

Abstract
MicroRNAs (miRNAs) are short endogenous RNAs known to post-transcriptionally repress gene expression in animals and plants. A microarray profiling survey revealed the expression patterns of 175 human miRNAs across 24 different human organs. Our results show that proximal pairs of miRNAs are generally coexpressed. In addition, an abrupt transition in the correlation between pairs of expressed miRNAs occurs at a distance of 50 kb, implying that miRNAs separated by <50 kb typically derive from a common transcript. Some microRNAs are within the introns of host genes. Intronic miRNAs are usually coordinately expressed with their host gene mRNA, implying that they also generally derive from a common transcript, and that in situ analyses of host gene expression can be used to probe the spatial and temporal localization of intronic miRNAs.
Keywords: noncoding RNA, miRNA, human gene expression, gene expression profiling, cistronic elements
INTRODUCTION
MicroRNAs are a group of short RNAs, initially identified in Caenorhabditis elegans as regulators of developmental timing (Lee et al. 1993; Bartel 2004). These RNAs have been identified in a broad range of animals, and some are conserved from C. elegans through Drosophila melanogaster to Homo sapiens (Pasquinelli et al. 2000; Lagos-Quintana et al. 2001; Lau et al. 2001; Lee and Ambros 2001). MicroRNA genes are abundant, encompassing ~1% of the genes of worms, flies, and humans (Lai et al. 2003; Lim et al. 2003a,b). Their known role as modulators of metazoan gene expression continues to expand. Examples include lsy-6, a miRNA gene in C. elegans that controls neuronal left/right asymmetry, and bantam, a miRNA in D. melanogaster that regulates cell proliferation (Brennecke et al. 2003; Johnston and Hobert 2003). In mammals, miRNAs have been implicated in the modulation of hematopoietic lineage differentiation (Chen et al. 2004), and recent computational predictions of miRNA target sites implicate their involvement in a broader regulatory network of gene interactions (Lewis et al. 2003; Kiriakidou et al. 2004).
RESULTS AND DISCUSSION
To analyze the global expression of miRNAs found in human tissues, a DNA oligonucleotide-based microarray was designed. Preparation and fluorophore (Cy3) labeling of biological samples for hybridization to the array were based on techniques previously developed to clone and sequence populations of small RNAs (Fig. 1 ▶; Lau et al. 2001). At each array feature, the signal from the biological sample was normalized to that of an internal reference. The internal reference oligonucleotides were labeled with a different fluorophore (Cy5) using the same methods as for the biological samples, starting from a uniform amount of synthetic oligonucleotides corresponding to each of the mi-RNAs probed on the array.
FIGURE 1.

Microarray sample preparation and reference oligonucleotide synthesis. Small RNAs were fractionated on a polyacrylamide gel, and oligonucleotide primers were then ligated to the 5′ and 3′ ends of the small RNA library (Lau et al. 2001). A cDNA library was generated through reverse transcription, and the product was amplified using PCR. Using a pair of modified oligonucleotide primers in a second PCR, the sense strand of the library was fluorescently labeled and the antisense strand was selectively lengthened (Williams and Bartel 1995). The sense strand of the asymmetric duplex was purified away from the antisense strand in a denaturing gel, and this purified dye-labeled ssDNA sample was used for hybridization and detection on the array. At each feature, the signal from the miRNA sample was compared to that from a reference sample, which had been generated by amplifying and labeling synthetic oligonucleotides using the same strategy as for the miRNA sample.
To explore array design options and hybridization conditions, a pilot array was designed that contained single and multiple mismatched probes. Using this array, miRNA sequences that differed by a single nucleotide could often be resolved under stringent hybridization conditions (Supplementary Fig. S1, http://web.wi.mit.edu/bartel/pub/bartel_publications.html). A more comprehensive array was printed, which included a nonredundant set of Homo sapiens, Mus musculus, and C. elegans miRNA probes. In an effort to achieve comparable stringency for each probe, the lengths of the probes were adjusted to bring the predicted melting temperature (Tm) of each sequence to a uniform temperature.
In principle, our method, which normalized to a uniform amount of each reference oligonucleotide, should reveal the relative levels of each miRNA in the biological sample. To test the correlation between microarray signal and miRNA abundance, total RNA from mixed-stage C. elegans was cloned and analyzed with the microarray. The observed scores from the microarray were in good accordance with the cloning frequencies of these miRNAs (Supplementary Fig. S2, http://web.wi.mit.edu/bartel/pub/bartel_publications.html). Deviations from a perfect correlation can be attributed to multiple factors, including potentially differing qualities of DNA synthesis for the oligonucleotides used to make the reference sample, and the potential crosshybridization of reference oligonucleotides, preferentially increasing the reference signal for miRNAs in closely related families. Cloning frequencies are in turn known to generally correlate with the molecular abundance of the miRNAs (Lim et al. 2003b). Nonetheless, our current methods are more reliable for comparing expression of the same miRNAs in different cells or conditions than for comparing the relative levels of different miRNAs. Other systems have recently been described for the expression profiling of miRNAs (Babak et al. 2004; Liu et al. 2004; Miska et al. 2004; Nelson et al. 2004; Thomson et al. 2004). The method of Miska et al. (2004), similar to ours, includes a PCR amplification step, which likely increases sensitivity but is not as convenient as direct labeling. The method of Thomson et al. (2004), like ours, uses a synthetic reference set, which provides a uniform positive control for hybridization and a valuable internal standard for normalization.
The array was used to profile the expression of 175 miRNAs from 24 different human organs and the HeLa S3 cell line, the broadest survey of miRNA expression to date (Fig. 2A ▶). Our results were consistent with and substantially expanded previous RNA blotting studies, which had examined smaller sets of miRNAs from 7–10 mammalian tissues (Lagos-Quintana et al. 2002, 2003; Sempere et al. 2004). For example, miRNAs originally identified in brain (miR-124a, miR-9) (Lagos-Quintana et al. 2002), liver (miR-122a) (Lagos-Quintana et al. 2002), muscle (miR-1b, miR-133) (Lee and Ambros 2001; Lagos-Quintana et al. 2002; Sempere et al. 2004), and bone marrow (miR-223) (Chen et al. 2004) are often cited as having highly specific expression in these organs, and our analysis of additional tissues further confirmed these specificities. Other miRNAs previously described as specifically expressed in the brain, lung, or spleen, based on a panel of seven organs (Sempere et al. 2004) were found to be expressed at equal or greater amounts in some of the newly examined tissues. In addition, we identified miRNAs preferentially expressed in breast and thymus tissue (miR-205), skeletal muscle (miR-206), pancreas (miR-216), colon (miR-194), ovary (miR-189), and in immune organs (Fig. 2B ▶). Most miRNAs, however, are more broadly expressed, although some of these could have very specific expression within the particular cell types common to the different organs.
FIGURE 2.

Hierarchical clustering of human miRNAs. (A) Colorgram depicts high (red), average (white), low (black), and absent (dark green) array scores for miRNAs. The highest score was 7095 (miR-143 in Uterus); the average score was 32. The background scores ranged between 0.4 and 3.5; for each tissue, features scoring below the background were set to the level of the background. MicroRNA scores used to generate the colorgram are reported as supplemental data (Supplementary Table 1, http://web.wi.mit.edu/bartel/pub/bartel_publications.html). (B) Examples of miRNAs preferentially expressed in specific tissues. Brain-specific miRNAs, muscle-specific miRNAs, and miRNAs associated with organs of the human immune system are highlighted.
The coexpression of closely clustered miRNAs has been used as evidence that they derive from a common primary transcript (Lau et al. 2001; Sempere et al. 2004), and for a few human and fly miRNA clusters, RT-PCR has confirmed the presence of a polycistronic transcript (Lee et al. 2002, 2004; Aravin et al. 2003). To explore the potential for co-expression among clusters of human miRNAs, we first mapped the distance between the miRNA precursors annotated in the July 2003 build of the human genome. The distances separating pairs of miRNAs on the same chromosome in this sample ranged from 101 nucleotides to >100 Mb. For each chromosome, pairwise comparisons were made between the expression profiles of all miRNAs oriented in the same direction, calculating for each pair a correlation coefficient ranging between -1 (anti-correlated) and 1 (perfectly correlated). The miRNAs were ranked by the distance separating each pair of miRNA genes from each other, and correlations were then plotted based on this ranking, with closer genes having lower rank numbers (Fig. 3 ▶). Most miRNA genes within 50 kb of each other have highly correlated expression patterns, consistent with the idea that they are processed from polycistronic primary transcripts. Beyond the 50-kb range, the correlation dropped abruptly to background levels, suggesting that the upper bound for a majority of miRNA polycistronic transcripts is 50 kb. Selective pressures might act to group some miRNAs into proximal clusters of coregulated elements. Interestingly, a plot of the distribution of distances between miRNAs found in the genome mirrors the transition that is observed in the correlative analysis; relatively few miRNAs are found between 50 kb and 500 kb of each other. These two data sets support the idea that clustered miRNAs are selected to fall within a range of 0.1 kb and 50 kb (the midpoint of the transition for both the correlative analysis and the mapped distances). This distance is similar to the average length of a human gene (55 kb) (from statistics on the first version of the human genome reference sequence, http://www.ncbi.nlm.nih.gov/Web/Newsltr/Spring03/human.html), supporting the idea that miRNAs are also transcribed by RNA polymerase II. It is also consistent with the observation that RNA polymerase II is the polymerase that transcribes the ~2.2-kb miR-23a-27a-24 primary transcript (Lee et al. 2004). There are exceptions to this 50-kb boundary; for example, miRNAs that are separated by >50 kb can be highly correlated, such as the miRNA pairs miR-7–miR-9 (correlation, 0.78) and miR-128–miR-138 (corr., 0.91), which are separated by >750 kb and 8 Mb, respectively (Fig. 2 ▶, points a,b). Each of these miRNAs is highly brain-specific, and the correlation between these miRNAs is likely the result of chance coexpression as opposed to being processed from the same primary transcript. Other miRNAs fall within the 50-kb range but are not highly correlated, such as the miR-23b–miR-24-1 pair (0.79 kb, corr., 0.19), and the miR-100–let-7a-2 pair (5.7-kb, corr., 0.14) (Fig. 2 ▶, points c,d). For each of these pairs, one of the miRNAs is represented by a duplicate miRNA at another locus, which if expressed differently than its paralog would explain the low correlation for the clustered pairs. However, other explanations, including differential processing or independent transcription, cannot be ruled out.
FIGURE 3.

Relationship between the distance separating miRNA loci and their coordinate expression in human tissues. Each of these miRNAs was paired with each of the others lying in the same orientation on the same chromosome. For each pair, the distance between the two loci was ranked, and the correlation coefficient for their expression was plotted according this rank (small points, left axis). A five-point moving average of correlation coefficients is also shown (♦), as are the distances between miRNAs in the human genome (○, right axis), and the average correlation coefficient for all the plotted pairs (dashed line). Outliers discussed in the text are annotated (a–d).
Most human miRNAs lie between protein-coding genes, whereas about one-third are within the introns of annotated mRNAs. These intronic miRNAs are usually in the same orientation as the pre-mRNA, and thus could be under the control of the promoter driving the primary mRNA transcript. Alternatively, they could be independently transcribed through an alternative promoter, and the common orientation could be explained by the prospect of colliding polymerases disfavoring opposing orientations. To test whether these intronic miRNAs are coordinately expressed with host mRNAs, we examined the correlation between the miRNA expression profiles observed from the microarray data (Fig. 2A ▶) and expression profiles of host genes, as monitored by existing Affymetrix data sets (Rebhan et al. 1998). Numerous strongly correlated expression profiles were observed between miRNAs and their host genes, whereas far fewer were observed between miRNAs and their upstream or downstream genes (Table 1 ▶), providing evidence that the miRNAs are processed from the same primary transcripts as their host genes. Similar findings for two miRNAs were also recently reported (Rodriguez et al. 2004).
TABLE 1.
Correlation between expression of intronic miRNAs and host gene
| Gene name | Ensembl IDa | MicroRNA | Host gene corr. | Upstream corr. | Downstream corr. | In situ |
| CROC4 | 125462 | hsa-miR-9-1 | 0.999 | 0.083 | −0.054 | Ab (Jeffrey et al. 2000) |
| PDE2A | 186642 | hsa-miR-139 | 0.990 | −0.293 | 0.493 | Ab (Sadhu et al. 1999) |
| C20orf166 | 174407 | hsa-miR-133a-2 | 0.988 | −0.105 | 0.185 | |
| hsa-miR-1-1 | 0.968 | −0.045 | 0.226 | |||
| PGSF1 | 176840 | hsa-miR-7-3 | 0.961 | −0.394 | −0.123 | |
| ABLIM2 | 163995 | hsa-miR-95 | 0.960 | −0.302 | −0.089 | |
| LOC254559 | LOC254559 | hsa-miR-9-3 | 0.950 | −0.122 | −0.150 | |
| AATYK | 181409 | hsa-miR-338 | 0.921 | 0.906 | −0.123 | Ab (Baker et al. 2001; Tomomura et al. 2001) |
| EGFL7 | 172889 | hsa-miR-126 | 0.888 | 0.727 | −0.246 | Ab, Nuc (Parker et al. 2004) |
| R3HDM | 48991 | hsa-miR-128a | 0.856 | −0.037 | −0.363 | |
| MCM7b | 166508 | hsa-miR-25 | 0.838 | −0.548 | −0.377 | |
| TRPM3 | 83067 | hsa-miR-204 | 0.796 | −0.047 | −0.240 | |
| TLN2 | 171914 | hsa-miR-190 | 0.663 | −0.269 | −0.172 | |
| PANK3 | 120137 | hsa-miR-103-1 | 0.638 | −0.203 | 0.079 | |
| PTPRN | 54356 | hsa-miR-153-1 | 0.626 | 0.579 | −0.178 | |
| CTDSP2 | 175215 | hsa-miR-26a-2 | 0.609 | 0.492 | 0.265 | |
| SMC4L1 | 113810 | hsa-miR-15b | 0.509 | −0.134 | −0.098 | |
| hsa-miR-16-2 | 0.504 | −0.128 | −0.021 | |||
| UREB1 | 86758 | hsa-miR-98 | 0.503 | −0.209 | −0.512 | |
| hsa-let-7f-2 | 0.379 | −0.242 | −0.194 | |||
| PTPRN2 | 155093 | hsa-miR-153-2 | 0.499 | 0.302 | 0.536 | |
| CTDSP1 | 144579 | hsa-miR-26b | 0.453 | 0.292 | −0.079 | |
| ARPP-21 | 172995 | hsa-miR-128b | 0.444 | 0.622 | −0.171 | |
| MEST | 106484 | hsa-miR-335 | 0.442 | 0.129 | −0.088 | Nuc (Mayer et al. 2000) |
| NFYC | 66136 | hsa-miR-30c-1 | 0.406 | −0.071 | 0.419 | |
| Q8TDA7 | 174496 | hsa-miR-99a | 0.335 | 0.217 | 0.221 | |
| hsa-let-7c | 0.067 | 0.712 | −0.053 | |||
| hsa-miR-125b-2 | 0.297 | −0.037 | 0.221 | |||
| PANK2 | 125779 | hsa-miR-103-2 | 0.270 | −0.138 | 0.450 | |
| PRO2730 | 164091 | has-let-7g | 0.268 | 0.516 | −0.288 | |
| RCL1 | 120158 | hsa-miR-101-2 | 0.226 | −0.494 | 0.196 | |
| DNM1 | 79805 | hsa-miR-199a-1 | 0.020 | −0.124 | −0.444 | |
| C9orf5 | 106771 | hsa-miR-32 | −0.22 | 0.111 | −0.290 | |
| CTDSPL | 144677 | hsa-miR-26a-1 | −0.285c | −0.202 | 0.099 |
aEnsembl ID numbers begin with the prefix “ENSG00000,” LOC254559 is a Genecards ID.
bA score of >16 in at least one tissue was required to be included in the analysis. MCM7 contains two other miRNAs, miR-106b and miR-93, which were excluded based on this criterion although their correlation coefficients are consistent with coordinate expression.
cThe anti-correlation between miR-26a-1 and CTDSPL can be explained by the CTDSP2 transcript being the primary source of miR-26a. CTDSP2, the host gene of miR-26a-2, is generally expressed at much higher levels than is CTDSPL, and its expression is anti-correlated (−0.106) with that of CTDSPL. Similar scenarios could explain the low correlations between the expression of other miRNAs and their host genes. Nuc, nucleic acid hybridization; Ab, antibody staining or immunofluorescence.
In situ analysis of miRNA primary transcripts has been successful in fly embryos but has not yet been reported in mammals (Kosman et al. 2004). The coexpression of intronic miRNAs and host genes implies that existing mRNA in situ expression data already provide high-resolution information regarding which cells express these intronic miRNAs. Based on the specific expression of some miRNAs in mammalian neurons, it has been proposed that miRNAs may regulate translation in mammalian neurons, and that these miRNAs could have roles in synaptic remodeling (Kim et al. 2004). The expression profile of miRNAs was highly correlated with some host genes. miR-9-1 is embedded in intron 2 of the human CROC-4 gene, and its expression highly correlates with the expression of this gene (corr., 0.99). CROC-4 is expressed in proliferating and migrating cells in mouse and is present in the developing forebrain. When observed at higher resolution, specific staining is observed in the olfactory epithelium lining the olfactory pit and in the medial nasal process. In rat, CROC-4 expression is associated with cells proliferating around the fourth ventricle and in the developing optic vesicle. It has been suggested, based on its localization with proliferating and migrating cells during early brain development, that CROC-4 participates in pathways involved in modulating cellular architecture during neuronal development and plasticity (Jeffrey et al. 2000). The same could be true of its resident miRNA, miR-9-1. In situ analysis exists for other correlated host genes; particularly interesting examples are AATYK (miR-338) and EGFL7 (miR-126) (Baker et al. 2001; Fitch et al. 2004).
In summary, these findings support the idea that clusters of proximal miRNAs are typically expressed as polycistronic, coregulated units and that intronic miRNAs are generally coexpressed with their host genes. Host gene-miRNA coexpression is illuminating in cases where detailed host gene in situ analysis is available and can reveal complex miRNA expression patterns. Microarray profiling of miRNA expression is a useful strategy for examining the global expression profiles of this abundant class of small RNAs. The contents of this array are easily expanded; as new miRNAs are discovered in other systems, probes for these miRNAs can be easily incorporated into the existing array. It will be interesting, in the future, to explore the dynamics of global miRNA expression in developing tissues or whole organisms and to probe the relationship between miRNA expression and biological function, thus providing essential information for placing these abundant riboregulators into the gene regulatory circuitry of the animal.
MATERIALS AND METHODS
Microarray design
Antisense probes for the oligonucleotide array were synthesized at a 0.01 μM scale (MWG biotech), modified at their 5′ ends with a 6-carbon linker and a primary amine (MWG biotech). Each probe oligonucleotide sequence was designed to have a predicted Tm of ~55°C (20 nM probe, 50 mM NaCl) when paired to its cognate DNA (Breslauer et al. 1986). Two strategies were employed to narrow the distribution of melting temperatures on the array to allow tighter control of specificity during sample hybridization. Probe oligonucleotides with calculated Tm’s exceeding 55°C were truncated to bring their Tm to ~55°C, while retaining the segment that afforded the greatest discrimination among known miRNAs. In a few cases, the predicted Tm did not approach 55°C, even when hybridizing to the entire mature miRNA. These probes were extended with up to five additional nucleotides on their 3′ ends to extend complementarities into the constant flanking sequence of the labeled single-stranded DNA. For example, a probe that is the antisense of hsa-miR-190, ACCTAATATATCAAACATATCA, has a calculated Tm of 44.8°C while its extended version, ACCTAATATATCAAACATATCATTTCA has a Tm of 53.5°C (underlined sequence, 3′ extension). Synthetic probe oligonucleotides were spotted onto activated slides according to the manufacturer’s instructions (Codelink, Amersham Biosciences).
Reference sample
A synthetic reference oligonucleotide was synthesized for every probe on the array (MWG biotech). Each oligonucleotide was synthesized as the sense strand of the miRNA sequence plus the constant sequences flanking each end that were used as primer sites for PCR amplification. For example, the reference oligonucleotide for hsa-miR-190 is: ATCGTAGGCACCTGAAATGAT ATGTTTGATATATTAGGTCTGTAGGCACCATCAAT (primer sites underlined). The reference sample was amplified from a mixture containing 0.22 nM synthetic oligonucleotides. The sample was labeled during 10 cycles of PCR amplification using a Cy5 dye-labeled oligonucleotide (Cy5-ATCGTAGGCACCTGAAA, IDT) as the sense-strand primer. The antisense oligonucleotide primer (ATTGATGGTGCCTACAG-C18-(A)20) contained a C18 spacer followed by a 20-nucleotide polyadenosine sequence. The shorter, Cy5-labeled strand of the amplified dsDNA product was purified from a denaturing polyacrylamide gel (6% polyacrylamide, 8 M urea).
MicroRNA samples
Samples for hybridization to the array were size-selected, ligated to adapter oligos, reverse-transcribed, and amplified as described for miRNA cloning (Lau et al. 2001). To prevent the nonuniform amplification that can happen during late PCR cycles, all amplifications in this process were stopped at a point where product first became visible on an ethidium-stained agarose gel (4% NuSieve, Cambrex). For all tissues except bone marrow, total RNA was purchased from Ambion. Bone marrow total RNA was extracted from human bone marrow cells (Cambrex) using the Tri reagent according to the manufacturer’s protocol (Sigma). Biological replicates of human hepatocytes isolated from two separate transplant grade livers were prepared using the same method (Supplementary Fig. S3, http://web.wi.mit.edu/bartel/pub/bartel_publications.html). Labeling and purification of the single-stranded DNA was as described for the reference sample, except that Cy5 was replaced with Cy3 (Cy3-ATCGTAGGCACCTGAAA, IDT).
Hybridization and analysis
Before use, microarrays were prehybridized (Pre-Hyb solution, 3.5 × SSC, 1% BSA, 0.1% SDS, 50°C, 45 min). For hybridization to the microarray, 10 pmol of the labeled ssDNA library was mixed with 10 pmol of the reference set in hybridization solution [25 μL final volume, 3.5 × SSC, 1% BSA, 0.1% SDS, 0.1 mg/mL herring sperm DNA (Sigma), 0.2 mg/mL yeast tRNA (Sigma), 0.4 mg/mL polyA RNA (Sigma), 50°C]. Hybridization was beneath lifter cover slips, in an aluminum chamber submerged in a water bath (57°C, 6 h). Slides were washed (2 × SSC, 0.1%, SDS 50°C, 5 min, 0.1 × SSC, 0.1% SDS, 10 min, wash 3× with 0.1 × SSC, 1 min), dried, and scanned (Genepix pro 4000B, Axon). Spots with an unacceptably low reference signal (defined as less than or equal to the median background plus two times its standard deviation) were eliminated from the analysis (dark green features in Fig. 2 ▶). Median local background was subtracted from the median spot intensity to generate background-corrected median intensities for all spots. For each chip with human samples, the sample background was set at a score (sample signal divided by reference signal) that exceeded that of 95% of the spots from the C. elegans section of the array (typically between 0.4 and 3.5). Scores less than the sample-background score were set to the sample-background score. The Cluster and Treeview packages developed by M. Eisen at Stanford University were used for hierarchical clustering (average linkage correlation) and generating expression maps. When calculating correlation coefficients, data sets with a maximum value of >16 (a level that is half the average expression score across all miRNAs and tissues examined) were included in the analysis. Linear correlation coefficients were calculated using the equation:
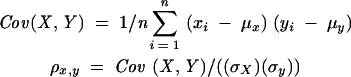 |
Acknowledgments
We thank M. Jones-Rhoades for gathering and organizing the initial miRNA data sets used in the design of this microarray, N. Lau for human bone marrow total mRNA, M. Axtell for many helpful discussions, and B. Chevalier for human liver samples used in Supplementary Figure S3. This work was supported by NIH grant DK068348.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.7240905.
REFERENCES
- Aravin, A.A., Lagos-Quintana, M., Yalcin, A., Zavolan, M., Marks, D., Snyder, B., Gaasterland, T., Meyer, J., and Tuschl, T. 2003. The small RNA profile during Drosophila melanogaster development. Drosophila. Drosophila. Cell 5: 337–350. [DOI] [PubMed] [Google Scholar]
- Babak, T., Zhang, W., Morris, Q., Blencowe, B.J., and Hughes, T.R. 2004. Probing microRNAs with microarrays: Tissue specificity and functional inference. RNA 10: 1813–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, S.J., Sumerson, R., Reddy, C.D., Berrebi, A.S., Flynn, D.C., and Reddy, E.P. 2001. Characterization of an alternatively spliced AATYK mRNA: Expression pattern of AATYK in the brain and neuronal cells. Oncogene 20: 1015–1021. [DOI] [PubMed] [Google Scholar]
- Bartel, D.P. 2004. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116: 281–297. [DOI] [PubMed] [Google Scholar]
- Brennecke, J., Hipfner, D.R., Stark, A., Russell, R.B., and Cohen, S.M. 2003. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113: 25–36. [DOI] [PubMed] [Google Scholar]
- Breslauer, K.J., Frank, R., Blocker, H., and Marky, L.A. 1986. Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. 83: 3746–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.Z., Li, L., Lodish, H.F., and Bartel, D.P. 2004. MicroRNAs modulate hematopoietic lineage differentiation. Science 303: 83–86. [DOI] [PubMed] [Google Scholar]
- Fitch, M.J., Campagnolo, L., Kuhnert, F., and Stuhlmann, H. 2004. Egfl7, a novel epidermal growth factor-domain gene expressed in endothelial cells. Dev. Dyn. 230: 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey, P.L., Capes-Davis, A., Dunn, J.M., Tolhurst, O., Seeto, G., Hannan, A.J., and Lin, S.L. 2000. CROC-4: A novel brain specific transcriptional activator of c-fos expressed from proliferation through to maturation of multiple neuronal cell types. Mol. Cell Neurosci. 16: 185–196. [DOI] [PubMed] [Google Scholar]
- Johnston, R.J. and Hobert, O. 2003. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature 426: 845–849. [DOI] [PubMed] [Google Scholar]
- Kim, J., Krichevsky, A., Grad, Y., Hayes, G.D., Kosik, K.S., Church, G.M., and Ruvkun, G. 2004. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc. Natl. Acad. Sci. 101: 360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriakidou, M., Nelson, P.T., Kouranov, A., Fitziev, P., Bouyioukos, C., Mourelatos, Z., and Hatzigeorgiou, A. 2004. A combined computational-experimental approach predicts human microRNA targets. Genes & Dev. 18: 1165–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosman, D., Mizutani, C.M., Lemons, D., Cox, W.G., McGinnis, W., and Bier, E. 2004. Multiplex detection of RNA expression in Drosophila embryos. Science 305: 846. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana, M., Rauhut, R., Lendeckel, W., and Tuschl, T. 2001. Identification of novel genes coding for small expressed RNAs. Science 294: 853–858. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer, J., Lendeckel, W., and Tuschl, T. 2002. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12: 735–739. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana, M., Rauhut, R., Meyer, J., Borkhardt, A., and Tuschl, T. 2003. New microRNAs from mouse and human. RNA 9: 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, E.C., Tomancak, P., Williams, R.W., and Rubin, G.M. 2003. Computational identification of Drosophila microRNA genes. Genome Biol. 4: R42–R42.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, N.C., Lim, L.P., Weinstein, E.G., and Bartel, D.P. 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858–862. [DOI] [PubMed] [Google Scholar]
- Lee, R.C. and Ambros, V. 2001. An extensive class of small RNAs in Caenorhabditis elegans. Science 294: 862–864. [DOI] [PubMed] [Google Scholar]
- Lee, R.C., Feinbaum, R.L., and Ambros, V. 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854. [DOI] [PubMed] [Google Scholar]
- Lee, Y., Jeon, K., Lee, J.T., Kim, S., and Kim, V.N. 2002. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 21: 4663–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y., Kim, M., Han, J., Yeom, K.H., Lee, S., Baek, S.H., and Kim, V.N. 2004. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23: 4051–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, B.P., Shih, I.H., Jones-Rhoades, M.W., Bartel, D.P., and Burge, C.B. 2003. Prediction of mammalian microRNA targets. Cell 115: 787–798. [DOI] [PubMed] [Google Scholar]
- Lim, L.P., Glasner, M.E., Yekta, S., Burge, C.B., and Bartel, D.P. 2003a. Vertebrate microRNA genes. Science 299: 1540. [DOI] [PubMed] [Google Scholar]
- Lim, L.P., Lau, N.C., Weinstein, E.G., Abdelhakim, A., Yekta, S., Rhoades, M.W., Burge, C.B., and Bartel, DP. 2003b. The micro-RNAs of Caenorhabditis elegans. Genes & Dev. 17: 991–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C.G., Calin, G.A., Meloon, B., Gamliel, N., Sevignani, C., Ferracin, M., Dumitru, C.D., Shimizu, M., Zupo, S., Dono, M., et al. 2004. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc. Natl. Acad. Sci. 101: 9740–9744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer, W., Hemberger, M., Frank, H.G., Grummer, R., Winterhager, E., Kaufmann, P., and Fundele, R. 2000. Expression of the imprinted genes MEST/Mest in human and murine placenta suggests a role in angiogenesis. Dev. Dyn. 217: 1–10. [DOI] [PubMed] [Google Scholar]
- Miska, E.A., Alvarez-Saavedra, E., Townsend, M., Yoshii, A., Sestan, N., Rakic, P., Constantine-Paton, M., and Horvitz, H.R. 2004. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 5: R68–R68.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, P.T., Baldwin, D.A., Scearce, L.M., Oberholtzer, J.C., and To-bias, J.W. 2004. Microarray-based, high-throughput gene expression profiling of microRNAs. Nature Methods 1: 155–161. [DOI] [PubMed] [Google Scholar]
- Parker, L.H., Schmidt, M., Jin, S.W., Gray, A.M., Beis, D., Pham, T., Frantz, G., Palmieri, S., Hillan, K., Stainier, D.Y., et al. 2004. The endothelial-cell-derived secreted factor Egfl7 regulates vascular tube formation. Nature 428: 754–758. [DOI] [PubMed] [Google Scholar]
- Pasquinelli, A.E., Reinhart, B.J., Slack, F., Martindale, M.Q., Kuroda, M.I., Maller, B., Hayward, D.C., Ball, E.E., Degnan, B., Srinivasan, A., et al. 2000. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408: 86–89. [DOI] [PubMed] [Google Scholar]
- Rebhan, M., Chalifa-Caspi, V., Prilusky, J., and Lancet, D. 1998. Gene-Cards: A novel functional genomics compendium with automated data mining and query reformulation support. Bioinformatics 14: 656–664. [DOI] [PubMed] [Google Scholar]
- Rodriguez, A., Griffiths-Jones, S., Ashurst, J.L., and Bradley, A. 2004. Identification of mammalian microRNA host genes and transcription units. Genome Res. 14: 1902–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhu, K., Hensley, K., Florio, V.A., and Wolda, S.L. 1999. Differential expression of the cyclic GMP-stimulated phosphodiesterase PDE2A in human venous and capillary endothelial cells. J. Histochem. Cytochem. 47: 895–906. [DOI] [PubMed] [Google Scholar]
- Sempere, L.F., Freemantle, S., Pitha-Rowe, I., Moss, E., Dmitrovsky, E., and Ambros, V. 2004. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 5: R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, J.M., Parker, J., Perou, C.M., and Hammond, S.M. 2004. A custom microarray platform for analysis of microRNA gene expression. Nature Methods 1: 47–53. [DOI] [PubMed] [Google Scholar]
- Tomomura, M., Fernandez-Gonzales, A., Yano, R., and Yuzaki, M. 2001. Characterization of the apoptosis-associated tyrosine kinase (AATYK) expressed in the CNS. Oncogene 20: 1022–1032. [DOI] [PubMed] [Google Scholar]
- Williams, K.P. and Bartel, D.P. 1995. PCR product with strands of unequal length. Nucleic Acids Res. 23: 4220–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]