

Abstract
A partition function calculation for RNA secondary structure is presented that uses a current set of nearest neighbor parameters for conformational free energy at 37°C, including coaxial stacking. For a diverse database of RNA sequences, base pairs in the predicted minimum free energy structure that are predicted by the partition function to have high base pairing probability have a significantly higher positive predictive value for known base pairs. For example, the average positive predictive value, 65.8%, is increased to 91.0% when only base pairs with probability of 0.99 or above are considered. The quality of base pair predictions can also be increased by the addition of experimentally determined constraints, including enzymatic cleavage, flavin mono-nucleotide cleavage, and chemical modification. Predicted secondary structures can be color annotated to demonstrate pairs with high probability that are therefore well determined as compared to base pairs with lower probability of pairing.
Keywords: RNA partition function, RNA secondary structure, statistical mechanics
INTRODUCTION
RNA plays many diverse roles in biology, including catalyzing peptide bond formation (Nissen et al. 2000; Hansen et al. 2002), catalyzing RNA splicing (Doudna and Cech 2002), localizing protein (Walter and Blobel 1982), and flagging development (Lagos-Quintana et al. 2001; Lau et al. 2001). New roles are being found for RNA, and the completion of whole genome projects (Goffeau et al. 1996; C. elegans Sequencing Consortium 1998; Adams et al. 2000; the Arabidopsis Genome Initiative 2000; Fraser et al. 2000; International Human Genome Sequencing Consortium 2001; Venter et al. 2001; Mouse Genome Sequencing Consortium 2002) provides the opportunity to find many new functional noncoding RNA sequences (Eddy 2001).
To understand the detailed mechanism of action of an RNA sequence, a model of the structure is required. The experimental methods used for determining structure, including NMR and X-ray crystallography, are time consuming and can depend on initial secondary structure models for developing constructs. Comparative sequence analysis (Pace et al. 1999) is the gold standard for RNA secondary structure in the absence of an all-atom model. A recent study demonstrated that > 97% of base pairs in ribosomal RNA secondary structures, predicted by comparative sequence analysis (Gutell et al. 2002), were subsequently demonstrated in high-resolution crystal structures (Ban et al. 2000; Schluenzen et al. 2000; Wimberly et al. 2000). Comparative sequence analysis, however, requires a large number of homologous sequences and is labor intensive.
In the absence of many homologous sequences, free energy minimization by dynamic programming can be used to predict the structure of a single sequence with an average of 73% accuracy (Mathews et al. 2004). This accuracy is sufficient to serve as a starting point for building an alignment for comparative sequence analysis or as an aid for designing RNA sequences (Mathews et al. 1997; Pappalardo et al. 1998; Diamond et al. 2001; Flamm et al. 2001), but improvements in the accuracy of base pair predictions would clearly be useful.
The predicted minimum free energy (MFE) structure provides a single best guess for the secondary structure, but it assumes that the secondary structure is at equilibrium, that there is a single conformation for the RNA, and that the thermodynamic parameters for evaluating conformation free energies are without error. One method to represent other possible or competing structures is to compute suboptimal secondary structures with free energies similar to the lowest free energy structure (Zuker 1989). Graphically, the lowest free energy structure possible for each base pair can be displayed in an energy dot plot (Zuker and Jacobson 1995). Information derived from the energy dot plot can also be used to color annotate base pairs in predicted secondary structures (Zuker and Jacobson 1998) to graphically demonstrate pairs that are contained in alternative low free energy structures.
The prediction of suboptimal secondary structures has been extended to predicting all suboptimal secondary structures within a given energy increment of the MFE structure (Wuchty et al. 1999). A natural extension of suboptimal secondary structure prediction would be to compute a partition function, which sums the contribution of all structures, weighted by their Boltzmann probabilities. However, this direct approach to calculating the partition function is impractical because the number of predicted secondary structures increases exponentially as a function of the size of the energy increment (Wuchty et al. 1999).
McCaskill (1990) pioneered a dynamic programming algorithm that can determine the partition function for secondary structure formation in O(N3) time and O(N2) storage, where N is the number of nucleotides in the sequence. This partition function calculation provides (among other things) the base pairing probability for each possible base pair in the sequence, which can be displayed in a probability dot plot. It has been implemented in the Vienna RNA package (Hofacker et al. 1994; Hofacker 2003) and has also been parallelized for use on computer clusters (Fekete et al. 2000). Previously it has been shown that base pairs in the MFE structure with few competing probable pairs (as measured by Shannon entropy) are more likely to be correctly predicted for small and large subunit rRNA (Huynen et al. 1997). Ding and Lawrence extended the partition function calculation to compute a statistically valid sample of secondary structures in the Boltzmann ensemble and calculate sampling statistics of structural features (Ding and Lawrence 2001, 2003).
In this study, I present an algorithm for computing RNA secondary structure partition functions using dynamic programming. This algorithm utilizes a recent set of nearest neighbor parameters for determining the free energy at 37°C for an RNA secondary structure (Mathews et al. 2004), including coaxial stacking of helices, and remains O(N3) in time and O(N2) in storage. In the predicted minimum free-energy structure, highly probable base pairs (as predicted by the partition function) are shown to be the pairs most likely to be in the known structure in a database of diverse RNA sequences. For example, on average, 91.0% of base pairs in the minimum free energy structure with a probability of 0.99 or greater of pairing are in the known structure based on comparative sequence analysis. Structures can be color annotated to show the probability of pairing and this annotation quickly demonstrates base pairs that are predicted with the highest confidence. A novel application of the partition function calculation is presented in which secondary structures, composed of highly probable pairs, are generated. This algorithm can also constrain the partition function using data determined by experiments, such as flavin mononucleotide (FMN) photocleavage (Burgstaller and Famulok 1997; Burgstaller et al. 1997), chemical modification (Ehresmann et al. 1987), and enzymatic cleavage (Knapp 1989). Predicted MFE RNA secondary structures with constraints determined by experiment are found to be more well determined than structures predicted without constraints.
RESULTS
A dynamic programming algorithm for the determination of an RNA secondary structure partition function, utilizing a current set of free energy parameters for secondary structure formation in RNA (Mathews et al. 2004), was written based on the method of McCaskill (1990) to predict the probability for each possible base pair in the sequence. This algorithm includes explicit coaxial stacking interactions, using experimentally determined free energy increments (Walter et al. 1994a, 1994b; Kim et al. 1996), but remains O(N3) in time and O(N2) in storage. Two types of coaxial stacking interactions are allowed, direct end-to-end stacking of adjacent helices and coaxial stacking mediated by a single mismatched pair between helices (Mathews et al. 2004). The Materials and Methods section presents the recursions used in the algorithm. In spite of the extra recursions required to include coaxial stacking interactions, the calculation is rapid for most sequences of interest. Table 1 shows the calculation time and memory requirements for sequences from 77 to 2904 nt.
TABLE 1.
Calculation size and time as a function of sequence length
| Length (nt) | RNA | Timea (h:min:sec) | Memory (MB) |
| 77 | E. coli arginine tRNA | <0:00:01 (<0:00:01) | 15.6 |
| 268 | Bacillus subtilis SRP | 0:00:04 (0:00:02) | 34.0 |
| 433 | Tetrahymena thermophila IVS LSU group I intron | 0:00:17 (0:00:06) | 39.6 |
| 631 | Saccharomyces cerevisiae A5 group II intron | 0:00:54 (0:00:15) | 49.9 |
| 1542 | E. coli small subunit rRNA | 0:19:25 (0:03:51) | 144.7 |
| 2904 | E. coli large subunit rRNA | 3:05:22 (1:07:45) | 430.3 |
aCalculated on a laptop computer with a Pentium 4, 3.06-GHz processor, and 1 GB of RAM using the Microsoft C++.NET compiler and Microsoft Windows XP Professional. In parentheses are calculation times when the recursions involving coaxial stacking are not used, although the memory for storage of the coaxial stacking contribution, Wcoax, is allocated. The calculation time involved in coaxial stacking is significant, but not prohibitive.
Because the same energy rules are used by the partition function algorithm and the secondary structure prediction algorithm, direct analysis of the predicted MFE structure using predicted base pairing probabilities, PBP, is possible. Of primary interest is the accuracy of RNA secondary structure prediction for sequences with known secondary structures. This can be measured in two ways, as sensitivity or positive predictive value. The sensitivity of base pair prediction for the predicted MFE structure has been reported previously (Mathews et al. 1999b, 2004) and is the percentage of base pairs in the structure determined by comparative analysis that are contained in the predicted MFE structure. The positive predictive value tabulates the results in the reverse fashion and is the percentage of base pairs in the predicted MFE structure that are in the structure determined by comparative sequence analysis. As shown in Table 2 for a diverse database of RNA sequences with structures determined by comparative sequence analysis (Larsen et al. 1998; Sprinzl et al. 1998; Brown 1999; Szymanski et al. 2000; Cannone et al. 2002), the average positive predictive value is 65.8% for the MFE structure prediction. This is lower than the average sensitivity of MFE structure prediction of 72.8% (Mathews et al. 2004) because it reflects an overprediction of base pairs by energy minimization. However, it also reflects a small underdetermination of base pairs by comparative sequence analysis. For example, the Sprinzl database of tRNA structures does not annotate base pairs in the variable loop region (Brennan and Sundaralingam 1976; Sprinzl et al. 1998). Therefore, any (correct) prediction of base pairs in the variable loop region of tRNA decreases the positive predictive value for predicted tRNA base pairs. Similarly, The Comparative RNA Web Site is conservative in assigning base pairs, requiring compensating base-pair changes for each annotated base pair (Cannone et al. 2002).
TABLE 2.
Sensitivity and positive predictive value for MFE structure prediction
| Positive predictive value | |||||||
| Type of RNA | MFE sensitivityh | MFE positive predictive value | PBP ≥ 0.99 | PBP ≥ 0.95 | PBP ≥ 0.9 | PBP ≥ 0.7 | PBP ≥ 0.5 |
| SSU rRNAa,c | 61.4 ± 23.1 | 54.5 ± 24.5 | 86.0 ± 23.3 | 78.1 ± 25.8 | 74.8 ± 25.8 | 68.1 ± 26.5 | 63.2 ± 24.9 |
| (44.2 ± 14.7) | (37.1 ± 14.4) | (78.3 ± 22.2) | (71.0 ± 19.3) | (67.6 ± 17.5) | (57.6 ± 15.7) | (52.0 ± 15.1) | |
| LSU rRNAa,c | 74.0 ± 12.3 | 65.8 ± 12.3 | 91.8 ± 11.4 | 87.6 ± 10.5 | 85.3 ± 9.6 | 79.5 ± 10.4 | 75.2 ± 12.3 |
| (55.2 ± 11.5) | (47.2 ± 11.7) | (78.0 ± 27.4) | (74.1 ± 23.0) | (72.8 ± 19.3) | (67.6 ± 14.5) | (64.0 ± 15.3) | |
| 5S rRNAd | 73.8 ± 26.7 | 64.6 ± 24.0 | 94.1 ± 14.4 | 86.1 ± 20.9 | 82.2 ± 21.8 | 72.6 ± 23.1 | 68.8 ± 23.1 |
| Group I intronc | 68.9 ± 14.5 | 61.4 ± 14.2 | 94.2 ± 12.1 | 90.4 ± 15.6 | 85.1 ± 16.7 | 76.0 ± 17.5 | 71.4 ± 16.4 |
| Group I intron–2b,c | (57.4 ± 13.2) | (54.2 ± 14.5) | (92.4 ± 11.8) | (89.1 ± 11.0) | (81.2 ± 15.9) | (70.8 ± 16.5) | (67.3 ± 16.0) |
| Group II intron | 87.6 ± 2.3 | 82.7 ± 6.7 | 89.9 ± 17.5 | 92.2 ± 13.6 | 90.8 ± 10.6 | 90.0 ± 7.5 | 87.4 ± 6.9 |
| RNase Pe | 63.3 ± 14.4 | 60.8 ± 13.2 | 96.0 ± 9.9 | 95.1 ± 7.9 | 86.7 ± 15.7 | 75.4 ± 13.8 | 72.1 ± 14.2 |
| RNase P–2b,e | (58.9 ± 7.6) | (56.6 ± 8.4) | (92.9 ± 12.4) | (92.0 ± 8.6) | (88.6 ± 10.1) | (78.7 ± 10.5) | (72.9 ± 11.8) |
| SRPf | 66.4 ± 26.1 | 50.9 ± 22.3 | 79.1 ± 20.3 | 70.2 ± 25.4 | 67.8 ± 25.0 | 60.4 ± 24.0 | 57.0 ± 24.0 |
| tRNAg | 87.0 ± 17.0 | 85.5 ± 20.0 | 96.6 ± 13.3 | 94.1 ± 14.2 | 93.1 ± 15.3 | 90.9 ± 16.0 | 88.8 ± 17.3 |
| Average | 72.8 ± 9.4 | 65.8 ± 12.4 | 91.0 ± 5.9 | 86.7 ± 8.6 | 83.2 ± 8.3 | 76.6 ± 10.3 | 73.0 ± 10.9 |
MFE sensitivity is the percentage of known base pairs that are correctly predicted in the MFE structure:
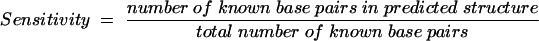 |
where the known base pairs are determined by comparative sequence analysis. MFE positive predictive value is the percentage of base pairs in the predicted MFE structure that are contained in the structure determined by comparative sequence analysis:
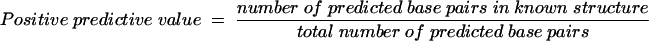 |
where the known structure is that determined by comparative sequence analysis. As explained in Materials and Methods, a predicted base pair is considered to be consistent with a known base pair if that predicted pair, or a pair slipped by up to one nucleotide on one side, occurs in the structure determined by comparative sequence analysis. The remaining columns are the positive predictive values for base pairs in the predicted MFE structure with probability above the shown probability threshold as predicted by the partition function calculation.
aThe large and small subunit rRNA sequences are divided into domains of less than 700 nt as determined by comparative sequence analysis (Mathews et al. 1999b). In parentheses are the results if the whole sequence is used in the calculations.
bThe group I introns and RNase P databases were divided into two sections and the second sections were withheld from the optimization of multibranch loop parameters (Mathews et al. 2004). The second section of each database is not included in the averages reported here.
Structures were acquired from the following databases: cCannone et al. 2002, dSzymanski et al. 2000, eBrown 1999, fLarsen et al. 1998, and gSprinzl et al. 1998.
hSensitivities are as reported previously for free energy minimization (Mathews et al. 2004).
The positive predictive value for base pairs in the predicted MFE structure can be increased by only considering base pairs with high probability as determined by the partition function calculation. Table 2 shows the positive predictive value for MFE structure base pairs with partition function predicted probability above specified thresholds. On average, base pairs in the MFE structure with probability of 0.99 and higher have a positive predictive value of 91.0 ± 5.9%. This falls off as base pairing probability threshold is reduced, so that, for a threshold of 0.9 and 0.5 probability, the positive predictive value is 83.2 ± 8.3% and 73.0 ± 10.9%, respectively.
As the base pair probability threshold is increased to consider only base pairs with higher positive predictive value, the fraction of base pairs in the predicted MFE structure that meet the requirement is reduced. Table 3 shows the mean and median base pairing probabilities for base pairs in the predicted MFE structure. On average for the database, the mean is 0.773 ± 0.050 and the median is 0.845 ± 0.052. Table 3 also shows the percentage of predicted MFE base pairs that meet specified thresholds of pairing probability. On average, 24.1 ± 5.7% of base pairs in the predicted MFE structure have base pairing probabilities of at least 0.99. As predicted by the partition function, 50.6 ± 6.5% and 81.6 ± 6.3% of base pairs are ≥ 0.9 and 0.5 probable, respectively. Interestingly, the average median is 0.845 and the average percentage of predicted base pairs above 0.9 probability is 50.6%. Although seemingly contradictory, this demonstrates that there is a subset of structures in each category of sequence for which a large portion of predicted pairs are predicted to be > 0.9 probable.
TABLE 3.
The distribution of base pairing probabilities (PBP) for base pairs in the predicted MFE structure
| Average PBP | % of base pairs in predicted MFE structure above pairing probability threshold | ||||||
| Type of RNAa | Mean | Median | PBP ≥ 0.99 | PBP ≥ 0.95 | PBP ≥ 0.9 | PBP ≥ 0.7 | PBP ≥ 0.5 |
| SSU rRNA | 0.728 ± 0.149 | 0.791 ± 0.227 | 22.9 ± 16.4 | 37.6 ± 19.6 | 46.0 ± 19.1 | 63.9 ± 18.4 | 77.0 ± 18.6 |
| (0.609 ± 0.133) | (0.641 ± 0.249) | (14.2 ± 9.7) | (26.3 ± 12.6) | (33.3 ± 13.6) | (49.9 ± 15.5) | (61.5 ± 16.5) | |
| LSU rRNA | 0.757 ± 0.088 | 0.869 ± 0.110 | 25.0 ± 13.5 | 41.5 ± 16.1 | 48.8 ± 15.9 | 68.6 ± 13.8 | 78.7 ± 11.4 |
| (0.621 ± 0.047) | (0.741 ± 0.101) | (18.5 ± 6.6) | (31.3 ± 6.3) | (37.6 ± 4.9) | (53.1 ± 6.3) | (62.4 ± 4.2) | |
| 5S rRNA | 0.793 ± 0.143 | 0.834 ± 0.188 | 26.9 ± 16.8 | 41.8 ± 20.4 | 51.3 ± 22.1 | 72.1 ± 22.9 | 84.3 ± 19.2 |
| Group I intron | 0.771 ± 0.082 | 0.869 ± 0.112 | 21.6 ± 12.7 | 38.7 ± 15.1 | 49.8 ± 15.8 | 69.5 ± 14.2 | 80.6 ± 10.1 |
| Group I intron–2 | (0.701 ± 0.091) | (0.801 ± 0.129) | (14.0 ± 9.2) | (25.9 ± 13.7) | (38.5 ± 15.0) | (60.4 ± 13.6) | (71.5 ± 14.2) |
| Group II intron | 0.826 ± 0.034 | 0.920 ± 0.034 | 14.6 ± 2.9 | 45.8 ± 8.7 | 56.7 ± 10.3 | 80.0 ± 4.0 | 89.3 ± 2.9 |
| RNase P | 0.709 ± 0.131 | 0.783 ± 0.173 | 22.5 ± 6.2 | 34.0 ± 5.9 | 42.2 ± 8.7 | 65.6 ± 15.2 | 73.5 ± 19.4 |
| RNase P–2 | (0.688 ± 0.106) | (0.782 ± 0.153) | (22.1 ± 9.2) | (37.0 ± 9.3) | (43.7 ± 13.2) | (57.9 ± 14.1) | (69.7 ± 15.1) |
| SRP | 0.743 ± 0.161 | 0.794 ± 0.236 | 24.3 ± 16.2 | 38.9 ± 20.5 | 47.0 ± 21.6 | 65.2 ± 22.3 | 77.7 ± 21.7 |
| tRNA | 0.857 ± 0.118 | 0.896 ± 0.149 | 34.8 ± 21.1 | 53.3 ± 23.8 | 62.7 ± 24.0 | 82.0 ± 19.5 | 91.4 ± 15.1 |
| Average | 0.773 ± 0.050 | 0.845 ± 0.052 | 24.1 ± 5.7 | 41.5 ± 5.9 | 50.6 ± 6.5 | 70.9 ± 6.8 | 81.6 ± 6.3 |
The mean and median base pairing probabilities are reported for each type of RNA sequence. Also reported is the percentage of base pairs in the MFE structure with 0.99%, 0.95%, 0.90%, 0.70%, and 0.50% probability and above.
aRefer to Table 2 for information about the databases of RNA sequences used in this study.
Another way the partition function can be applied to secondary structure prediction is to construct structures that only contain base pairs above a certain threshold of predicted pairing probability. If the pairing probability threshold is above 0.5, the resulting structure contains only nonconflicting pairs, that is, no nucleotide can be involved in more than one base pair with greater than 0.5 probability of pairing. Although a structure predicted by this method is not saturated with base pairs like a structure predicted by free energy minimization, it is composed of pairs only with a higher than average positive predictive value.
Table 4 gives the sensitivity and positive predictive value for structures constructed of pairs with predicted high probability. For a threshold of base pairing probability of 0.99, the positive predictive value is 90.9 ± 6.0%, but the sensitivity is only 24.4 ± 5.8%. By decreasing the threshold, a higher sensitivity is achieved at the cost of positive predictive value. A threshold of 0.5 probability of base pairing produces a structure with sensitivity of 70.0 ± 10.0% and positive predictive value of 70.3 ± 11.8%.
TABLE 4.
Sensitivity and positive predictive value for structures constructed of highly probable base pairs
| PBP ≥ 0.99 | PBP ≥ 0.9 | PBP ≥ 0.7 | PBP ≥ 0.5 | |||||
| Type of RNAa | Sensitivity | PPV | Sensitivity | PPV | Sensitivity | PPV | Sensitivity | PPV |
| SSU rRNA | 22.7 ± 17.1 | 86.0 ± 23.3 | 40.7 ± 21.5 | 74.5 ± 26.4 | 52.3 ± 22.5 | 67.0 ± 26.8 | 60.6 ± 23.5 | 61.7 ± 25.0 |
| (13.6 ± 10.3) | (78.2 ± 22.4) | (27.8 ± 13.8) | (67.0 ± 17.6) | (37.3 ± 14.6) | (55.2 ± 16.5) | (44.3 ± 15.8) | (47.5 ± 16.3) | |
| LSU rRNA | 25.2 ± 13.0 | 92.4 ± 11.4 | 46.4 ± 15.5 | 85.6 ± 9.5 | 62.9 ± 13.0 | 78.8 ± 10.6 | 71.4 ± 14.1 | 72.7 ± 13.9 |
| (17.9 ± 9.9) | (78.0 ± 27.4) | (33.9 ± 11.0) | (72.2 ± 18.4) | (46.1 ± 11.9) | (65.4 ± 13.1) | (53.7 ± 12.3) | (57.5 ± 13.9) | |
| 5S rRNA | 28.5 ± 17.6 | 94.1 ± 14.4 | 47.2 ± 22.9 | 82.0 ± 22.1 | 59.7 ± 25.7 | 71.5 ± 23.3 | 68.1 ± 26.1 | 66.2 ± 23.4 |
| Group I intron | 22.3 ± 13.1 | 93.9 ± 12.0 | 47.9 ± 15.4 | 85.0 ± 16.7 | 60.1 ± 15.7 | 75.5 ± 17.5 | 67.7 ± 15.4 | 70.4 ± 16.1 |
| Group I intron–2 | (13.7 ± 9.3) | (92.4 ± 11.8) | (33.6 ± 15.4) | (81.2 ± 15.9) | (47.0 ± 12.2) | (69.5 ± 14.5) | (56.4 ± 13.4) | (65.9 ± 13.5) |
| Group II intron | 13.9 ± 3.7 | 89.9 ± 17.5 | 54.5 ± 10.9 | 90.8 ± 10.6 | 76.3 ± 5.0 | 89.7 ± 8.2 | 83.8 ± 3.4 | 85.5 ± 10.4 |
| RNase P | 22.5 ± 7.1 | 96.0 ± 9.8 | 37.8 ± 9.2 | 86.5 ± 16.1 | 55.3 ± 13.1 | 72.1 ± 18.6 | 61.6 ± 16.8 | 66.5 ± 17.4 |
| RNase P–2 | (21.5 ± 10.2) | (92.9 ± 12.4) | (40.4 ± 12.5) | (88.6 ± 10.1) | (50.1 ± 10.4) | (77.0 ± 12.7) | (58.9 ± 8.8) | (68.4 ± 13.8) |
| SRP | 25.3 ± 17.7 | 78.7 ± 20.6 | 42.4 ± 24.0 | 66.5 ± 25.8 | 53.1 ± 26.6 | 58.0 ± 24.4 | 60.6 ± 27.6 | 52.0 ± 23.1 |
| tRNA | 34.4 ± 20.8 | 96.5 ± 13.2 | 59.5 ± 23.7 | 93.0 ± 15.4 | 76.8 ± 20.0 | 90.6 ± 16.1 | 85.8 ± 17.0 | 87.6 ± 17.8 |
| Average | 24.4 ± 5.8 | 90.9 ± 6.0 | 47.1 ± 7.2 | 83.0 ± 8.7 | 62.1 ± 9.6 | 75.4 ± 11.0 | 70.0 ± 10.0 | 70.3 ± 11.8 |
aRefer to Table 2 for information about the databases of RNA sequences used in this study.
This partition function algorithm has been written to accommodate constraints determined by experiment. Nucleotides that are single stranded or double stranded as determined by enzymatic cleavage (Knapp 1989), U’s that are in GU pairs as determined by flavin mononucleotide (FMN) cleavage (Burgstaller and Famulok 1997; Burgstaller et al. 1997), or nucleotides accessible to chemical modification can be specified (Ehresmann et al. 1987). Chemically modified nucleotides are allowed at helix ends, in loops, and in or adjacent to G-U pairs anywhere (Mathews et al. 2004). The fact that chemical modification can occur at these specific paired nucleotides necessitates added complexity in the recursions in order to use these data as structure prediction constraints (Mathews et al. 2004). Chemical modification, however, offers important experimental advantages, such as the small size of the reagents (Ehresmann et al. 1987), compared to enzymes, and the fact that the technique can be used to probe secondary structure in vivo (Zaug and Cech 1995; Mathews et al. 2004). Other constraints, forcing base pairs and prohibiting base pairs, can be specified that are not directly observable by experiment, but can be hypothesized, for example, on the basis of comparative sequence analysis. In the partition function calculation, conformations that violate the constraints are not allowed to contribute to the partition function.
To examine the effect of experimental constraints on the base pairing probabilities, RNA sequences with structures determined by comparative analysis that have been studied experimentally were assembled from the literature (Speek and Lind 1982; Miura et al. 1983; Kean and Draper 1985; Moazed et al. 1986; Egebjerg et al. 1987; Kwakman et al. 1990; Andreazzoli and Gerbi 1991; LaGrandeur et al. 1994; Tranguch et al. 1994; Zaug and Cech 1995; Burgstaller et al. 1997; Costa et al. 1998; Odell et al. 1998; Chamberlin and Weeks 2003; Mathews et al. 2004). A large number of correct constraints should improve the accuracy of secondary structure prediction. But, enzymatic methods are unable to determine every nucleotide that is paired or every nucleotide that is unpaired, largely because not all regions of a structure are accessible to the enzyme. FMN does not determine all U’s in GU pairs. Furthermore, in the case of the group II intron, 12 of 127 chemical modifications are inconsistent with the structure determined by comparative sequence analysis (Michel et al. 1989; Kwakman et al. 1990). This is possibly a result of multiple conformations for the RNA in the in vitro modification conditions (Mathews et al. 2004).
Previously, it has been shown that the sensitivity of the predicted MFE structure is improved by adding experimental constraints for structures that are poorly predicted, that is, < 50% sensitivity, without constraint (Mathews et al. 1999b, 2004). In general, structures well predicted, that is, greater than 50% sensitivity, without constraint remain well predicted when constraints are used in the MFE structure prediction, although little improvement in sensitivity is observed (Mathews et al. 1999b, 2004).
Experimental constraints applied to structure prediction also improve the fidelity of structure prediction as shown in Table 5. On average, experimental constraints improved the positive predictive value (PPV) for the predicted MFE structure from 63.3 ± 23.4% to 72.5 ± 13.1%. Furthermore, the percentage of predicted MFE base pairs with > 0.95 probability as calculated by the partition function is increased from an average of 38.8 ± 11.1% to 47.0 ± 8.7% with the application of experimental constraints. The Shannon entropy, S, introduced by Huynen et al. (1997), is a measure of well-definedness for the structure:
TABLE 5.
Accuracy and well-determinedness for RNA sequences without and with constraints derived from experiment
| Unconstrained | Constrainedw | ||||||||||
| Sequence | Type of data | Sensitivityx | PPV | % MFE base pairs PBP ≥ 0.95 | PPV base pairs PBP ≥ 0.95 | Shannon entropy | Sensitivity | PPV | % MFE base pairs PBP ≥ 0.95 | PPV base pairs PBP ≥ 0.95 | Shannon entropy |
| Dog SRPa | Modificationg,v | 18.2 | 15.8 | 36.6 | 27.0 | 0.432 | 84.1 | 74.0 | 45.9 | 77.8 | 0.289 |
| E. coli 5S rRNAb | In vivo modificationh, enzymatici | 26.3 | 25.6 | 17.9 | 100 | 0.355 | 86.8 | 89.2 | 56.8 | 100 | 0.113 |
| E. coli SSU rRNAc | Modificationj, enzymatick | 39.0 | 34.6 | 21.9 | 83.5 | 0.547 | 63.0 | 57.1 | 36.1 | 78.4 | 0.342 |
| C. vinosum RNase Pd | Modificationl | 53.5 | 51.3 | 57.1 | 70.6 | 0.159 | 53.5 | 51.3 | 58.0 | 71.0 | 0.151 |
| B. subtilis RNase Pd | Modificationm | 56.3 | 52.9 | 38.7 | 82.6 | 0.381 | 56.3 | 52.9 | 39.5 | 83.0 | 0.339 |
| E. coli RNase Pd | Modificationl | 57.3 | 58.7 | 41.3 | 100 | 0.356 | 63.7 | 63.7 | 40.3 | 100 | 0.312 |
| Yeast RNase Pd | Modificationn, enzymaticn | 59.3 | 55.2 | 38.8 | 97.8 | 0.493 | 70.4 | 64.4 | 46.6 | 89.1 | 0.331 |
| Tetrahymena | In vivo | 65.8 | 58.1 | 51.2 | 68.2 | 0.156 | 65.8 | 58.1 | 51.2 | 68.2 | 0.142 |
| telomerasee | modificationo | ||||||||||
| Yeast group I intron | Modificationp | 78.2 | 70.5 | 40.9 | 100 | 0.268 | 77.3 | 71.3 | 45.7 | 100 | 0.257 |
| Tetrahymena group | In vivo | 82.9 | 75.0 | 42.4 | 100 | 0.368 | 82.9 | 75.0 | 43.1 | 100 | 0.351 |
| I intronc | modificationo | ||||||||||
| T4 td group I intronc | FMN cleavageq | 85.0 | 89.5 | 47.4 | 100 | 0.393 | 83.8 | 87.0 | 67.5 | 100 | 0.156 |
| Yeast aI5c group II intronf | Modificationr | 86.1 | 87.0 | 41.0 | 100 | 0.254 | 77.7 | 83.1 | 39.7 | 100 | 0.255 |
| C. albicans 5S rRNAb | In vivo modificationh | 87.5 | 84.8 | 30.3 | 100 | 0.378 | 87.5 | 84.8 | 45.5 | 100 | 0.280 |
| E. coli 23 LSU rRNA domain 1c | Modifications | 88.9 | 75.2 | 46.3 | 95.7 | 0.239 | 88.9 | 75.7 | 48.6 | 95.8 | 0.225 |
| P. littoralis group II intronf | Modificationt | 89.7 | 88.3 | 47.3 | 100 | 0.296 | 89.7 | 88.3 | 35.1 | 100 | 0.276 |
| Mouse 5S rRNAb | Modificationu | 94.4 | 89.5 | 21.1 | 100 | 0.385 | 88.9 | 84.2 | 52.6 | 80.0 | 0.224 |
| Average | 66.8 ± 23.8 | 63.3 ± 23.4 | 38.8 ± 11.1 | 89.1 ± 19.8 | 0.341 ± 0.108 | 76.3 ± 12.4 | 72.5 ± 13.1 | 47.0 ± 8.7 | 90.2 ± 11.8 | 0.253 ± 0.078 |
Structures derived from comparative sequence analysis were derived from aGorodkin et al. 2001, bSzymanski et al. 2000, cCannone et al. 2002, dBrown 1999, eRomero and Blackburn 1991, ten Dam et al. 1991, and fMichel et al. 1989.
Experimental constraints were derived from gAndreazzoli and Gerbi 1991, hMathews et al. 2004, iSpeek and Lind 1982, jMoazed et al. 1986, kKean and Draper 1985, lLaGrandeur et al. 1994, mOdell et al. 1998, nTranguch et al. 1994, oZaug and Cech 1995, pDMS modification (Chamberlin and Weeks 2003), qBurgstaller et al. 1997, rKwakman et al. 1990, sEgebjerg et al. 1987, and tnative conditions chemical modification (Costa et al. 1998), and uMiura et al. 1983.
vThe experimental constraints from protein-bound RNA were used.
wFor a base pair to be forced single or double stranded on the basis of enzymatic cleavage, cleavage is required on both sides of a nucleotide by the same enzyme (Mathews et al. 1999b). Strong and moderate chemical modifications are used as constraints when data are stratified by intensity (Mathews et al. 2004).
xWhen there is more than one structure with the lowest free energy, the first structure predicted by the dynamic programming algorithm is scored.
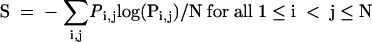 |
where N is the length of the sequence and Pi,j is the predicted probability of pairing of nucleotides i and j. Well-defined structures have lower Shannon entropy as compared to structures with many alternative, competing base pairs. On average, the Shannon Entropy improved from 0.341 ± 0.108 to 0.253 ± 0.078 with the use of experimentally derived constraints.
The largest improvements in fidelity occur when constraints are applied for sequences with low, that is, < 50%, sensitivity and positive predictive value MFE structure predictions without constraint (Mathews et al. 2004). This is observed as the marked improvement of the percentage of predicted MFE base pairs with 0.95 or greater chance of pairing as predicted by the partition function and of well-definedness as measured by Shannon entropy. For example, the percentage of highly probable pairs (greater than or equal to 0.95) for the Escherichia coli 5S rRNA increases from only 17.9% to 56.8% with constraints. The Shannon entropy improves from 0.355 to 0.113. Although not as dramatic, this improvement in Shannon entropy is also observed for RNA sequences that have well-predicted secondary structures without constraint. For example, the Candida albicans 5S rRNA is well predicted both without and with constraints derived from in vivo chemical modification (Mathews et al. 2004), but the constraints still serve to improve the fidelity of prediction. The Shannon entropy improves from 0.378 to 0.280.
The experimental constraints do not affect the positive predictive value for probable base pairs in predicted MFE structures. On average, without and with experimental constraints, the positive predictive value for base pairs above 0.95 probability is 89.1 ± 19.8% and 90.2 ± 11.8%, respectively. This demonstrates that highly probable base pairs in the predicted MFE structure have a high positive predictive value, regardless of the average positive predictive value for all pairs in that structure.
Figure 1 illustrates the improved fidelity of secondary structure prediction with experimental constraints. The E. coli 5S rRNA is shown predicted without and with constraints derived from experiments. The structures are color annotated according to base pairing probability using a color scheme derived from Zuker and Jacobson (1995). The enzymatic and chemical modification constraints improve the average positive predictive value for base pairs, that is, many more of the predicted pairs are in the structure determined by comparative sequence analysis, and also increase the base pairing probabilities, indicating that, on average, the base pairs are more well determined. For example, in the unconstrained structure, only one stem–loop region (the only correctly predicted region of the structure) contains pairs with 0.95 or greater probability of pairing. After the structure is predicted with the chemical modification and enzymatic constraints (Speek and Lind 1982; Mathews et al. 2004), many pairs (56.8% of all predicted pairs) are predicted to pair with 0.95 or greater probability. These highly probable pairs are all correctly predicted as compared to the structure from comparative sequence analysis (Szymanski et al. 2000).
FIGURE 1.

Secondary structure prediction of E. coli 5S rRNA. The secondary structure predicted without (A) and with (B) constraints from chemical modification (Mathews et al. 2004) and enzymatic cleavage (Speek and Lind 1982). Base pairs in the structure determined by comparative sequence analysis (Szymanski et al. 2000) are shown with a heavy line. Structures are color annotated to indicate base pairing probabilities as shown in C. The structures were drawn using XRNA (http://rna.ucsc.edu/rnacenter/xrna/xrna.html). The predicted free energies are -53.0 and -46.4 kcal/mole for the nonconstrained structure (A) and the constrained structure (B), respectively.
DISCUSSION
The RNA secondary structure nearest neighbor parameters used here for structure prediction are an approximation based on a finite set of experiments (Xia et al. 1998; Mathews et al. 1999b, 2004). The parameters average some sequence-specific effects. Physiological salt conditions are approximated with 1 M NaCl (Xia et al. 1999; Turner 2000) and therefore the parameters may neglect some magnesium-dependent effects. Furthermore, they are folding free energies at 37°C, although many organisms live at other temperatures, including hyperthermic temperatures. In spite of these limitations, 73% of known base pairs are correctly predicted for a diverse set of RNA sequences (Mathews et al. 1999b, 2004). This work demonstrates that base pairing probabilities determined by the partition function calculation can serve to overcome the limitations of the parameters by providing confidence intervals for predicted pairs. Base pairs predicted to have a probability of 0.99 and above have a positive predictive value of 91.0% (Table 2). Base pairs predicted to have a probability of < 0.5 have a much lower positive predictive value of < 73%.
Another limitation of the predicted MFE structure is the assumption that the RNA sequence has a single conformation in solution. For some RNA sequences, either natural or designed, more than one secondary structure is required for function (Zavanelli and Ares 1991; Baumstark et al. 1997; Michiels et al. 2000; Schultes and Bartel 2000; Flamm et al. 2001; Hoffman et al. 2003). For other sequences, such as mRNA, it is possible that many structures are populated in solution. These secondary structures cannot be adequately described by a single predicted MFE secondary structure, but the probability dot plot from the partition function calculation can display alternative base pairs to those in the secondary structure. Color annotated MFE structures can also point to the base pairs with low pairing probability, suggesting that they may fold into alternative pairs.
This partition function algorithm explicitly considers the contribution of terminal mismatches, dangling ends, and coaxial stacking in the stability of multibranch loops and exterior loops, that is, loops that contain the ends of the sequence. Each of these stabilizing effects is mutually exclusive, and for some helices that terminate in multibranch loops or exterior loops, each of these stabilizations is possible. In this work, each of these stabilizations is considered to be a unique secondary structure conformation and each therefore contributes to the total partition function. This approach does have a tendency to increase the probability of base pairs that contribute to the formation of multi-branch or exterior loops as compared to the Vienna RNA Package (Hofacker et al. 1994; Hofacker 2003), which uses an alternative method for handling multibranch loops. The Vienna RNA Package simplifies the energy rules so that 3′ dangling ends are assumed for each helix termination. This allows significantly faster calculation times, but may blur the distinction between competing secondary structures. For example, some helices will receive a 3′ dangling end contribution when no such interaction is possible, falsely increasing the probability of pairs that contribute to that conformation. For other multibranch loops, however, this approach simulates the coaxial stacking increment with the dangling end free energy, with much less computational overhead than that used here.
To remain O(N3) in time and O(N2) in storage, the partition function calculation described by this work does not allow pseudoknots. Rivas and Eddy (1999) introduced a dynamic programming method for predicted pseudoknotted structures by free energy minimization that is O(N6) in time and O(N4) in storage. This approach has been extended to a partition function calculation in O(N5) time and O(N4) storage (Dirks and Pierce 2003), by considering a smaller subset of pseudoknots than considered in the energy minimization algorithm (Rivas and Eddy 1999).
The difference in scaling from O(N3) to O(N5) or O(N6) means that many sequences of interest cannot be reasonably considered by an algorithm that can predict pseudoknots explicitly by dynamic programming. An alternative for finding pseudoknots is to examine the energy dot plot (Gaspin and Westhof 1995) or the probability dot plot. For example, the Tetrahymena Nuclear LSU rRNA Group I intron contains a pseudoknot defined by helices P3 and P7. The probabilities of base pairing for five of the six base pairs in P7 are 0.378, 0.392, 0.393, 0.376, and 0.221. The sixth pair is considered isolated (by a bulge loop from the rest of the helix) and is not allowed (Mathews et al. 1999b). The six canonical pairs in the P3 helix have probabilities of 0.524, 0.525, 0.525, 0.524, 0.516, and 0.494. Therefore, both helices are displayed in a probability dot plot of all pairs with conservative threshold of probability greater than 0.01. Interestingly, the predicted MFE structure contains the P3 helix, although the P7 helix has higher probability of pairing according to the partition function. Therefore, a quick scan of probable pairs that are not in the predicted MFE structure would reveal the potential pseudoknot. Structures containing pseudoknots could be constructed from base pair probability data using an iterated loop matching algorithm (Ruan et al. 2004).
Finally, the results presented here for RNA secondary structures predicted with constraints (Table 5) show that experimental constraints not only improve the average accuracy of structure prediction as shown previously (Mathews et al. 1999b, 2004), but also improve the well-definedness of the structure. Therefore, experiments that examine secondary structure can improve the confidence in predicted base pairs in the MFE structure.
This article presents a new partition function algorithm, which was used to predict base pairing probabilities. This new algorithm represents an advance over previous algorithms because it utilizes the complete set of thermodynamic parameters for predicting RNA secondary structure stabilities, including coaxial stacking, and can constrain the prediction of the partition function using data derived from chemical modification experiments.
The partition function calculation presented in this work does not replace MFE structure prediction, but instead adds to its utility by providing implicit confidence estimates in base pairs. This article shows that the most probable base pairs, as determined by the partition function calculation, are more likely to be contained in the known structure as determined by comparative sequence analysis. Color annotation of a predicted MFE secondary structure provides a quick method for scanning for base pairs in a predicted structure that are more likely to be in the actual solution structure. The base pairs that are not as probable, < 0.5 probability, are base pairs that should be the focus of experimental studies that can test structural hypothesis, such as site-directed mutagenesis. These, less probable, pairs may also demonstrate the locations of pseudoknots or base pairs that are dynamic. The computational tools presented here are available for download from the World Wide Web as part of the RNAstructure software package for Microsoft Windows.
MATERIALS AND METHODS
Partition function calculation
The goal of the calculation is to determine base pairing probabilities from the partition function, Q:
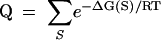 |
[1] |
where ΔG is the conformational Gibb’s Free Energy change, R is the gas constant, T is the absolute temperature, and S, over which the summation is performed, is the set of possible secondary structures. This partition function is determined by nonredundant and exhaustive recursions, expanded from those used by McCaskill (1990) to accommodate coaxial stacking.
Six N × N arrays are needed for the nonredundant calculation that includes coaxial stacking, where N is the number of nucleotides in the sequence. For free energy minimization, three N × N arrays are sufficient (Mathews et al. 2004) because the recursions contain redundancies, that is, the same conformation can be implicitly considered multiple times during the minimization, but this does not alter the result of minimization. The partition function, on the other hand, sums contributions by different conformations, and therefore each conformation must only be counted once. In free energy minimization, the total free energy is found as the sum of free energy terms. The analogy for the partition function calculation is the total equilibrium constant, which is the product of equilibrium constants for each motif. The equilibrium constant for a motif is simply e−ΔG(motif)/RT.
For the partition function, the N × N arrays are V(i,j), W(i,j), WL(i,j), WMB(i,j), WMBL(i,j), and Wcoax(i,j). V(i,j) is the partition function for the fragment from nucleotides i to j, inclusive, with i paired to j. W(i,j) and WL(i,j) are the partition functions for the fragment from nucleotides i to j, inclusive, such that this fragment will be incorporated in a multibranch loop and it has one single helical branch. WL(i,j) also requires that nucleotide j terminates the helical branch as either a paired nucleotide, a 3′ dangling end, or a nucleotide in a terminal mismatch. Wcoax(i,j) is the partition function from nucleotides from i to j, inclusive, such that there are two coaxially stacked branches. Nucleotides i and j must either be paired or be in a single mismatch separating the two helices. WMB(i,j) and WMBL(i,j) are the partition function from nucleotides i to j, inclusive, such that this fragment will be incorporated into a multibranch loop and it contains two or more branches. WMBL(i,j) also requires that nucleotide j be paired or associated with a helix as either a 3′ dangling end, a nucleotide in a terminal mismatch, or a nucleotide in a mismatch between two coaxially stacked helices. Figure 2 illustrates the structural requirements of each of these arrays. In addition, two linear arrays of size N are used. W5(i) is the partition function for the nucleotide fragment from the 5′ end of the sequence to and including nucleotide i. W3(i) is the partition function from and including nucleotide i to the 3′ end of the sequence.
FIGURE 2.

The structural requirements for the two-dimensional arrays W, WL, Wcoax, WMB, and WMBL. Branches are represented as a stem-loop, but any arrangement of nucleotides is allowed, that is, the stem-loop can be extended by a helix, internal loop, bulge loop, or multibranch loop. Shaded items are not required, but allowed. For example, WMBL(i,j) can be started with any number of branches and terminates with j as either a paired nucleotide, a 3′ dangling end, a terminal mismatch, or a mismatched nucleotide mediating a coaxial stack. W(i,j) can have any number of unpaired nucleotides 5′ or 3′ to a single helix. WL(i,j) can have any number of unpaired nucleotides 5′ to a single helix. WMB(i,j) can have any number of unpaired nucleotides 5′ or 3′ to two or more helices. WMBL(i,j) can have any number of unpaired nucleotides 5′ to two or more helices.
The calculations are performed so that, for the two dimensional arrays, i < j, j < 2N, and j − N < i. When j > N, the fragment being considered contains the ends of the sequence (the excluded fragment), that is, nucleotides i to N and 1 to j − N, inclusive. By definition, the probability of any given secondary structure conformation in the ensemble of structures is:
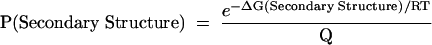 |
[2] |
The probability of a given base pair from i to j, Pi,j, is the sum of probabilities of all secondary structures that contain that pair:
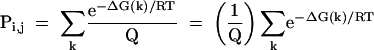 |
[3] |
where the index k counts each structure with the i to j base pair. But the summation in equation 3 is of the same form as a partition function, shown in equation 1, and is the partition function constrained to structures with the i to j base pair. Therefore, the probability of a base pair involving nucleotides i and j is:
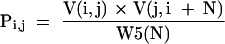 |
[4] |
because W5(N) = Q and V(i,j) × V(j,i + N) represents the total contribution to Q of secondary structure conformations that contain the base pair of i to j. This equation, 4, for predicting base pair probabilities is simple compared to that used by McCaskill (1990), but does not represent any improvement in computation time. By calculating the restricted partition functions for the excluded fragment that contains the ends of the sequence, that is, j > N, more calculation time is used up front. This time is then saved by this simple formulation for Pi.j as compared to the O(N3) recursions used by McCaskill for calculating Pi,j.
V is the sum of terms involving a pair between i and j, base pair stacking, hairpin loop closure, internal loop closure, and multibranch loop closure:
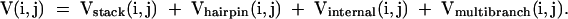 |
[5] |
For j > N, a term for exterior loop closure, Vexterior(i,j), also needs to be included in the sum. The hairpin contribution is defined as:
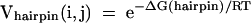 |
[6] |
where ΔG(hairpin) is calculated for the hairpin closed by a base pair between nucleotides i and j according to the nearest neighbor parameters of Mathews et al. (2004). The stacking contribution (of a base pair on a previous pair) is:
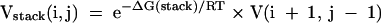 |
[7] |
ΔG(stack) is stacking nearest neighbor parameter for a canonical base pair i to j stacked on the base pair i + 1 to j − 1, derived by Xia et al. (1998) for Watson–Crick pairs and Mathews et al. (1999b) for G-U pairs. The internal loop term, which also considers bulge loops, requires a search over i′ and j′:
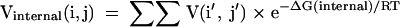 |
[8] |
where the sums are over all i′ and j′ such that i < i′ < j′ < j except where i′ = i + 1 and j′ = j + 1 simultaneously, that is, the base pair stacking case. ′G(internal) is the term for the free energy of closing a loop with base pairs i to j and i′ to j′. These free energies are calculated using the length and sequence-specific terms in Mathews et al. (2004). The search is limited to internal loops of total size of ≤30 nt, that is, i′ − i + j − j′- 2 ≤ 30, to limit the algorithm to O(N3). It has been shown that the energy function for internal loops allows an O(N3) solution that does not need to limit the size of internal loops (Lyngsø et al. 1999). This has been implemented in a partition function that includes pseudoknots (Dirks and Pierce 2003), but was not implemented for this algorithm, so that the same energy function as the MFE structure prediction algorithm (Mathews et al. 2004) is used.
Vmultibranch(i,j) is defined as:
 |
[9] |
where sums are over k for all i < k < j. The dangle, terminal mismatch, and coaxial stacking free energies, ΔG(dangle), ΔG(terminal mismatch), and ΔG(coaxial stacking), are sequence specific and are compiled by Mathews et al. (1999b). The terms a′ = e−a/RT, b′ = e−b/RT, and c′ = e−c/RT are factors for closing a multibranch loop, adding an unpaired nucleotide to a multibranch loop, and adding a branching helix to a multibranch loop, respectively (Mathews et al. 2004). Note that when using the current thermodynamic parameters, b is zero (Mathews et al. 2004), although it is presented here because nonzero b is supported in the computer code. The calculation of Vmultibranch(i,j) defines the O(N3) time requirement because for each i and j, a sum of j − i elements is performed. The six coaxial stacking terms are required to consider all cases of coaxial stacking on the helix terminated with the pair of i and j. For example, the last term considers the coaxial stacking of a helix terminated with the pair of k and j − 2 with an intervening mismatch of nucleotides j − 1 and i + 1. Similarly, Vexterior(i,j) is defined as:
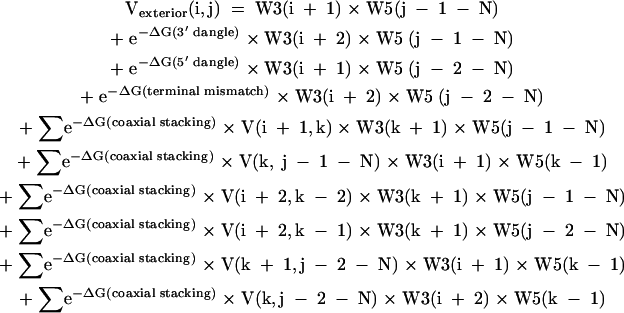 |
[10] |
where the sums are over k. For cases where k is indexing W3, i < k ≤ N. For cases where k is indexing W5, 1 ≤ k < (j − N).
W5(i), the nucleotide fragment from 1 to i, inclusive, is calculated as:
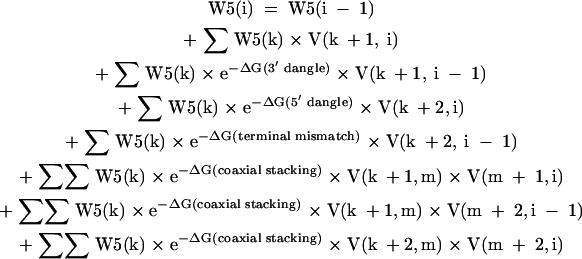 |
[11] |
where sums are over k and m for all 0 ≤ k < i and k < m < i. W5(0) is initialized as 1. W3(i), the nucleotide fragment from i to N, inclusive, is calculated similarly with the initialization of W3(N + 1) = 1. By initializing W5(0) = W3(N + 1) = 1, the state with no base pairs, ΔG = 0, is included in the partition function.
The remaining terms are:
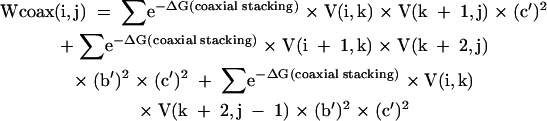 |
[12] |
where sums are over k for all i < k < j.
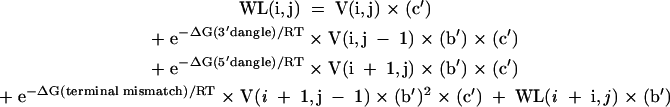 |
[13] |
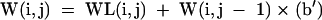 |
[14] |
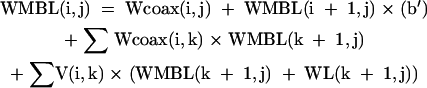 |
[15] |
where sums are over k for all i < k < j.
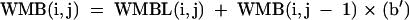 |
[16] |
Applying chemical modification constraints
Chemical modification constraints are applied similarly to MFE structure prediction as described in Mathews et al. (2004). Briefly, modified nucleotides are not allowed to stack in base pairs on previous base pairs, unless the pair being considered or the previous pair is a G-U base pair. However, to allow a terminal base pair that contains a modified nucleotide, at each reference to V(i,j) in loop closure, V′(i,j) must be considered for i or j modified where:
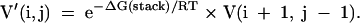 |
[17] |
The recursions involving V′(i,j), to be checked when either i or j is modified, are expanded in detail in the computer code for efficiency. During the calculation of base pair probabilities, for i or j modified:
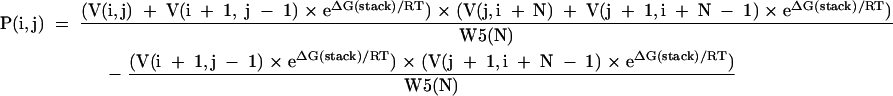 |
[18] |
The first term has added back the contributions of the base pair stacking for both the interior and exterior fragment and the second term removes the contribution where the pair is buried in a helix from both directions (which is not allowed).
Statistics
A predicted base pair is considered to be consistent with the comparative sequence analysis structure if that base pair or a base slipped by one position on one side of the helix occurs in the comparative sequence analysis structure (Mathews et al. 1999b). So, for a predicted pair i − j to be consistent with a pair in the known structure, the comparative analysis structure must contain a pair i to j, (i + 1) to j, (i − 1) to j, i to (j + 1), or i to (j − 1).
Error limits, as reported in Tables 2–4, are single standard deviations. For each category of RNA in Tables 2 and 3, the reported average and standard deviation is of accuracy calculated on each sequence separately. For the overall averages, the categories of RNA are averaged, excluding the second database of group I intron and RNase P RNA sequences. This overall average gives each category of RNA sequence equal weight regardless of the number of sequences or nucleotides in each category.
Availability
The partition function calculation has been incorporated into the RNAstructure suite of algorithms for sequence analysis (Mathews et al. 1999a, 2004; Mathews and Turner 2002; Matveeva et al. 2003), which is available for download from the World Wide Web (http://rna.chem.rochester.edu/RNAstructure). RNAstructure is a user-friendly interface for Microsoft Windows and is an executable program that requires no compilation. RNAstructure runs under Wine (version December 12, 2003), a Windows emulator, on Red Hat Linux 9. C++ code for compilation on other platforms is also available by request to the author.
Acknowledgments
I gratefully appreciate helpful discussions with David A. Case. This work received partial support from National Institutes of Health (NIH) grant RR12255 to D.A.C.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.7650904.
REFERENCES
- Adams, M.D., Celniker, S.E., Holt, R.A., Evans, C.A., Gocayne, J.D., Amanatides, P.G., Scherer, S.E., Li, P.W., Hoskins, R.A., Galle, R.F., et al. 2000. The genome sequence of Drosphila melanogaster. Science 287: 2185–2195. [DOI] [PubMed] [Google Scholar]
- Andreazzoli, M. and Gerbi, S.A. 1991. Changes in 7SL RNA conformation during the signal recognition particle cycle. EMBO J. 10: 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Ban, N., Nissen, P., Hansen, J., Moore, P.B., and Steitz, T.A. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289: 905–920. [DOI] [PubMed] [Google Scholar]
- Baumstark, T., Schröder, A.R.W., and Riesner, D. 1997. Viroid processing: Switch from cleavage to ligation is driven by a change from tetraloop to a loop A conformation. EMBO J. 16: 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan, T. and Sundaralingam, M. 1976. Structure of transfer RNA molecules containing the long variable loop. Nucleic Acids Res. 3: 3235–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J.W. 1999. The ribonuclease P database. Nucleic Acids Res. 27: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgstaller, P. and Famulok, M. 1997. Flavin-dependent photocleavage of RNA at G•U base pairs. J. Am. Chem. Soc. 119: 1137–1138. [Google Scholar]
- Burgstaller, P., Hermann, T., Huber, C., Westhof, E., and Famulok, M. 1997. Isoalloxazine derivatives promote photocleavage of natural RNAs at G•U base pairs embedded within helices. Nucleic Acids Res. 25: 4018–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium. 1998. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282: 2012–2018. [DOI] [PubMed] [Google Scholar]
- Cannone, J.J., Subramanian, S., Schnare, M.N., Collett, J.R., D’Souza, L.M., Du, Y., Feng, B., Lin, N., Madabusi, L.V., Muller, K.M., et al. 2002. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3. [DOI] [PMC free article] [PubMed]
- Chamberlin, S.I. and Weeks, K.M. 2003. Differential helix stabilities and sites pre-organized for tertiary interactions revealed by monitoring local nucleotide flexibility in the bI5 group I intron RNA. Biochemistry 42: 901–909. [DOI] [PubMed] [Google Scholar]
- Costa, M., Christian, E.L., and Michel, F. 1998. Differential chemical probing of a group II self-splicing intron identifies bases involved in tertiary interactions and supports an alternative secondary structure model of domain V. RNA 4: 1055–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond, J.M., Turner, D.H., and Mathews, D.H. 2001. Thermodynamics of three-way multibranch loops in RNA. Biochemistry 40: 6971–6981. [DOI] [PubMed] [Google Scholar]
- Ding, Y. and Lawrence, C. 2001. Statistical prediction of single-stranded regions in RNA secondary structure and application to predicting effective antisense target sites and beyond. Nucleic Acids Res. 29: 1034–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2003. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 31: 7280–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks, R. and Pierce, N. 2003. A partition function algorithm for nucleic acid secondary structure including pseudoknots. J. Comput. Chem. 24: 1664–1677. [DOI] [PubMed] [Google Scholar]
- Doudna, J. and Cech, T. 2002. The chemical repertoire of natural ribozymes. Nature 418: 222–228. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. 2001. Non-coding RNA genes and the modern RNA world. Nat. Rev. 2: 919–929. [DOI] [PubMed] [Google Scholar]
- Egebjerg, J., Leffers, H., Christensen, A., Andersen, H., and Garrett, R.A. 1987. Structure and accessibility of domain I of Escherichia coli 23 S RNA in free RNA, in the L24-RNA complex and in 50 S subunits. J. Mol. Biol. 196: 125–136. [DOI] [PubMed] [Google Scholar]
- Ehresmann, C., Baudin, F., Mougel, M., Romby, P., Ebel, J., and Ehresmann, B. 1987. Probing the structure of RNAs in solution. Nucleic Acids Res. 15: 9109–9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekete, M., Hofacker, I.L., and Stadler, P.F. 2000. Prediction of RNA base pairing probabilities on massively parallel computers. J. Comput. Biol. 7: 171–182. [DOI] [PubMed] [Google Scholar]
- Flamm, C., Hofacker, I.L., Maurer-Stroh, S., Stadler, P.F., and Zehl, M. 2001. Design of multistable RNA molecules. RNA 7: 254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, C., Eisen, J., and Salzberg, S. 2000. Microbial genome sequencing. Nature 406: 799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspin, C. and Westhof, E. 1995. An interactive framework for RNA secondary structure prediction with a dynamical treatment of constraints. J. Mol. Biol. 254: 163–174. [DOI] [PubMed] [Google Scholar]
- Goffeau, A., Barrell, B.G., Bussey, H., Davis, R.W., Dujon, B., Feldmann, H., Galibert, F., Hoheisel, J.D., Jacq, C., Johnston, M., et al. 1996. Life with 6000 genes. Science 274: 546, 563–567. [DOI] [PubMed] [Google Scholar]
- Gorodkin, J., Knudsen, B., Zwieb, C., and Samuelsson, T. 2001. SRPDB (signal recognition particle database). Nucleic Acids Res. 29: 169–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell, R.R., Lee, J.C., and Cannone, J.J. 2002. The accuracy of ribosomal RNA comparative structure models. Curr. Opin. Struct. Biol. 12: 301–310. [DOI] [PubMed] [Google Scholar]
- Hansen, J.L., Schmeing, T.M., Moore, P.B., and Steitz, T.A. 2002. Structural insights into peptide bond formation. Proc. Natl. Acad. Sci. 99: 11670–11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker, I.L. 2003. Vienna RNA secondary structure server. Nucleic Acids Res. 31: 3429–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker, I.L., Fontana, W., Stadler, P.F., Bonhoeffer, L.S., Tacker, M., and Schuster, P. 1994. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 125: 167–168. [Google Scholar]
- Hoffman, B., Mitchell, G.T., Gendron, P., Major, F., Anderson, A.A., Collins, R.A., and Legault, P. 2003. NMR structure of the active conformation of the Varkud satellite ribozyme cleavage site. Proc. Natl. Acad. Sci. 100: 7003–7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynen, M., Gutell, R., and Konings, D. 1997. Assessing the reliability of RNA folding using statistical mechanics. J. Mol. Biol. 267: 1104–1112. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Kean, J.M. and Draper, D.E. 1985. Secondary structure of a 345-base RNA fragment covering the S8/S15 protein binding domain of Escherichia coli 16S ribosomal RNA. Biochemistry 24: 5052–5061. [DOI] [PubMed] [Google Scholar]
- Kim, J., Walter, A.E., and Turner, D.H. 1996. Thermodynamics of coaxially stacked helices with GA and CC mismatches. Biochemistry 35: 13753–13761. [DOI] [PubMed] [Google Scholar]
- Knapp, G. 1989. Enzymatic approaches to probing RNA secondary and tertiary structure. Methods Enzymol. 180: 192–212. [DOI] [PubMed] [Google Scholar]
- Kwakman, J.H.J.M., Konings, D.A.M., Hogweg, P., Patel, H.J., and Grivell, L.A. 1990. Structural analysis of a group II intron by chemical modifications and minimal energy calculations. J. Biomol. Struct. Dyn. 8: 413–430. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana, M., Rauhut, R., Lendeckel, W., and Tuschl, T. 2001. Identification of novel genes coding for small expressed RNAs. Science 294: 853–858. [DOI] [PubMed] [Google Scholar]
- LaGrandeur, T.E., Hüttenhofer, A., Noller, H.F., and Pace, N.R. 1994. Phylogenetic comparative chemical footprint analysis of the interaction between ribonuclease P RNA and tRNA. EMBO J. 17: 3945–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, N., Samuelsson, T., and Zwieb, C. 1998. The signal recognition particle database (SRPDB). Nucleic Acids Res. 26: 177–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, N.C., Lim, L.P., Weinstein, E.G., and Bartel, D.P. 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858–862. [DOI] [PubMed] [Google Scholar]
- Lyngsø, R., Zuker, M., and Pederson, C. 1999. Fast evaluation of internal loops in RNA secondary structure prediction. Bioinformatics 15: 440–445. [DOI] [PubMed] [Google Scholar]
- Mathews, D.H. and Turner, D.H. 2002. Dynalign: An algorithm for finding the secondary structure common to two RNA sequences. J. Mol. Biol. 317: 191–203. [DOI] [PubMed] [Google Scholar]
- Mathews, D.H., Banerjee, A.R., Luan, D.D., Eickbush, T.H., and Turner, D.H. 1997. Secondary structure model of the RNA recognized by the reverse transcriptase from the R2 retrotransposable element. RNA 3: 1–16. [PMC free article] [PubMed] [Google Scholar]
- Mathews, D.H., Burkard, M.E., Freier, S.M., Wyatt, J.R., and Turner, D.H. 1999a. Predicting oligonucleotide affinity to nucleic acid targets. RNA 5: 1458–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews, D.H., Sabina, J., Zuker, M., and Turner, D.H. 1999b. Expanded sequence dependence of thermodynamic parameters provides improved prediction of RNA secondary structure. J. Mol. Biol. 288: 911–940. [DOI] [PubMed]
- Mathews, D.H., Disney, M.D., Childs, J.L., Schroeder, S.J., Zuker, M., and Turner, D.H. 2004. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. 101: 7287–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveeva, O.V., Mathews, D.H., Tsodikov, A.D., Shabalina, S.A., Gesteland, R.F., Atkins, J.F., and Freier, S.M. 2003. Thermodynamic criteria for high hit rate antisense oligonucleotide design. Nucleic Acids Res. 31: 4989–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaskill, J.S. 1990. The equilibrium partition function and base pair probabilities for RNA secondary structure. Biopolymers 29: 1105–1119. [DOI] [PubMed] [Google Scholar]
- Michel, F., Umesono, K., and Ozeki, H. 1989. Comparative and functional anatomy of group II catalytic introns—A review. Gene 82: 5–30. [DOI] [PubMed] [Google Scholar]
- Michiels, P.J.A., Schouten, C.H.J., Hilbers, C.W., and Heus, H.A. 2000. Structure of the ribozyme substrate hairpin of Neurospora VS RNA: A close look at the cleavage site. RNA 6: 1821–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura, K., Tsuda, S., Ueda, T., Harada, F., and Kato, N. 1983. Chemical modification of guanine residues of mouse 5 S ribosomal RNA with kethoxal. Biochim. Biophys. Acta 739: 281–285. [DOI] [PubMed] [Google Scholar]
- Moazed, D., Stern, S., and Noller, H.F. 1986. Rapid chemical probing of conformation in 16S ribosomal RNA and 30S ribosomal subunits using primer extension. J. Mol. Biol. 187: 399–416. [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
- Nissen, P., Hansen, J., Ban, N., Moore, P.B., and Steitz, T.A. 2000. The structural basis of ribosomal activity in peptide bond synthesis. Science 289: 920–930. [DOI] [PubMed] [Google Scholar]
- Odell, L., Huang, V., Jakacka, M., and Pan, T. 1998. Interaction of structural modules in substrate binding by ribozyme from Bacillus subtilis RNase P. Nucleic Acids Res. 26: 3717–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, N.R., Thomas, B.C., and Woese, C.R. 1999. Probing RNA structure, function, and history by comparative analysis. In The RNA world, 2nd ed. (eds. R.F. Gesteland et al.), pp. 113–141. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Pappalardo, L., Kerwood, D.J., Pelczer, I., and Borer, P.N. 1998. Three-dimensional folding of an RNA hairpin required for packaging HIV-1. J. Mol. Biol. 282: 801–818. [DOI] [PubMed] [Google Scholar]
- Rivas, E. and Eddy, S.R. 1999. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J. Mol. Biol. 285: 2053–2068. [DOI] [PubMed] [Google Scholar]
- Romero, D.P. and Blackburn, E.H. 1991. A conserved secondary structure for telomerase RNA. Cell 67: 343–353. [DOI] [PubMed] [Google Scholar]
- Ruan, J., Stormo, G.D., and Zhang, W. 2004. An iterated loop matching approach to the prediction of RNA secondary structures with pseudoknots. Bioinformatics 20: 58–66. [DOI] [PubMed] [Google Scholar]
- Schluenzen, F., Tocilj, A., Zarivach, R., Harms, J., Gluehmann, M., Janell, D., Bashan, A., Bartels, H., Agmon, I., Franceschi, F., et al. 2000. Structure of functionally activated small ribosomal subunit at 3.3 Å resolution. Cell 102: 615–623. [DOI] [PubMed] [Google Scholar]
- Schultes, E.A. and Bartel, D.P. 2000. One sequence, two ribozymes: Implications for emergence of new ribozyme folds. Science 289: 448–452. [DOI] [PubMed] [Google Scholar]
- Speek, M. and Lind, A. 1982. Structural analyses of E. coli 5S RNA fragments, their associates and complexes with proteins L18 and L25. Nucleic Acids Res. 10: 947–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzl, M., Horn, C., Brown, M., Ioudovitch, A., and Steinberg, S. 1998. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 26: 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski, M., Barciszewska, M.Z., Barciszewski, J., and Erdmann, V.A. 2000. 5S ribosomal RNA database Y2K. Nucleic Acids Res. 28: 166–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Dam, E., van Belkum, A., and Pleij, K. 1991. A conserved pseudoknot in telomerase RNA. Nucleic Acids Res. 19: 6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranguch, A.J., Kinderberger, D.W., Rohlman, C.E., Lee, J., and Engelke, D.R. 1994. Structure-sensitive RNA footprinting of yeast nuclear ribonuclease P. Biochemistry 33: 1778–1787. [DOI] [PubMed] [Google Scholar]
- Turner, D.H. 2000. Conformational changes. In Nucleic Acids (eds. V. Bloomfield et al.), pp. 259–334. University Science Books, Sausalito, CA.
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304–1351. [DOI] [PubMed] [Google Scholar]
- Walter, P. and Blobel, G. 1982. Signal recognition particle contains a 7S RNA essential for protein translocation across the endoplasmic reticulum. Nature 299: 691–698. [DOI] [PubMed] [Google Scholar]
- Walter, A.E., Turner, D.H., Kim, J., Lyttle, M.H., Müller, P., Mathews, D.H., and Zuker, M. 1994a. Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc. Natl. Acad. Sci. 91: 9218–9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, A.E., Wu, M., and Turner, D.H. 1994b. The stability and structure of tandem GA mismatches in RNA depend on closing base pairs. Biochemistry 33: 11349–11354. [DOI] [PubMed] [Google Scholar]
- Wimberly, B.T., Brodersen, D.E., Clemons, W.M., Morgan-Warren, R.J., Carter, A.P., Vonrhein, C., Hartsch, T., and Ramakrishnan, V. 2000. Structure of the 30S ribosomal subunit. Nature 407: 327–329. [DOI] [PubMed] [Google Scholar]
- Wuchty, S., Fontana, W., Hofacker, I.L., and Schuster, P. 1999. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers 49: 145–165. [DOI] [PubMed] [Google Scholar]
- Xia, T., SantaLucia Jr., J., Burkard, M.E., Kierzek, R., Schroeder, S.J., Jiao, X., Cox, C., and Turner, D.H. 1998. Parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson–Crick pairs. Biochemistry 37: 14719–14735. [DOI] [PubMed] [Google Scholar]
- Xia, T., Mathews, D.H., and Turner, D.H. 1999. Thermodynamics of RNA secondary structure formation. In Prebiotic chemistry, molecular fossils, nucleosides, and RNA (eds. D.G. Söll et al.), pp 21–47. Elsevier, New York.
- Zaug, A.J. and Cech, T.R. 1995. Analysis of the structure of Tetrahymena nuclear RNAs in vivo: Telomerase RNA, the self-splicing rRNA intron, and U2 snRNA. RNA 1: 363–374. [PMC free article] [PubMed] [Google Scholar]
- Zavanelli, M.I. and Ares Jr., M. 1991. Efficient association of U2 sn-RNPs with pre-mRNA requires an essential U2 RNA structural element. Genes & Dev. 5: 2521–2533. [DOI] [PubMed] [Google Scholar]
- Zuker, M. 1989. On finding all suboptimal foldings of an RNA molecule. Science 244: 48–52. [DOI] [PubMed] [Google Scholar]
- Zuker, M. and Jacobson, A.B. 1995. “Well-determined” regions in RNA secondary structure predictions. Applications to small and large subunit rRNA. Nucleic Acids Res. 23: 2791–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1998. Using reliability information to annotate RNA secondary structures. RNA 4: 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]