mixture on ice and UV irradiating for 20 min. The longer the incubation time was set under cleavage reaction conditions, the smaller the quantity of cross-linked complexes were detected with the S+4s4U substrate (Fig. 4C ▶). In other words, the lesser the amount of RzS complex detected (i.e., more ribozyme–product, RzP2), the lesser the cross-linking occurred. This indicates that the cross-link occurred before the cleavage reaction (i.e., within the RzS complex). Note that an overexposition of the autoradiogram was required to detect the cross-linked complex (Fig. 4C ▶, inset). Due to the overexposition, the band corresponding to the product appears to be already saturated, which is not the case. This was confirmed using a shorter exposition. When the experiment was performed using a 5′-end-labeled substrate and omitting the visualization of a ribozyme–product cross-link, the results were virtually identical: a major cross-linked complex was detected and its concentration observed to decrease with the incubation time of the cleavage reaction (data not shown). The decrease in concentration of the cross-linked complex was exponential according to a scatter curve graph. Such a representation was not appropriate because the time shown on the x-axis must represent the exact cleavage reaction time. Also, despite the experimental conditions that are set at 37°C, some cleavage might occur due to UV irradiatio′n, which provides a limited amount of heat. This hypothesis receives physical support from the detection of some cleavage product in the first lane of the autoradiogram in Figures 4C ▶ (inset) and 3A ▶ (these lanes correspond to t = 0). Consequently, to avoid misinterpretation, a histogram has been used to illustrate the variation of cross-linked complexes detected as a function of the time of the cleavage reaction.
Finally, the metal ion dependency of the cross-link was compared to that of the cleavage reaction. Both the cross-linking and cleavage reactions were performed in the presence of 10 mM of various metal and nonmetal ions (Table 2 ▶). All bivalent cations that support the catalysis of the delta ribozyme exhibited the cross-linking reaction. The order of reactivity of the bivalent metal ions for the cleavage catalyzed by the ribozyme was in good agreement with that of another study (Wrzensinski et al. 2001). In addition, the cobalt hexamine and monovalent ions (ammonium and sodium), which do not support catalysis, allowed detection of the cross-linked complexes. These results suggest that the requirement of the magnesium observed in the initial cross-linking experiments was for a structural ion. Identical results were obtained when the experiments were repeated using the SC−1s4U (Table 2 ▶).
TABLE 2.
Cross-linking and cleavage intensities observed with the various ions.
| S+4s4U | SC−1s4U | |||
| Ions | cleavage | cross-link | cleavage | cross-link |
| MgCl2 | +++ | +++ | +++ | +++ |
| CaCl2 | +++ | +++ | +++ | ++ |
| SrCl2 | + | ++ | + | ++ |
| MnCl2 | ++ | ++ | ++ | ++ |
| BaCl2 | − | ++ | − | ++ |
| Co(NH3)6Cl3 | − | + | − | + |
| CoCl2 | − | − | − | − |
| TbCl3 | − | − | − | − |
| CdCl2 | − | − | − | − |
| FeCl2 | − | − | − | − |
| ZnCl2 | − | − | − | − |
| PbCl2 | − | − | − | − |
| CsCl | − | − | − | − |
| NH4Cl | − | + | − | + |
| NaCl | − | + | − | + |
Structure of the P1 stem
The cross-link involving the 4-thiouridine in the middle of the P1 stem (i.e., S+4s4U) provides direct evidence of a structural proximity between the middle of both the substrate and the ribozyme. For this event to occur we would expect that this uridine is not part of a Watson-Crick base pair. To investigate this possibility the P1 stem was probed using partial ribonuclease (RNase) hydrolysis. Unfortunately, the probing of a trans-acting delta ribozyme is a lot more complex than that of the equivalent cis-acting version for several reasons, including the fact that, even in the presence of an excess of substrate, a significant proportion of the 5′-end 32P-labeled ribozyme remains unbound. As a consequence, the P1 strand region of these molecules will be hydrolyzed by single-stranded specific nucleases, thereby producing a background on the autoradiograms. Moreover, it is almost impossible to probe the short substrate strand (11 nt) of the P1 stem (J. Ouellet and J.P. Perreault, un-publ.). To avoid these limitations, an equivalent cis-acting version was probed Fig. 5 ▶). The kinetic behavior of this cis-acting delta ribozyme has been previously characterized, and is comparable to that of the trans-acting version used in this work (Ananvoranich and Perreault 2000). As opposed to what is observed with the trans-acting ribozyme, where the ribozyme mutant C76A is completely devoid of any cleavage activity, the equivalent cis-acting molecule (ribozyme C76A) exhibited limited self-cleavage activity. However, a mutant including a guanosine residue at this position (ribozyme C76G; Fig. 5A ▶) did not allow the detection of any self-cleavage activity, even after an extended incubation period. This mutant was probed as an ideal mimic of a precleavage complex.
FIGURE 5.

Nuclease mapping of cis-acting delta ribozymes. (A) Predicted secondary structure and nucleotide sequence of the inactive cis-acting ribozyme used to probe a precleavage version (i.e., ribozyme C76G). The homopurine base pair at the top of the P4 stem is represented by two large dots (G••G), while the Wobble base pair is represented by a single large dot (G•U). (B) Typical autoradiogram of a 7% PAGE gel of a nuclease mapping experiment performed with a 5′-end-labeled ribozyme. Alkaline hydrolysis of the ribozyme was performed to determine the location of each position (lane 1). Lanes 2–6 are incubations of the 5′-end-labeled ribozyme in the presence of RNase T1, T2, V1, A, and U2, respectively. RNase V1 preferentially hydrolyzes double-stranded nucleotides, regardless of the identity of the bases. RNase T1, RNase A, RNase U2, and RNase T2 hydrolyse after single-stranded nucleotides with specificities for guanosines, pyrimidines (C or U), adenines, or regardless of the identity of the base, respectively. Lane 7 is an incubation of the 5′-end-labeled ribozyme alone. The secondary structure motifs of delta ribozyme are indicated on the right. (C, D) Summaries of the nuclease mapping of the P1 stem regions of the pre- and post-cleavage versions, respectively. The inset shows the legend of the nuclease cleavages. Closed symbols represent the most susceptible sites.
To favor folding into a homogenous conformation, the 5′-end 32P-labeled ribozymes were initially heat-denatured and snap-cooled, and then were preincubated in the presence of 10 mM MgCl2. Various RNases were then added and aliquots removed at different time intervals and analyzed on sequencing gels. RNAses V1 and T2 preferentially hydrolyze nucleotides in double-stranded and single-stranded regions, respectively, regardless of the base identity. RNases T1, A and U2 hydrolyze the phosphodiester bond located 3′ of guanosines, pyrimidines (U or C), and adenosines, respectively, preferentially in single-stranded regions. A typical autoradiogram is shown in Figure 5B ▶. This autoradiogram shows that the overall architecture of the ribozyme is respected. For example, the nucleotides forming both the P1 and P3 stems were hydrolyzed by RNase V1, while those of both the L4 loop and J1/2 junction were not. Instead, the latter nucleotides were susceptible to the cleavage activity of RNase T2, while both the P1 and P3 stems were not. Moreover, according to the hydrolysis pattern for C24 and C25, the P1.1 stem appears to be present in a small percentage of the molecules. The hydrolysis pattern of the P1 stem, the region of interest, is summarized in Figure 5C ▶. From the bottom to the top, the nucleotides that form the first two and last three base pairs appear to be significantly hydrolyzed by RNase V1. Conversely, the two nucleotides that form the third base pair (positions 3 and 37) appear largely as single-stranded residues, while those forming the fourth base pair (positions 4 and 36) adopted either one or the other of the conformations depending on the particular molecule. According to this banding pattern, the residues of these two central base pairs are single-stranded in some conformations and base-paired in others, while those in the flanking regions are double-stranded. It might be possible that within the RzS complex resulting from the formation of the P1 stem that these central nucleotides are base-paired, while they become single-stranded in the RzS′ complex after the conformational transition. More importantly, these results confirm the hypothesis that the uridine in position +4 of the substrate has the ability to participate in a tertiary interaction.
To verify if this particular conformation of the P1 stem exists after the cleavage (i.e., post-cleavage), nuclease mapping of the self-cleaved product of an active cis-acting ribo-zyme was performed. The P1 stem is virtually identical, with the exception of the fourth base pair that appears to be exclusively in a single-stranded conformation (Fig. 5D ▶). In general, the nuclease-based secondary structure of the self-cleaved product appeared to be virtually identical to that of the precleavage form, except that the catalytic core (i.e., P1 stem, P3–L3 stem–loop, P1.1 pseudoknot, and J4/2 junction) is less susceptible to all nucleases, indicating that its structure is more compact. Specifically, the P1.1 stem is preferentially folded in the post-cleavage conformation, but not in the precleavage one.
We attempted to perform cross-linking experiments using a 3′-end-product including a s4U in position +4 (7 nt). Only a very small amount of cross-linked complex was observed. The alkaline hydrolysis of this complex supports a cross-link involving the uridine residue at position U17. Based on the nuclease results, the observation of a very small amount of cross-linked complex (RzP2) was not due to U+4 being in a double-stranded structure. This suggests that the s4U of most of the population of RzP2 complexes does not have a partner with which it interacts in a tertiary manner, supporting the notion that the RzS and RzP2 complexes adopt different tertiary structures.
Mutations influencing the cross-links with the S+4s4U substrate
To further characterize the cross-links obtained in the presence of the S+4s4U substrate, the experiments were repeated using different mutated ribozymes. Mutants were synthesized to analyze the nature of the residue in position U17 of the ribozyme (i.e., that which was the most intensively cross-linked by the S+4s4U). The base pair U17-A83 was mutated to either C17–G83 or G17–C83, thereby changing the nature of the residue in position 17, but preserving the base pair Fig. 6 ▶, mutants U17C–A83G and U17G–A83C, respectively). Both of these ribozymes catalyzed the cleavage of the small model substrate with the same efficiency. The cross-linking experiments in the presence of the U17C–A83G mutant led to the observation of a drastic decrease in the information of the complex ×1, which involves the residue in position 17 of the ribozyme. Conversely, the quantity of both the ×2 and ×3 complexes, which involve C22 and U23, increased significantly. The observation of this inverted relationship suggests that the replacement of U17 reduced the importance of the misfolded complex. Similar results were observed in the presence of U17G–A83C (see Fig. 6 ▶), although a smaller proportion of cross-linked complexes with the residue in position 17 was expected because an s4U residue cross-links less efficiently with a purine than a py-rimidine (Favre et al. 1998).
FIGURE 6.

Relative cleavage activity and cross-linking efficiency, with S+4s4U, of site-directed mutants. The relative levels of cleavage and cross-linking are indicated using + symbols. The cross-linked positions were determined by electrophoretic mobility. The star (*) for the cross-link in the presence of the P1.1-UUUU mutant indicated that the RzS complex included a bound with the C24 and not the C22 or U23.
Three mutants were synthesized to permit some characterization of the cross-link involving C22. The base pair G30–C22 was mutated to C30–G22, A30–U22, or G30–U22 Fig. 6 ▶, mutants C22G–G30C, C22U–G30A, and C22U, respectively). The two first mutants exhibited a drastically reduced level of cleavage activity, while no cleavage was detectable for the third mutant. This supports the earlier demonstration of the importance of this base pair in the formation of the P3 stem (for a review, see Been and Wickham 1997). Cross-linking experiments revealed that the level of complex ×1 was either very small or not detectable, while the levels of the ×2 and ×3 complexes were unchanged. These results showed that the nature of the base pair at the bottom of the P3 stem appears to be important for both the cross-linking with S+4s4U and the cleavage activity. More specifically, they demonstrate that it is possible to avoid the cross-linking of the nonproductive complex. Subsequently, two mutants were synthesized and used to characterize the cross-linked U23, the first nucleotide of the L3 loop (mutants U23A and U23C). In both cases, cleavage activity was abolished, in agreement with previous data showing the importance of U23 (which forms a reversed Wobble-type base pair with G28; Tanaka et al. 2002b). These two mutants resulted in the detection of only one cross-linked complex (×1), which corresponded to the misfolded complex. The absence of the ×2 and ×3 complexes suggests that the RzS complex was not properly folded, thereby explaining the absence of cleavage activity.
Finally, two mutants were synthesized with the goal of clarifying when the cross-linking reactions occur along the folding pathway. Initially, the experiment was performed using an inactive ribozyme in which the cytosine in position 76 was mutated to an adenosine Fig. 6 ▶, C76A). This cytosine has been shown to be responsible for the acid-base catalysis mechanism of the ribozyme (Perrotta et al. 1999; Nakano et al. 2000). The cross-linked products were identical to those found with the original ribozyme, but were present (as it was with the original ribozyme). This result suggests that the nature of the base in position 76 is not responsible for the positioning of the scissile phosphate in the catalytic center. Second the experiment was performed using a ribozyme in which the P1.1 pseudoknot cannot be formed Fig. 6 ▶, mutant P.1.1-UUUU). This mutant ribozyme is completely devoid of any catalytic activity (Deschênes et al. 2003). It has been demonstrated that the adoption of the P1.1 pseudoknot requires the previous binding of both the magnesium and the substrate (i.e., folding of the P1 stem) before it is formed (Ananvoranich and Perreault 2000). The P1.1-UUUU mutant ribozyme gives a cross-linked product corresponding to a bond formed between U+4 (from S+4s4U) and C24 Fig. 6 ▶). Taken together, our results indicate that the cross-linking reactions occurred after the binding of both the substrate and magnesium (Fig. 3A ▶), and before both the formation of the P1.1 pseudoknot and the chemical cleavage step.
DISCUSSION
The distinctive features of the delta ribozyme suggest that an understanding of its structure/function relationship will provide unique information that will be of value in the study of other RNAs. Even though delta ribozyme is a small catalytic RNA, its molecular mechanism appears to be complex, and our understanding of it remains limited. Many steps must occur prior to the chemical step. The starting point is the formation of the P1 stem that brings the substrate and ribozyme together through the formation of a seven base pair helix (RzS complex). This step is magnesium independent and requires at least the presence of monovalent ions (Ananvoranich and Perreault 2000). Based on mutagenesis studies it has been proposed that, upon the addition of magnesium, the P1 stem is involved in an essential structural transition (Ananvoranich et al. 1999). This step was proposed to be dependent on the formation of tertiary interactions between nucleotides of the middle of the P1 stem and chemical groups within the catalytic core of the ribozyme. The nuclease probing data presented in this report support the idea that the middle base pairs of the P1 stem of the RzS complex are dynamic, and exists in at least two different conformations, specifically one in which they are single-stranded and one in which they are double-stranded Fig. 5 ▶). Importantly, these data show that the substrate uridine in position +4 has the ability to participate in a tertiary interaction. Based on this observation, cross-linking experiments using the photoagent 4-thiouridine were undertaken to investigate the possibility of distance proximities between bases of the substrate and ribozyme, as well as to understand the dynamic positioning of the substrate within the catalytic core.
The cross-linked complexes
Substrates including an s4U residue in position −1, +4, or +8 were synthesized. The kinetic data obtained for the cleavage activity of these substrates indicated that the introduction of the s4U did not alter the ribozyme’s cleavage activity Fig. 2 ▶; Table 1 ▶). Regardless of the position of the s4U, the formation of cross-linked complexes required the presence of the Rz, the S, and MgCl2. More exhaustive characterization of the metal ion dependence for both the S+4s4U and SC−1s4U substrates showed a perfect correlation between the bivalent cations supporting the catalysis by the ribozyme and the occurrence of the cross-links (Table 2 ▶). However, it was observed that cobalt hexamine supports the occurrence of cross-linked complexes without supporting the catalytic activity. This suggests that outersphere ions can modulate the structure without necessarily behaving as a catalytic ion. This differs from the conclusion of Nakano et al. (2003), who suggested that innersphere ions act as structural ions while outersphere ions act as catalytic ions in a multichannel reaction pathway. However, the latter study was performed with a genomic version of the delta ribozyme that reacts differently with divalent ions when compared to the antigenomic version used in this work. The differences are most likely the result of the presence of the bulged CAA in the upper part of the P4 stem (Wadkins et al. 2001). Results from metal ion-induced cleavages indicated that a magnesium ion was located along the J4/2 junction within the precleavage RzS complex, and at the bottom of the P2 stem in the post-cleavage complex (Lafontaine et al. 1999). Moreover, the crystal structure of a self-cleaved product of the genomic ribozyme revealed the presence of a loosely bound ion near the middle of the P1 stem (Ferré d’Amaré and Dounda 2000). In addition, the results of imino proton NMR analysis suggest that the catalytic magnesion ion binds to a pocket formed by the P1 stem and the L3 loop (Tanaka et al. 2002a). Precise localization of the metal ions within the ribozyme core would be instructive in this regard.
On one hand, some of the cross-linked complexes exhibited self-cleavage activity (Fig. 4A ▶). For example, the species including the cross-link with residues C22 and U23 (S+4s4U), and U31 (predominant species with S+8s4U) and U77 (predominant species with SC−1s4U), appeared as productive complexes. It is tempting to suggest that the latter correspond to active ternary RzS complexes. However, it is not impossible that these complexes arose from misfolded complexes that can refold into active conformations. On the other hand, the species including a cross-link with U17 (the predominant species with the S+4s4U) did not self-cleave, suggesting that this nonproductive complex was a mis-folded species that is, in the extreme, a kinetic trap. This conclusion received additional physical support from the demonstration that the presence of urea did unfold this misfolded RzS complex. In the course of this work, we also observed that the cross-link between SC−1s4U and U68 corresponds to another misfolded RzS complex (data not shown).
To provide a model representation of these data, a three-dimensional structure of the delta ribozyme was developed using the software MC-Sym (Major et al. 1991). As the presence of the P1.1 pseudoknot was shown to be undesirable for the cross-link to occur Fig. 6 ▶), the RzS complex model was developed without this stem. Moreover, the cross-linking data was not included as distance constraints in the modeling. One of the structures obtained is illustrated in Figure 7A ▶ (see also Supplemental Representation Fig. 8 at http://132.210.163.235/jo/Figure8.html to view a 360° rotation around the y-axis of the structure). This provides a model structure for the initial RzS complex without magnesium ions. Note that in this conformation the U+4 of the substrate is distant from both the C22 and U23 of the ribozyme. From this model structure, the Steered Molecular Dynamics software package (SMD, with the VMD and NAMD software; Humphrey et al. 1996; Kalé et al. 1999) was used to bring the uridine residue in position +4 of the substrate in close proximity (~3 Å, corresponding to the length of a bound) to either C22 and U23, or U17, of the ribozyme (Fig. 7B,C ▶, respectively). When the U+4 of the substrate is near the bottom of the P3 stem, the substrate C−1 and U+8 are in close proximity to the ribozyme U77 and G31, respectively (Fig. 7B ▶). This observation supports the idea that these cross-links are part of complexes compatible with being active species present along the folding pathway. According to this model structure, the catalytic cytosine in position 76 is very close to the scissille phosphate, as has been observed by crystal analysis (Ferré D’Amaré 1998, 2000). Conversely, the cross-link between U+4 and U17 cannot be formed in this complex. In contrast, when U+4 comes near U17 of the P2 stem, neither of the other interactions appears feasible (Fig. 7C ▶). In this structure the P1 stem is almost completely disrupted. Note that this misfolded complex is only a hypothetical visualization, most likely this RzS* complex is, in fact, misfolded to a greater extent.
FIGURE 7.

Three-dimensional representations of the ribozyme–substrate complexes. (A) Initial ribozyme–substrate complex (RzS). (B) Active ribozyme–substrate complex (RzS′) where the U+4 of the substrate is stacked between both the C22 and U23 of the ribozyme. (C) Inactive ribozyme–substrate complex (RzS*) where the U+4 of the substrate is stacked to the U17 of the ribozyme. The P1 stem is orange, the P2 blue, the P3 purple, and the P4 cyan. The J1/4 junction is white, the J4/2 green, and the L3 loop yellow. The nucleotides U+4 of the substrate and C22 and U23 of the ribozymes are red. The U17 of the ribozyme is blue (C). The C−1 of the substrate and the U77 of the ribozyme are pink. Finally, the C76 of the ribozyme is green. Supplementary representations including a 360° rotation are available at http://132.210.163.235/jo/Figure8.html.
More generally, these three-dimensional structures constitute interesting models of the antigenomic delta ribozyme precleavage complexes. It is important to note that the presence of the P1.1 stem is not yet formed in these structures, although its presence has been shown to be absolutely required for the chemical reaction to occur (Wadkins et al. 1999; Nishikawa and Nishikawa 2000; Deschênes et al. 2003). Most likely, the P1.1 stem is formed almost simultaneously with the chemical step. The formation of this small pseudoknot might be the event that completes the formation of the network of interactions within this highly structured catalytic center.
Cross-linking versus the kinetic pathway
According to the data obtained, cross-linking required the presence of magnesium, and occurred either after, or simultaneously with, the conformational transition involving the P1 stem (Ananvoranich and Perreault 2000). Furthermore, it was shown that the cross-link occurred before both the chemical step and the folding of the P1.1 stem (Figs. 4C ▶, 6 ▶). Considering the minimal kinetic pathway described previously (Mercure et al. 1998; Chadalavada et al. 2002; Shih and Been 2002), the cross-linking data can be explained using the following scheme:
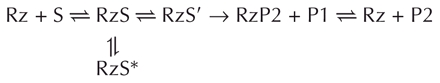 |
where the P1 and P2 correspond to the 5′-end and 3′-end products, respectively. Upon the addition of magnesium, two types of cross-linked complexes were formed (i.e., RzS* or RzS′). The RzS* are nonproductive complexes like that including the cross-link between U+4 of the substrate and U17 of the ribozyme. Conversely, the RzS′ complexes correspond to complexes compatible with being active intermediates present along the folding pathway. The cross-links between the substrate SC−1s4U and U77, as well as the substrate S+4s4U and either the C22 or U23 of the ribozyme, correspond to this second type of complex. Furthermore, it was observed that when the correctly folded RzS is rearranged to the RzS′ state, the substrate is then cleaved. The latter would drive the equilibrium from the nonproductively RzS* to the productive folded RzS according to Le Chatelier’s principle only if the misfolded complex can adopt an active conformation. Clearly, the delta ribozyme has an heterogeneity of intermediates that have the potential to cleave the substrate.
Previous biochemical analyses have revealed that the limiting step of this kinetic pathway is the conformational rearrangement from the RzS to the RzS′ complex (Mercure et al. 1998). More specifically, it was suggested that the slow folding of the P1.1 pseudoknot is the rate-limiting step (Chadalavada et al. 2002). The ground state destabilization of the RzS complex, and the formation of a very stable RzP2 complex, were proposed to be two of the forces driving the kinetic pathway (Shih and Been 2002). RzP2 includes several features, such as the P1.1 pseudoknot (Ferré D’Amaré et al. 1998; Wadkins et al. 1999; Deschênes et al. 2003) and the U23-G28 base pair (Tanaka et al. 2002b), that are stabilizing and have not yet been described as being part of the RzS′ complex. Structural differences between the pre- and the post-cleavage conformations were revealed using various approaches. For example, 2-aminopurine fluorescence quenching experiments showed a local conformational change in the catalytic core of delta ribozyme along the pathway from precursor to product (Harris et al. 2002). More specifically, it was shown that the trefoil turn near the catalytic cytosine is found within the post-cleavage complex (Ferré D’Amaré and Doudna 1998), but not in the precleavage one (Harris et al. 2002). This trefoil turn motion is kinetically coincidental to a global structural change as monitored by florescence resonance energy transfert (Pereira et al. 2002). Moreover, the nuclease probing data presented above indicate that the post-cleavage complex was significantly more compact than the precleavage one Fig. 5 ▶).
At this point, the pathway cannot be subdivided into more steps. However, it is tempting to make some speculations, including: (1) initial interactions occur between the middle of the P1 stem and the bottom of the P3 stem, thereby bringing the P1 stem inside the catalytic center; and, (2) subsequent interactions between C−1 of the substrate and the ribozyme’s catalytic C76 take place. This would position the scissile phosphate in the appropriate orientation for transesterification. Clearly, one of the next exciting steps in the study of the molecular mechanism of delta ribozyme would be to divise a system capable of dividing the folding pathway into many consecutive steps. This should allow a more thorough description of the intimate details encompassed in the behavior and folding of catalytic as well as noncatalytic RNAs.
MATERIALS AND METHODS
DNA construct
The DNA templates for the original trans-acting ribozyme, as well as those for the derived mutants were synthesized as described previously (Mercure et al. 1998). Briefly, pairs of complementary and overlapping DNA oligonucleotides corresponding to the T7 RNA promoter followed by the full-length ribozyme were synthesized, annealed, and cloned into PstI and SphI codigested pUC19. The sequences of all mutant ribozyme genes were confirmed by DNA sequencing. The construction of the cis-acting delta ribozymes has been reported previously (Ananvoranich and Perreault 2000).
RNA synthesis
Ribozymes were synthesized by in vitro run-off transcription as described previously (Deschênes et al. 2003). In brief, the transcriptions were performed in solution (100 μL) using linearized recombinant plasmids (17 μg) reported previously in the presence of RNA Guard (27 units, Amersham Biosciences), 80 mM HEPES-KOH pH 7.5, 24 mM MgCl2, 2 mM spermidine, 40 mM DTT, 5 mM of each NTP, 0.01 units yeast pyrophosphatase (Roche Diagnostic), and 10 μg of purified T7 RNA polymerase at 37°C for 2 h. The reactions were stopped by adding 5 units of DNase I (RNase free, Promega) and incubating at 37°C for 30 min. Formamide dye buffer (95% formamide, 10 mM EDTA, 0.025% bromophenol blue and 0.025% xylene cyanol) was added and the ribozymes fractionated on long 8% denaturing PAGE gels (19:1 ratio of acryl-amide to bisacrylamide) in buffer containing 45 mM Tris-borate pH 7.5, 7 M urea, and 1 mM EDTA. The reaction products were visualized by UV shadowing and the bands corresponding to the ribozymes were cut out. The transcripts were eluted from the gel slices overnight at room temperature in a solution containing 0.1% SDS and 0.5 M ammonium acetate. The resulting mixtures were passed through G-50 Sephadex spun columns (Amersham Biosciences) and the RNA precipitated by the addition of 0.1 volumes of 2 M sodium acetate pH 4.5 and 2.5 volumes of ethanol. After washing in 70% ethanol and drying, the pellets were dissolved in ultrapure water and the quantity of RNA determined by spectrophotometry at 260 nm.
Substrates and analogs
The original 11-nt substrate was synthesized by in vitro transcription as described previously (Deschênes et al. 2003). Briefly, complementary and overlapping oligonucleotides were annealed in 20 μL of buffer containing 10 mM Tris-HCl pH 7.5, 10 mM MgCl2 and 50 mM KCl by incubating at 95°C for 2 min and then allowing the solution to slowly cool to 37°C. The resulting DNA duplexes (500 pmole) were then used as templates for in vitro transcription reactions, and the RNA purified using 20% PAGE gels as described above. For the production of S+4s4U, the100 mM UTP was replaced by 100 mM s4UTP (TriLink BioTechnologies). SC−1s4U and S+8s4U were synthetized with 2′ACE chemistry by Dharmacon Inc. and deprotected as recommended by the manufacturer. Formamide dye buffer was then added and the modified RNA purified on denaturing 20% PAGE gels and recovered as described above.
End-labeling of RNA
Labeling with [γ-32P]-ATP
Either purified substrates or ribozymes (40 pmole) were dephos-phorylated in a final volume of 50 μL containing 200 mM Tris-HCl, pH 8.0, 10 units RNA Guard, and 0.2 units of calf intestinal alkaline phosphatase (Amersham Biosciences) at 37°C for 30 min. The reactions were purified by extracting twice with phenol:chloroform, the RNA then precipitated with ethanol, washed with 70% ethanol, and dried. Dephosphorylated substrates (6 pmole) were 5′-end-labeled in a final volume of 10 μL containing 3.2 pmole [γ-32P]-ATP (6000 Ci/mmole, Amersham Biosciences), 10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 50 mM KCl, and 3 units of T4 polynucleotide kinase (Amersham Biosiences) at 37°C for 90 min. The reactions were stopped by the addition of formamide dye buffer (5 μL), and the mixtures fractionated through denaturing 20% PAGE gels. The bands containing the appropriate 5′-end-labeled RNAs were excised, and the nucleic acid recovered as described above. The labeling of the ribozymes was made at a higher concentration to obtain more radioactive cross-linked complexes for further purification. The dephosphorylated ribozymes (15 pmole) were 5′-end-labeled in a final volume of 20 μL containing 11.2 pmole [γ-32P]-ATP (6000 Ci/mmole) in the same reaction buffer as described previously. The reaction was incubated at 37°C for 180 min, and the ribozyme then purified by extracting twice with phenol/chloroform, precipitating with ethanol, washing with 70% ethanol and drying.
Labeling with [γ32P]Cp
For 3′-end-labeling, both the substrates (10 pmole) and the ribozymes (20 pmole) were incubated overnight at room temperature in a final volume of 10 μL containing 50 mM Tris-HCl pH 7.8, 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 10% dimethylsulfoxide, 10 pmole [32P]Cp (3000 Ci/mmole, Amersham Biosciences) and 10 U of T4 RNA ligase (New England Biolabs). The labeled RNAs were then purified on 7M urea 20% PAGE gels and recovered as described above.
Cleavage reactions and kinetic assays
Unless otherwise stated, cleavage reactions were carried out in 20 μL reaction mixtures containing 50 mM Tris-HCl, pH 7.5, and 10 mM MgCl2 at 37°C under single turnover conditions ([Rz] >> [S]; Fiola and Perreault 2002). Prior to the reaction, trace amounts of 5′-end-labeled substrates (<1 nM) and nonradioactive ribozymes (200 nM) were mixed together, heated at 70°C for 1 min, snap-cooled on ice for 3 min, and then incubated at 37°C for 5 min. Following this preincubation step, the cleavage reactions were initiated by the addition of MgCl2. Aliquots (2–3 μL) were removed either at various times up to 1 h or until the end point of the cleavage was reached, and were quenched by the addition of ice-cold formamide dye buffer (8 μL). The mixtures were fractionated on denaturing 20% PAGE gels and exposed to PhosphorImager screens (Molecular Dynamics). The extent of cleavage was determined from measurements of the radioactivity present both in the substrate and in the 5′ product bands at each time point using the ImageQuant software. The fractions of cleaved substrate were determined, and the rate of cleavage (kobs) obtained by fitting the data to the equation At = Ao(1−e−kt), where At is the percentage of cleavage at time t, Ao is the maximum percent cleavage (or the end point of cleavage), and k is the rate constant (kobs). Each rate constant was calculated from at least two independent measurements. Kinetic assays were performed using various concentrations of ribozyme (5 nM to 400 nM). The values of kobs obtained were plotted in relation to ribozyme concentration to determine the other kinetic constants (k2, KM, and k2/KM).
Cross-linking experiments
Typical cross-linking experiments were performed in a final volume of 20 μL containing 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 0.2 pmole of ribozyme, and a trace amount of 5′-end-labeled substrate (i.e., single turnover conditions). Once the ribozyme and substrate were mixed together, but prior to the addition of magnesium, they were heat denatured, snap-cooled, and then prein-cubated with the Tris at 37°C. The reactions were then chilled on ice for 15 min in a 96-well plate cover. Following the addition of MgCl2 to a final concentration of 10 mM, the samples were rapidly irradiated in a UV cross-linker (UV Stratalinker 2400, Stratagene) equipped with a 15W 366 nm bulb for 20 min on ice. Exceptionally, the substrate was cleaved at 37°C for different intervals of time before the UV irradiation (e.g., the experiment presented in Fig. 4C ▶). Furthermore, to unfold misfolded RNA structures, some cross-linking experiments were performed in the presence of urea (Fig. 4B ▶). In that case, various concentrations of urea were added followed by incubation at 37°C for 5 min prior to chilling the mixtures on ice and performing the UV irradiation. Formamide dye buffer (10 μL) was then added to each sample prior to fractionation by denaturing 20% PAGE gel electrophoresis. The resulting bands were visualized using a PhosphorImager. When performing cross-linking experiments in the presence of various ions, a final concentration of 10 mM was used in all cases.
With the purpose of performing self-cleavage experiments, the 5′-end-labeled S+4s4U substrate (~10 pmole) was cross-linked to the ribozyme (1 pmole) and the resulting complexes were cut out of the gel and eluted as described above. The purified complexes were snap-cooled and subjected to self-cleavage with the addition of magnesium (10 mM). The cleavage reactions were performed for 60 min at 37°C, then formamide dye buffer (10 μL) was added and the samples were fractionated on a 20% denaturing PAGE gel and visualized using a PhosphorImager.
Mapping the cross-linking sites
To map the sites of the cross-links, the experiments were performed under multiple turnover conditions ([S] > [Rz]) using a nonradioactive substrate (15 pmole) and either 5′- or 3′-end-labeled ribozymes (5 pmole). The experiments were performed as described above except that the samples were analyzed on 8% PAGE gels. The gels were visualized by autoradiography, the bands corresponding to the RzS complexes were cut out and the RNA species recovered as described previously. The resulting pellets were dissolved in ultrapure water (8 μL), and half of the samples were submitted to alkaline hydrolysis (NaOH) for 1 min. The reactions were quenched on ice by the addition of formamide dye (4 μL) and 1 M Tris-HCl pH 7.5 (2 μL), and then analyzed on denaturing 10% PAGE gels. The gels were prerun for 1 h at 60 W with 1× TBE buffer in both chambers, then the buffer in the lower chamber was changed to 1 M NaOAc and the gel prerun for a further 30 min before loading the samples. To permit the localization of the cross-linking sites, partial alkaline and ribonuclease T1 (i.e., using 2.5 U RNase T1, Boehringer Mannheim) hydrolyses of noncross-linked, labeled ribozymes were performed and analyzed on the same gels.
Nuclease digestions and alkaline hydrolysis
Trace amounts (~0.01 pmole) of 5′-end-labeled ribozymes were successively incubated for 2 min at 95°C, 2 min on ice, and 5 min at 37°C in a 10 μL of a mixture containing 20 mM Tris-HCl, pH 7.5, 100 mM NH4Cl, and either in the absence or the presence of 10 mM MgCl2. Either RNase T1 (0.6 U), RNase T2 (0.015 U), RNase V1 (0.0002 U), RNase A (2.5 × 10−6 U) or RNase U2 (1 U) was then added and the mixture incubated at 37°C for 2 min. Reaction aliquots (3 μL) were removed and quenched by the addition of loading solution (7 μL, 97% formamide, 5 mM EDTA, 0.05% xylene cyanol). For alkaline hydrolysis, the 5′-end-labeled ribozyme was resuspended in water (5 μL) and 1 N NaOH (1 μL) added. The reaction was incubated at room temperature for 1–5 min, and then quenched by the addition of 1 M Tris-HCl, pH 7.5 (3 μL) and loading buffer (7 μL). The resulting mixtures were analyzed on denaturating 7% PAGE gels.
Molecular modeling
Models of the ribozyme–substrate complex were generated using the constraint-satisfaction program MC-Sym 3.3.2 (Major et al. 1991). Then a selected structure was modified by Steered Molecular Dynamics (SMD) with the softwares VMD 1.8.2 and NAMD 2.5 (Humphrey et al. 1996; Kalé et al. 1999). Figures were prepared using nuccyl 1.1 software (www.mssm.edu/students/jovinl02/research/nuccyl.html) to simplify the representation of bases as cylinders into the PyMOL 0.93 software (http://pymol.sourceforge.net). The coordinates of the pdb are available on demand from the corresponding author.
Acknowledgments
The authors thank Mr. Dominique Lévesque for helpful technical assistance in the nuclease probing. This work was supported by a grant from the Canadian Institute of Health Research (CIHR) to J.P.P. The RNA group is supported by grants from both the CIHR and Fonds FCAR (Québec). J.O. was the recipient of a predoctoral fellowship from Fonds FCAR. J.P.P. is an Investigator from the CIHR.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.7230604.
REFERENCES
- Ananvoranich, S. and Perreault, J.P. 2000. The kinetic and magnesium requirements for the folding of antigenomic δ ribozymes. Biochem. Biophys. Res. Commun. 270: 600–607. [DOI] [PubMed] [Google Scholar]
- Ananvoranich, S., Lafontaine, D.A., and Perreault, J.P. 1999. Mutational analysis of the antigenomic trans-acting delta ribozyme: The alterations of the middle nucleotides located on the P1 stem. Nucleic Acids Res. 27: 1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Been, M.D. and Wickham, G.S. 1997. Self-cleaving ribozymes of hepatitis delta virus RNA. Eur. J. Biochem. 247: 741–753. [DOI] [PubMed] [Google Scholar]
- Bergeron Jr., L., Ouellet, J., and Perreault, J.P. 2003. Ribozyme-based gene-inactivation systems require a fine comprehension of their substrates specificities; The case of delta ribozyme. Curr. Med. Chem. 10: 2589–2597.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua, P.C., Brown, T.S., Nakano, S., and Yajima, R. 2003. Catalytic roles for proton transfer and protonation in ribozymes Biopolymers 73: 90–109. [DOI] [PubMed] [Google Scholar]
- Blount, K.F. and Uhlenbeck, O.C. 2002. Internal equilibrium of the hammerhead ribozyme is altered by the length of certain covalent crosslinks. Biochemistry 41: 6834–6841. [DOI] [PubMed] [Google Scholar]
- Bravo, C., Lescure, F., Laugaa, P., Fourrey, J.L., and Favre, A. 1996. Folding of the HDV antigenomic ribozyme pseudoknot structure deduced from long-range photocrosslinks. Nucleic Acids Res. 24: 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadalavada, D.M., Senchak, S.E., and Bevilacqua, P.C. 2002. The folding pathway of the genomic hepatitis delta virus ribozyme is dominated by slow folding of the pseudoknots. J. Mol. Biol. 317: 559–575. [DOI] [PubMed] [Google Scholar]
- Deschênes, P., Ouellet, J., Perreault, J., and Perreault, J.P. 2003. Formation of the P1.1 pseudoknot is critical for both the cleavage activity and substrate specificity of an antigenomic trans-acting hepatitis delta ribozyme. Nucleic Acids Res. 31: 2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre, A., Saintome, C., Fourrey, J.L., Clivio, P., and Laugaa, P. 1998. Thio nucleobases as intrinsic photoaffinity probes of nucleic acid structure and nucleic acid protein interactions. J. Photochem. Photobiol. 42: 109–124. [DOI] [PubMed] [Google Scholar]
- Ferré D’Amaré, A.R., Zhou, K., and Doudna, J.A. 1998. Crystal structure of a hepatitis δ virus ribozyme. Nature 395: 567–574. [DOI] [PubMed] [Google Scholar]
- Ferré D’Amaré, A.R. and Doudna, J.A. 2000. Crystallization and structure determination of a hepatitis delta virus ribozyme: Use of the RNA-binding protein U1A as a crystallization module. J. Mol. Biol. 295: 541–556. [DOI] [PubMed] [Google Scholar]
- Fiola, K. and Perreault, J.-P. 2002. Kinetic and binding analysis of the catalytic involvement of ribose moieties of a trans-acting delta ribozyme. J. Biol. Chem. 277: 26508–26516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, D.A., Rueda, D., and Walter, N.G. 2002. Local conformational changes in the catalytic core of the trans-acting hepatitis delta virus ribozyme accompany catalysis. Biochemistry 41: 12051–12061. [DOI] [PubMed] [Google Scholar]
- Hiley, S.L., Sood, V.D., Fan, J., and Collins, R.A. 2002. 4-Thio-U crosslinking identifies the active site of the VS ribozyme. EMBO J. 21: 4691–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey, W., Dalke, A., and Schulten, K. 1996. VMD—Visual molecular dynamics. J. Mol. Graphics 14: 33–38. [DOI] [PubMed] [Google Scholar]
- Kalé, L., Skeel, R., Bhandarkar, M., Brunner, R., Gursoy, A., Krawetz, N., Phillips, J., Shinozaki, A., Varadarajan, K., and Schulten, K. 1999. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 151: 283–312. [Google Scholar]
- Lafontaine, D.A., Ananvoranich, S., and Perreault, J.P. 1999. Presence of a coordinated metal ion in a trans-acting antigenomic delta ribozyme. Nucleic Acids Res. 27: 3236–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major, F., Turcotte, M., Gautheret, D., Lapalme, G., Fillion, E., and Cedergren, R. 1991. The combination of symbolic and numerical computation for three-dimensional modeling of RNA. Science 253: 1255–1260. [DOI] [PubMed] [Google Scholar]
- Mercure, S., Lafontaine, D., Ananvoranich, S., and Perreault, J.-P. 1998. Kinetic analysis of delta ribozyme cleavage. Biochemistry 37: 16975–16982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, S.I., Chadalavada, D.M., and Bevilacqua, P.C. 2000. General acid-base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science 287: 1493–1497. [DOI] [PubMed] [Google Scholar]
- Nakano, S.I., Cerrone, A.L., and Bevilacqua, P.C. 2003. Mechanistic characterization of the HDV genomic ribozyme: Classifying the catalytic and structural metal ion sites within a multichannel reaction mechanism. Biochemistry 42: 2982–2994. [DOI] [PubMed] [Google Scholar]
- Nishikawa, F. and Nishikawa, S. 2000. Requirement of the canonical base pairing in the short pseudoknot structure of genomic hepatitis delta virus ribozyme. Nucleic Acids Res. 28: 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, J., Thirumalai, D., and Woodson, S.A. 1997. Folding of RNA involves parallel pathways. J. Mol. Biol. 273: 7–13. [DOI] [PubMed] [Google Scholar]
- Pereira, M.J.B., Harris, D.A., Rueda, D., and Walters, N.G. 2002. Reaction pathway of the trans-acting hepatitis delta virus ribozyme: A conformational change accompanies catalysis. Biochemistry 41:730–740. [DOI] [PubMed] [Google Scholar]
- Perrotta, A.T., Shih, I., and Been, M.D. 1999. Imidazole rescue of a cytosine mutation in a self-cleaving ribozyme. Science 286: 123–126. [DOI] [PubMed] [Google Scholar]
- Pinard, R., Heckman, J.E., and Burke, J.M. 1999. Alignment of two domains of the hairpin ribozyme–substrate complex defined by interdomain photoaffinity crosslinking. J. Mol. Biol. 287: 239–251. [DOI] [PubMed] [Google Scholar]
- Saenger, H.L., Klotz, G., Riesner, D., Gross, H.L., and Kleinschmidt, A.K. 1976. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-laired rod-like structures. Proc. Natl. Acad. Sci. 73: 3852–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih, I.H. and Been, M.D. 2002. Catalytic strategies of the hepatitis delta virus ribozymes. Annu. Rev. Biochem. 71: 887–917. [DOI] [PubMed] [Google Scholar]
- Tanaka, Y., Hori, T., Tagaya, M., Sakamoto, T., Kurihara, M., and Uesugi, S. 2002a. Imino proton NMR analysis of HDV ribozymes: Nested double pseudoknot structure and Mg2+ ion-binding site close to the catalytic core in solution. Nucleic Acids Res. 30: 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, Y., Tagaya, M., Hori, T., Sakamoto, T., Kurihara, Y., Katahira, M., and Uesugi, S. 2002b. Cleavage reaction of HDV ribozymes in the presence of Mg2+ is accompanied by a conformational change. Genes Cells 7: 567–579. [DOI] [PubMed] [Google Scholar]
- Wadkins, T.S., Perrotta, A.T., Ferré D’Amaré, A.R., Doudna, J.A., and Been, M.D. 1999. A nested double pseudoknot is required for self-cleavage activity of both the genomic and antigenomic hepatitis delta virus ribozymes. RNA 6: 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadkins, T.S., Shih, I.H., Perrotta, A.T., and Been, M.D. 2001. A pH-sensitive RNA tertiary interaction affects self-cleavage activity of the HDV ribozymes in the absence of added divalent metal ion. J. Mol. Biol. 305: 1045–1055. [DOI] [PubMed] [Google Scholar]
- Wrzesinski, J., Legiewick, M., Smolska, B., and Ciesiolka, J. 2001. Catalytic cleavage of cis- and trans-acting antigenomic delta ribozymes in the presence of various divalent metal ions. Nucleic Acids Res. 29: 4482–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]