

Abstract
The ribonucleoprotein enzyme telomerase ensures the stability and fidelity of linear chromosome ends by elongating the telomeric DNA that is lost during each round of DNA replication. All telomerases contain a catalytic protein component homologous to viral reverse transcriptases (TERT) and an RNA (TR) that provides the template sequence, acts as the scaffold for ribonucleoprotein assembly, and activates the enzyme for catalysis. Vertebrate telomerase RNAs contain three highly conserved structural and functional domains: the template domain, the “CR4-CR5” or “activation” domain essential for activation of the enzymatic activity, and a 3′-terminal “box H/ACA”–homology domain responsible for ribonucleprotein assembly and maturation. Here we report the NMR structure of a functionally essential RNA structural element derived from the human telomerase RNA CR4-CR5 domain. This RNA, referred to as hTR J6, forms a stable hairpin interrupted by a single nucleotide bulge and an asymmetric internal loop. Previous work on telomerase has shown that deletion of the hTR J6 asymmetric internal loop results in an RNA incapable of binding the enzymatic protein component of the RNP and therefore an inactive RNP without telomerase activity. We demonstrate here that the J6 internal loop introduces a twist in the RNA structure that may position the entire domain into the catalytic site of the enzyme.
Keywords: NMR, residual dipolar couplings, ribonucleoprotein, RNA structure, telomerase, telomeres
INTRODUCTION
The linear chromosomes of eukaryotic organisms require protection of their terminal ends (telomeres) from fusion, recombination, spurious DNA repair, and the progressive truncation due to incomplete copying of linear DNA (Lingner and Cech 1998). Several hundred to several thousands of G-rich repeats are added at the telomeres by the ribonucleoprotein (RNP) enzyme telomerase. These repeats allow the genetic information near these ends to be buffered from degradation and to nucleate the chromatin structures that cap the ends, preventing recombination and unnecessary DNA repair (McElligott and Wellinger 1997).
Telomerase is an RNA-dependent DNA polymerase (Lingner et al. 1997), and it is therefore functionally homologous to retroviral reverse transcriptases. The RNA component (TR) and the enzymatic protein component (TERT) can fully reconstitute telomerase activity in vitro, but additional protein components, many still uncharacterized, regulate telomerase RNA processing and aid in RNP assembly and maturation. The TERT reverse transcriptase domain provides the catalytic activity; TR provides the template for reverse transcription but also enhances and regulates enzymatic activity by ensuring both processive nucleotide addition and repeat amplification (Xia et al. 2000; Chen et al. 2002; Moriarty et al. 2002, 2004; Chen and Greider 2003; Mason et al. 2003). TR has two biochemically distinct domains essential for telomerase enzymatic activity: Its 5′-end provides the template and enhances repeat amplification processivity, while a second domain called the “CR4-CR5” or also “activation” domain (Fig. 1A), supports catalytic activity by enhancing nucleotide addition processivity (Tesmer et al. 1999; Chen et al. 2000; Mitchell and Collins 2000). TERT interacts with TR via two regions in addition to the catalytic domain, RID-1 and RID-2 (Xia et al. 2000; Moriarty et al. 2002, 2004). These highly conserved protein domains interact with the template (RID-1) (Moriarty et al. 2004) and CR4-CR5 domains (RID-2) (Lai et al. 2001), respectively. A third TR RNA domain present in vertebrates closely resembles box H/ACA scaRNAs and associates with several proteins shared with box H/ACA RNPs (Mitchell et al. 1999; Pogacic et al. 2000; Fu and Collins 2003; Jady et al. 2004). This eukaryotic-specific domain (H/ACA-homology domain) and its associated proteins are important for RNA processing and RNP assembly.
FIGURE 1.

(A) A schematic of the secondary structure and domain structure of human telomerase RNA. The template, H/ACA, and CR4-CR5 domains are indicated by blue, green, and red boxes, respectively. (B) The sequence of the RNA studied and regions of secondary structure are indicated as paired regions 6a and 6b (P6a and P6b) and bulge loop 6 (L6). The numbering scheme matches that of wild-type hTR.
The focus of the present study is the CR4-CR5, enzyme activation domain of human telomerase RNA. This region of the RNA is a true functional and structural domain: It can assemble into a functional particle when added in trans after being transcribed separately from the remainder of TR (Tesmer et al. 1999; Mitchell and Collins 2000; Chen et al. 2002). This region is essential for processive nucleotide addition (Chen et al. 2000) and forms key interactions with the enzyme TERT; when these interactions are disrupted, the enzymatic activity is essentially abolished (Mitchell and Collins 2000). Since telomerase activation is a common step in immortalization in many tumor types (Delange 1994; Kelland 2000), this domain is also a promising target for structure-aided discovery of new antitumor drugs that effect telomerase activity.
As part of a systematic effort to dissect the structural requirements for telomerase activity, we previously reported the NMR structure of an essential small hairpin from this region (Leeper et al. 2003). In the present work, we present the NMR structure of an adjacent essential region (Fig. 1A,B): the asymmetric internal loop “J6” from the CR4-CR5 domain of human telomerase (Mitchell and Collins 2000; Chen and Greider 2004). We have used residue-specific labeling, multinuclear NMR, and residual dipolar coupling (RDC) refinement to determine a high precision NMR structure of this 32-nucleotide RNA to a root mean square deviation (RMSD) of 0.79 Å. We discuss the implications of the structure for the mechanism of telomerase activation.
RESULTS
Structure determination
The solution structure of the J6 region of the CR4-CR5 activation domain of human telomerase RNA was determined by utilizing NOE, dihedral, and RDC restraints. Representative NMR data are shown in Figure 2. Of the 100 structures that were calculated, 40 had zero NOE or dihedral violations >0.5 Å or >5° and were deemed to be the converged set of structures. The converged structures were ranked by lowest energy, and the best 10 coordinate sets were selected as members of the final ensemble representing the conformation of this fragment of human telomerase RNA. As shown in Table 1, a relatively large number of restraints were used. Approximately 23 significant constraints per residue were obtained; in other words, each residue was constrained by an average of 15.6 distance restraints, five dihedrals, and two RDCs.
FIGURE 2.

NMR spectra of the hTR J6 containing RNA. (A) The anomeric to aromatic region of a 300-msec mixing time NOESY recorded at 750 MHz with sequential H1′ to aromatic peaks labeled “s,” intraresidue peaks are labeled “i,” and H5/6 cross-peaks for the pyrimidines are indicated in gray. (B) The aromatic region of a constant time 13C HSQC.
TABLE 1.
Structural statistics
| Total number of restraints | 726 (∼23/nt) |
| NOE-derived restraints | 429 |
| Dihedral restraints | 159 |
| Dipolar coupling restraints | 65 |
| Hydrogen bonds | 62 |
| Planarity | 11 |
| Averge RMS deviations from experimental restraints | |
| Distance | 0.06 Å |
| Dihedral | 0.6° |
| RDC | 0.95 Hz |
| Average RMS deviations from ideal geometries | |
| Bonds | 0.0056 Å |
| Angles | 0.89° |
| Impropers | 0.41° |
| Heavy atom rms deviation from mean structure (Å) | |
| Watson-Crick paired residues | 0.66 |
| P6a, including bulged C262 | 0.45 |
| P6b with UUCG tetraloop | 0.35 |
| Bulge J6 and flanking pairs | 0.89 |
| All residues | 0.79 |
The overlay of the 10 lowest energy members of the ensemble of converged structures is shown in Figure 3. The RMSD to the average structure of all heavy atoms for base-paired residues is 0.66 Å. For each stem segment, the RMSD was 0.45 Å for residues 257–265 and 292–299, and 0.35 Å for residues 268– 288. The asymmetric internal loop that is the core of the entire structure had an RMSD of 0.89 Å. Structural statistics are presented in the bottom part of Table 1. Overall, these values indicate a structure of very high quality. This statement remains true for the internal loop region, although this region has fewer NOEs per nucleotide compared with the remainder of the structure, and hydrogen bonding or planarity restraints were not employed.
FIGURE 3.

Superimposition of the 10 best-scoring members of the ensemble of hTR J6 RNA structures. Residues are colored according to the diagram in Figure 1B, except that the backbone is traced in grey to highlight the distorted curvature and ζ-like conformation induced by the J6 internal loop.
Structure of the apical stem–loop of the enzyme-activating domain of human telomerase RNA
The stem regions adopts canonical A-form RNA structure within the double-helical regions and are capped by a familiar UUCG tetraloop conformation (Allain and Varani 1995): a sheared GU pair with a U271 2′OH hydrogen bond and the base of G285 flipped into the syn conformation. The bases of U272 and C284 fan out and down, and C284 cross-strand stacks above U271; the sugars of residues 272 and 284 both have strong 2′-endo character.
Nucleotides 257–265 and 292–299 form an 8-bp helix interrupted by a single bulged C. A base pair present between U261/A295 is undoubtedly more consistent with our NMR data than a C262/A295 pair, because of many very clear NOE-determined distances observed within this region: The G260 H2′ to C262 H6 NOE is not present, and C262 2′ to G263 H8 is quite weak and only present at long mixing times. In contrast, the NOE seen between G260 H2′ and U261 H6 is strong, as would be expected if U261were paired and A-form. Furthermore, three “cross-bulge” NOEs, U261 H1′, H2′, and H6 to G263 H8, are more consistent with a paired U261 than with a paired C262 conformation. The broad imino proton of U261 also correlates to the N1 of an adenine in HNN-COSY spectra. However, a C262/A295 base pair was predicted at this site based on phylogenetic data (Chen et al. 2000; Chen and Greider 2004). C262 has strong conservation; position 295 is A in humans but G in all other species, strongly supporting a C262/G295 base pair. Furthermore, nucleotide U261 is highly variable and is even deleted in one species, the Quoll. However, the phylogenetic data are to some extent in conflict with biochemical data. When hTR was probed with base-modifying chemicals, C262 was moderately susceptible to DMS in vitro and in vivo, whereas under the same conditions U261 was not reactive to CMCT (Antal et al. 2002). It was also found that A295 was weakly susceptible to DMS. Perhaps the base pairs that form in this region are not very stable and may be easily disrupted by the reverse transcriptase protein when it binds to this RNA. Alternatively, U261 may be bulged out in most species, when the C262/G295 base pair forms, while in hTR C262 is bulged out and substitutes for U261 functionally.
The most functionally interesting region of the structure is the J6 internal loop (Fig. 4). C266 and A289 at the 5′ ends of each strand in the internal loop stack precisely on top of their stem-associated 5′ neighboring nucleotides. The conformation of C267, C290, and U291 are less precisely defined by the NMR data, but clear trends are nonetheless apparent. C267 displays two populations of structures, either stacking upon C266 or lying nearly coplanar with the G268/C288 base pair (Fig. 4B). The second set of conformations resemble a base triple but are not fully consistent with established hydrogen bonding patterns seen for other base triples (Walberer et al. 2003), in that the O2 carbonyl of C267 is in relatively close proximity to the O6 carbonyl of G268. If a base triple is present, it may be water mediated. The Watson-Crick and major groove faces of C267 are both exposed to solvent and might be used for contacts with TERT or the rest of TR (Fig. 4B,D). While there is a slight preference for the C267/G268/C288 triple in ensemble members, both conformations of C267 are consistent with the NMR data, and perhaps both are present in conformational exchange. Furthermore, C267 is involved in nearly the same number of NOEs as are the more ordered residues C266 and A289, suggesting that a low NOE density for this residue is not the sole explanation for the apparent conformational heterogeneity. In fact, residue C267 has a total of 19 NOEs, 13 of which are sequential NOEs to C266 or G268; residue C266 has 21 NOEs, of which 14 are sequential; residue A289 also has 19 NOEs of which 13 are sequential and one is a long-range cross-strand NOE involving the H2. Additionally, the peak shapes for C266 and C267 are similar, indicating qualitatively similar local dynamics. On the opposite strand, C290 is predominantly stacked under A289. U291 partially stacks below C290 and more clearly stacks above G292. While the distances between atoms in the base of C266 and the base of U291 are larger than what would be expected for a C/U pair, their topological position implies that some fraction of the time these two nucleotides are in paired conformation (Fig. 4C).
FIGURE 4.

(A) Stereo views of the J6 bulge region from the lowest energy conformer of the hTR J6 structure, highlighting the juxtaposition of C266 and U291, the putative base triple (indicated by a red arrow), and the stacking of A289 and C290 under the P6b stem. (B) A superimposition of the nucleotides involved in this proposed base triple clearly shows the two populations of the C267 position (red arrow) for the 10-structure ensemble, with the less frequently observed stacked conformation displayed in pale green. Note that the perspective for B is rotated by ∼180° with respect to A. (C) The C266/U291 pair consistent with phylogenetic conservation is shown with possible hydrogen bonds indicated by the red dashed lines. (D) A possible hydrogen-bonding network stabilizing the base triple is indicated by dashed red lines.
Dynamics of the structure
During refinement of the structure with residual dipolar coupling (RDC) data, separate grid searches were performed to optimize Da and R by considering each of the individual stems as separate units; in addition, a third simulation was conducted that considered the entire molecule as a single rigid unit. This was done to establish whether the internal loop introduces a discontinuity in the structure that leads to independent motions for the two helical domains on either side of the loop. The presence of dynamics of a two-domain molecule can be established from the variation in Da and R between simulations that treat each domain independently of the other. When the Da and R for the two subdomains of a molecule are the same, the molecule is very likely to tumble together as a single rigid unit and the two domains experience little relative reorientation relative to one another. When Da is very different between the two domains, however, but R is the same, the degree of motion between the domains is directly proportional to the difference in Da. In this case, the motion is pseudo-isotropic and can be modeled as diffusion within the envelope of a cone (Tolman et al. 2001; Al-Hashimi et al. 2002). When both Da and R vary together, in contrast, the motion between the two domains is highly anisotropic. It can be modeled as motion along an arc, and the magnitude of this motion can be evaluated by the combined contributions of the differences in Da and R (Tolman et al. 2001). Because R is highly sensitive to motions of this second type, even relatively large deviations in optimal R of 0.1 or 0.2 represent relatively small deviations in orientation.
As described in detail in the Materials and Methods section, we systematically compared the differences in Da and R obtained for the three different simulations conducted for J6 RNA with published estimates of degree of motions along an arc (Tolman et al. 2001). This analysis established that the two helical stems sample relatively small rearrangements, of ≤10°, and that these motions are discreetly organized in an anisotropic trajectory. Most likely, a motion along an arc represents fairly accurately the underlying reorientation of the two helical stems with respect to one another. However, we wish to emphasize that it is very clear that the amplitude of these motions is relatively small: In fact, we can choose single values of Da and R to refine the entire molecule because the differences for the three simulations are so small. Therefore, the final conformational ensembles represent an average of the small dynamic motions along a relatively limited trajectory. The implication of these results is that while the bulge may act as a subtly plastic motif, it has a more rigid conformation than other bulges in RNA that are often highly dynamic, such as the bulge in the free TAR RNA (Aboul-ela et al. 1995; Al-Hashimi et al. 2002; Zhang et al. 2003).
DISCUSSION
The CR4-CR5 or activation domain of hTR RNA is one of two functional domains of hTR that are required for telomerase enzymatic activity in vitro and in vivo. Removal of the entire domain or truncations that eliminate the J6 internal loop essentially abolish telomerase activity. These effects are at least in part due to the abolition of TERT binding to this region of the RNA, because there is a strong correlation between TERT binding and telomerase activity for mutants within this region (Mitchell and Collins 2000). The CR4-CR5 region of hTR is a bona fide functional and structural domain: It can be provided in trans and activates the enzyme when provided on a separate molecule from the remainder of the RNA (Tesmer et al. 1999; Mitchell and Collins 2000). Indeed, active telomerase can be functionally assembled with hTERT and two inactive domains of hTR comprising the template and activation domain, respectively (Tesmer et al. 1999). Truncation studies have established that the functionally essential regions within the CR4-CR5 domain include the three-way junction and the L6.1 loop, as well as the region up to and including the J6 internal loop (Fig. 1). While removal of the internal loop J6 abolishes activity, additional deletions further up the terminal stem–loop have no effect on TERT binding or enzymatic activity, establishing the boundary of the functional region of CR4-CR5 (Mitchell and Collins 2000). These results demonstrate that TERT has two separate interactions with hTR, the template and activation domain, including the J6 region studied here. Intriguingly, functional and structural homology have led to the suggestion that these two domains constitute a minimal ancestral telomerase RNA (Chen et al. 2000; Mitchell and Collins 2000; Sperger and Cech 2001).
The essential structural features of the P6a/J6/P6b region presented in this work can be summarized as follows. The loop region forms a stable secondary structure, although phylogeny could not confirm base pairing and this region was first drawn as a large, unstructured loop (Mitchell et al. 1999; Chen et al. 2000, 2002; Mitchell and Collins 2000). The two paired regions P6a and P6b form standard A-form stems, but P6a is interrupted by a bulged cytosine. Within the asymmetric J6 internal loop region, we observe a possible C/U pair stacking over P6a. Two populations of structures are observed for nucleotide C267. In one ensemble, C267 stacks over C266; in the second, it forms a novel base triple with the G268/C288 base pair of P6b. The first two nucleotides of the 3′ strand, A289 and C290, stack directly under the first G/C pair of P6b. The third nucleotide on the 3′ strand (U291) is stacked above G292 and only partially stacked under C290; it is topologically juxtaposed with nucleotide C266, forming a C/U pair. These local distortions affect the overall conformation of the entire region. The helical axis of the two paired regions are not coaxial: The bulge introduces a strong over-twist that gives the RNA an unusual profile, vaguely resembling the lowercase Greek character zeta, ζ (Fig. 3). This distortion is most clearly demonstrated by an analysis of the helical parameters (Lavery and Sklenar 1988). The best-fit helical axis to the P6a and P6b stems show a rotation in the helical angles of 20° and a deflection from coaxial orientation of ∼3 Å (Fig. 5).
FIGURE 5.

Two schematic views of the structure derived from the output of the CURVES program (Lavery and Sklenar 1988), rendered with RASMOL (Sayle and Milnerwhite 1995), rotated by ∼90° with respect to each other. These images highlight the angular bend (A; 20°) and deflection (B; 3 Å) between the two stem segments, P6a and P6b, induced by the internal loop J6.
The J6 internal loop is common to all mammalian telomerases (Chen et al. 2000). This motif is absent in birds, but it is present in fish and half of all reptiles. In organisms where the J6 internal loop is present, the first C and the last U are conserved, except for chinchillas and guinea pigs, which have G substitutions at both positions. The conservation of these two nucleotides supports the unusual C/U pair seen in our structural ensemble. However, it is not clear how a G/G pair can replace this C/U pair in species where these substitutions occur. A purine is always present in the first position of the 3′ strand of the loop; the increased stability afforded by purine 3′ stacking (Freier et al. 1986; Ohmichi et al. 2002) is fully consistent with the stacking of A289 under P6b. The middle position of the 3′ strand varies, but it is never a G. This negative selection can be explained by the requirement to avoid pairing with the conserved Cs on the opposite strand. The resulting C/G pair would disrupt the C266/U291 pair and/or the possible C267 base triple. It is very likely that a G/C pair here would simply zip-up the internal loop, thereby generating a nearly uninterrupted helical structure. The resulting structure would eliminate any subtle flexibility and the long-range structural distortions introduced by the internal loop, as we observe, that we believe are functionally important. There is some phylogenetic evidence in support of the potential triple involving C267, G268, and C288. The GC pair that terminates the loop and initiates the double-helical segment P6b is absolutely conserved: No other pair is seen at this position, not even a CG pair. Furthermore, either C or U is present at the position 267 that would complete the putative triple, but never a purine. In the proposed base triple, the 2′OH of C267 (U267 in some organisms) is clearly poised to hydrogen bond with G268 N7 rather than the G268 ribose O4′, as is observed in regular A-form RNA. However, the hydrogen bonds involving the O2 are less obviously defined by the present data. This group may be hydrogen bonded with G268 N7 via a water molecule, or it may share a bifurcated hydrogen bond to the G268 O6 with the C288 amino (Fig. 4C). Unfortunately, it is notoriously very difficult to directly observe water molecules in RNA and unambiguously establish water-mediated formation of hydrogen bonds.
A surface plot of the fitted ensemble envelope for a representative J6 conformation (Fig. 6) reveals a tunnel in the asymmetric J6 internal loop representing a region of space unoccupied by any atom and accessible to solvent. This feature is unusual and indicates that the J6 region may be a receptor for some fragment of RNA or TERT protein, perhaps the RID-2 domain of hTERT. Consistent with this suggestion is the observation that the N3 of C290 (red in Fig. 6) is reactive to DMS in vitro in the absence of TERT (Antal et al. 2002), but not in vivo in the presence of the TERT protein. This observation validates the significance of this part of the structure. If this atom was completely buried, DMS modification would be inhibited. The small cavity in the J6 bulge shows promise as a drug target. Because the J6 bulge region is essential for CR4-CR5 domain RNA to interact with hTERT, a small molecule docked into this cavity could disrupt this interaction and abolish telomerase activity.
FIGURE 6.
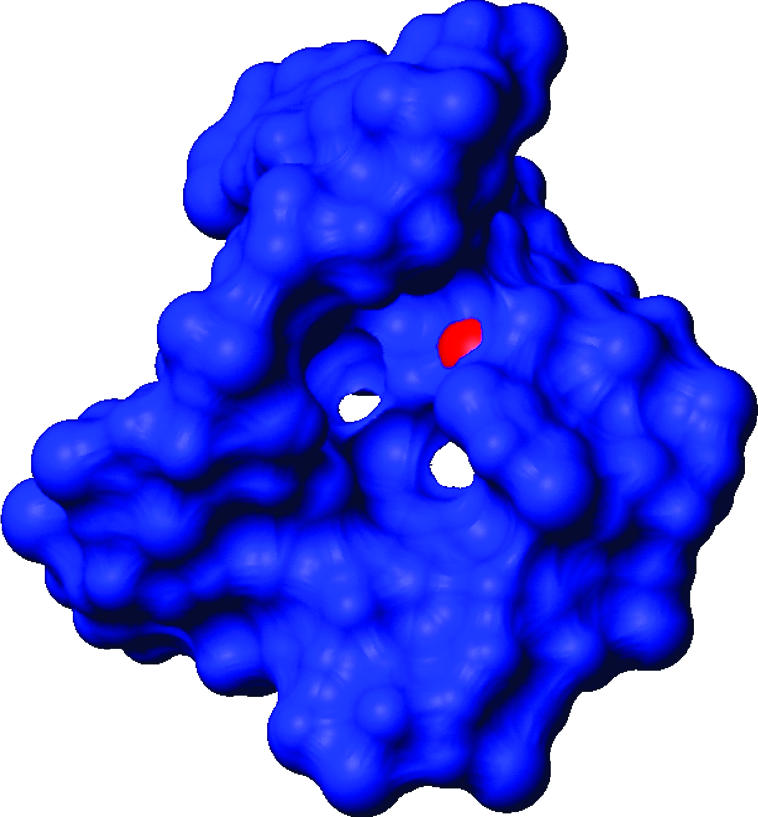
The solvent accessible surface of the hTR J6 structure, showing a tunnel through the core of the J6 region. The N3 atom of C290 (red) is only reactive to dimethyl sulfate (DMS) in the absence of the hTERT protein (Antal et al. 2002). This region is a likely binding determinant for hTERT, and we speculate that this cavity is a protein-docking site.
Substitutions within the J6 internal loop have varying but substantial affects upon telomerase activity in vitro (Mitchell and Collins 2000). Deletion of this loop completely abolished the ability of the CR4-CR5 domain to interact with hTERT and to activate telomerase function. On the 3′-strand, substitutions from ACU to UUA only partially reduced activity; residues C266 and C267 can be substituted with AA and still retain activity. Examining these results in the light of the current structure, the potential C/U pair would be changed to an A/U pair by this mutation. This change would be rather conservative with regard to affecting J6 loop structure. However, the C267 to A267 change would be expected to change dramatically the conformation of this region of the RNA. Because individual nucleotides can be substituted without generally abolishing the domain’s function (contrary to what is observed for the L6.1 loop) (Chen et al. 2002; Leeper et al. 2003), we suggest that the key functional feature of this region is the distortion in the structure introduced by the internal loop. Consistent with this pronounced local backbone distortion is the presence of a reverse transcriptase pause at this site (Antal et al. 2002). We hypothesize that the over-twisting introduced by the internal loop allows the CR4-CR5 domain to fold onto itself or against the hTERT active site surface to generate the global structure required for activation of the enzymatic activity. This directional change may be the major role of the J6 internal loop. We propose that the predominant role of this internal loop is structural with regard to establishment of interaction between this region of hTR and the hTERT protein. In contrast, loop L6.1 is more likely to be directly involved in enzymatic activity, because the enzyme is very sensitive to mutations of surface-exposed nucleotides.
With this second communication, we have made significant progress toward a structure of the full CR4/CR5 domain. Figure 7 schematically demonstrates the strides we have made toward solving the entire structure of this domain, but also the remaining challenges. The structural analysis of the three-helix junction just following P5 is the next step toward completing the structure of this essential element of human telomerase.
FIGURE 7.

Progress toward the complete NMR structure of the full CR4/CR5 domain. Atomic coordinates replace nucleotide sequence for the two regions we have determined by NMR, and represent 64% of the functional sequence (47/73 nucleotides). Residues with ≥80% sequence conservation are colored red; many of these residues are present in the RNA structures that we have determined.
MATERIALS AND METHODS
RNA synthesis
The P6a and P6b regions of the J6 RNA sequence were slightly modified from the hTR sequences at the 5′ and 3′ ends to aid synthesis, and in P6b to aid assignments and reduce the size of the molecule. The changes made to P6b were based on the results of the deletion of the apical part of P6b that can support telomerase activity in hTR (Mitchell and Collins 2000). The conserved G268/C288 pair of P6b was left unchanged. The resulting stem–loop construct “J6” (Fig. 1B) was synthesized by using T7 polymerase and in vitro transcription from unlabeled and 13C/15N-labeled nucleotide triphosphates (NTPs) using standard methods (Price et al. 1998). The 13C/15N-labeled NTPs were purchased from Silantes and used without further purification. The J6 RNA samples were gel purified, electroeluted, ethanol precipitated, and extensively dialyzed into NMR buffer: 10 mM sodium phosphate (pH 6.5). The RNA was annealed by heating it to 90° C followed by snap-cooling on ice, and then was lyophilized and dissolved in 100% D2O. For samples to be studied in water, the J6 RNA was redried and redissolved with 8% D2O/92% H2O. Two samples were prepared with nucleotide-specific 13C/15N labels at A and C (“AC”) or G and U (“GU”), respectively. These samples were used to help resolve regions of spectral overlap seen with uniformly labeled samples and to confirm spectral assignments.
RNA samples used for RDC measurements were dialyzed against 10 mM sodium succinate (pH 6) in D2O for collecting reference spectra. Following dialysis, a 225 μL volume of this sample was mixed with 83 μL of 56 mg/mL Pf1 phage (Hansen et al. 1998) purchased from ASLA Ltd. that had simultaneously been dialyzed against the same buffer. Homogenous deuterium splitting of this sample was determined to be 15 Hz at 300 MHz with no apparent residual center peak.
NMR spectroscopy
NMR data sets were collected on a Bruker 500-MHz spectrometer with Bruker HCN or HCP probes with triple-axis gradients, as needed. In addition, two-dimensional (2D) NOESY and TOCSY spectra were collected at 750 MHz using a Bruker HCN probe with single-axis Z-gradient. Most data sets were processed with Xwinnmr, but three-dimensional (3D) data sets were processed instead with nmrPipe (Delaglio et al. 1995). NMR spectra were visualized and resonances analyzed with Sparky (Kneller and Kuntz 1993).
Spectral assignments were based on standard NMR methods (Varani et al. 1996). Briefly, NOESY spectra collected on unlabeled samples with 80-, 120-, and 300-msec mixing times recorded at 750 MHz were analyzed to generate initial proton assignments. Experiments in water clearly identified the adenine H2 protons and allowed assignment of exchangeable protons (Varani et al. 1995). HCCH-COSY (Pardi and Nikonowicz 1992) and 3D 13C-NOESY-HSQC experiments were used to aid the proton assignment process and to assign the carbon resonances. HNN-COSY experiments (Dingley and Grzesiek 1998; Pervushin et al. 1998) were used to confirm the base pairing, including the presence of a non-NN base pair, assigned to a sheared GU pair in the UUCG tetraloop, where the N1 of G285 hydrogen bonds with the O2 of U271. The AC and GU samples were analyzed by using 3D NOESY-HSQC experiments; there was very good agreement between the chemical shifts of the uniformly and partially labeled samples.
In-Phase-Anti-Phase HSQC spectra (IPAP) (Andersson et al. 1998) were collected at 500 Mz and 25°C. Couplings were measured in the proton dimension to take advantage of the enhanced digital resolution relative to the carbon dimension. The large carbon spectral widths and differing 13C-13C coupling constants required measuring aromatic and ribose RDCs in separate spectra. IPAP data were processed with nmrPipe and analyzed with Sparky.
Structure determination
One hundred structures were calculated in Xplor-NIH by using torsion angle dynamics and simulated annealing (Schwieters et al. 2003). A single extended structure was used as the starting point, and initial velocities were randomized to sample conformational space. The annealing schedule and force constants were identical to standard values used previously for RNA (Clore and Kuszewski 2003). The same input script was used, but “rama” and “orient” restraints were omitted. In our experience, it is best to use NOE and dihedral data first, with iterative analysis of the few initial NOE violations highlighting mistakes in NOE peak assignments or trivial errors of typographical origin. In nearly all cases, NOE violations could be clearly sorted out as mistaken assignments associated with regions of overlap and were subsequently corrected or removed, before further refinement of the structure. A few violated restraints were confirmed to be correct; when emphasized by duplication, they revealed genuine errors elsewhere in the constraint file. RDCs were finally added only after the majority of converged structures were consistent with the NOE and dihedral data. Only RDCs obtained from well-ordered residues, 257–261, 263–265, 268–288, and 292–299, were used in the refinement. The energy scaling factors applied to the RDC restraints during the “dynamics internal” steps were changed to start at 0.001 and ramped to 1.0. This change provided better agreement with NOE and dihedral restraints while at the same time optimally fitting the RDCs.
Distance restraints were obtained from 2D NOESY spectra recorded at 750 MHz at the shorter mixing times (80 and 120 msec). Additional restraints were obtained from the 3D 13C-NOESY-HSQC recorded at 120-msec mixing time acquired at 500 MHz. Approximately 85% of the NOE restraints were derived from the two-dimensional data sets. Distances corresponding to strong, medium, or weak NOEs were given bounds of 2.6 ± 1, 3.6 ± 1, and 4.0 ± 1.4 Å, respectively. Restraints involving the exchangeable proton resonances were obtained from the two-dimensional water NOESY based on the Watergate water suppression scheme (Piotto et al. 1992). Weak base-pair planarity and standard hydrogen-bonding restraints were used for unambiguously established base pairs. The imino proton of U261 was too broad to identify reliable distance restraints involving this proton. However, the U261/A295 base pair was determined to be present based upon HNN-COSY and from the consistent positioning of these two bases across from one another based solely upon other experimentally determined distances (i.e., without hydrogen bond or planarity restraints). During refinement, loose (±0.4 Å) hydrogen bonding restraints were added, but planarity restraints were omitted for this pair. We see no evidence for NOEs between nucleotides C262 and G260, indicating that the proposed putative structure containing a C262/A295 pair and a bulged U261 (Chen and Greider 2004) is not present. However, the genetic data regarding this site strongly indicate that a CA or CG pair should occupy this site. It is possible that the bulged C262 is unique to human hTR and may be substituted by a similar conformation and adjacent bulged U in other species.
Dihedral angle restraints were applied to the backbone and glycosidic angles as follows. The glycosidic angles were loosely set to anti (−158° ± 40) for nucleotides with a clearly weak H1′ to aromatic NOE as observed at 80-msec mixing times. The glycosidic angle of G285 (with a very strong H1′ to aromatic NOE) was instead restrained to the syn conformation (0° ± 90). The remaining glycosidic angles (C262, C266, C267, U271, C284, C288, A289, C290, U291) were left unrestrained. The sugar conformation was constrained to C3′-endo (ν1 = −20° ± 10°, ν2 = 35° ± 10°, and δ = 80° ± 20°) for riboses lacking visible H1′ to H2′ cross-peaks in DQF-COSY and three-dimensional 13C separated TOCSY spectra, and to C2′-endo (ν1 = 35° ± 10°, ν2 = −35° ± 10°, and δ = 145° ± 20°) for residues with strong H1′–H2′ correlations in both spectra (C262, U272, C284). The sugar puckers could not be clearly determined for U261, C267, A269, and U291 and were therefore left unrestrained. The remaining backbone torsion angles were restrained by using established methods (Varani et al. 1996). For residues with canonical 31P chemical shift values, α and ζ were set to exclude the trans conformation. β was restrained to trans and γ to gauche+ for residues with measurable 4JH4′/iP correlations in the 1H/31P HETCOR spectra. The angle ɛ was restrained to trans when an H2′ to P(i + 1) peak was observed in either HCP or HETCOR spectra.
Residual dipolar couplings (RDCs) for 13C-attached protons were obtained from IPAP 13C HSQC spectra collected in the presence of 15 mg/mL Pf1 phage. Reference one-bond J-couplings were determined under the same buffer conditions in the absence of orienting media. Apparent one-bond couplings were measured in the directly detected ω2 dimension to take advantage of the increased proton digital resolution. RDCs were calculated as the difference in apparent one-bond coupling constants between oriented and unoriented samples (Jw/pf1 − Jw/outpf1 = RDC). Errors in RDCs were estimated from the differences in RDCs obtained for the uniformly labeled and nucleotide-specific labeled samples and ranged from 1.8 Hz–2.6 Hz. Resonances that were close to the residual water line or that were broad due to exchange were given more generous errors (2.4–3.2 Hz). RDCs for well-restrained residues were applied as susceptibility anisotropy (“SANI”) restraints. Thus no RDC restraints were used for the residues of the large bulge as they might have interfered with determining a precise conformation in this region due to mildly differing local dynamics.
Initial values for the magnitude of the alignment tensor (Da) and the rhombicity (R) were estimated from the powder pattern–like distribution of the RDCs, giving rise to initial values of Da = −48 and R = 0.3 (Clore et al. 1998a). The asymmetry of the alignment tensor is difficult to predict accurately for RNA via a powder pattern plot, since its bond vectors do not fully sample conformational space, and hence the plot is incomplete (Clore et al. 1998b). In order to find optimal values of Da and R, systematic grid searches were conducted around the values established from the powder pattern. A “grid search” of Da and R entails calculating an ensemble of structures for each combination of Da and R within the range of values for each parameter that are to be explored (Clore et al. 1998b). A “grid point” is a given value of Da and R, such as −46 and 0.5 or −38 and 0.05, for example. The RDC energies of the ensemble members at each of these grid points were averaged and ranked to find an optimal pair of values for both Da and R. Twenty structures were calculated per grid point to sample Da values between −40 and −54 in 2-Hz increments; values of R between 0.05 and 0.65 were sampled in steps of 0.05. This range of values was chosen to flank the predicted values of Da estimated by the powder pattern plot, and R was sampled over the full discrete range below the maximum possible value of 0.667. The SANI energy represents the penalty attributed to a structure with a bond vector orientation that deviates from two conical paths calculated from that individual spin-system’s measured RDC. Thus, reduced or minimized SANI energies were used as the metric for agreement with the RDC data. The contribution of SANI restraints was adjusted by scaling down their initial and final force constants to 0.001 and 0.5 from the standard values of 0.01 and 1.0 to improve agreement with NOE and dihedral data at suboptimal Da/R grid points. At suboptimal grid-point values, with the standard SANI force constants, the SANI term becomes very large and subsequently competes with covalent geometry terms and other NMR constraint terms for determining the total energy of the molecule. In these cases, spurious violations of topology and distance constraints result, which can perturb the structure in clearly erroneous ways. Although there are multiple solutions to any RDC fit, very distorted structures are common for suboptimal values of Da and R. Thus, a balance must be obtained to find an optimal SANI force constant that does not dramatically perturb the structure at these suboptimal values yet that has a significant influence upon the interdomain orientation. We chose to use reduced SANI force constants to begin at 0.001 and ramping up to 0.5 during the Da and R determination to weigh the NOE and dihedral data more strongly early in the simulation, as well as stereochemical constraints. However, once an optimal pair of values was determined, subsequent ensembles were calculated with a higher final SANI force constant of 1.0 to allow the RDCs to have a more significant impact upon the final fold.
Separate grid searches of the parameters Da and R were performed for each independently treated double-helical stem (257–265/292–299 and 268–288, respectively) and for the entire RNA treated as a single rigid molecule. These three sets of grid search calculations on the individual stem regions and the whole RNA were conducted in order to evaluate whether the internal loop introduces extensive relative motions of the separate double-helical regions. If the Da and R parameters were revealed to be very different between the two small subregions of structure, then the molecule would very likely be experiencing significant interdomain motions. Once this analysis was conducted, it became clear that the extent of reorientation about the internal loop was small, because the optimal values of both Da and R were very similar for all three simulations. Optimal Da and R parameters for the entire RNA (treated as a rigid molecule) and for the individual stem regions (P6a and P6b) were determined to be −48/0.25, −44/0.2, and −46/0.25, respectively. Since there are only very small differences (well within the uncertainty) between the values of Da and R determined for the two separate stems and for the entire molecule, a single-axis system and alignment tensor was used for refining this RNA structure. Furthermore, a plot of the RDCs distributed over topological arrangement (Al-Hashimi et al. 2002) revealed that the spread in RDC magnitude was similar for both helical stems (Supplementary Material: http://faculty.washington.edu/varani/tleeper/suppl.RNA.2005.pdf). Since the degree of alignment scales with the generalized degree of order, this observation qualitatively confirms the presence of similar dynamics for the two stems. Thus, both the topological distribution of RDCs and the results of the grid searches justified and supported the use of a single set of Da and R values for this molecule.
In the course of the refinement, we noticed that the H2′ RDCs tended to be more strongly violated than any other category of RDC, and they were subsequently given the most generous error category (3.2 Hz). In RNA helices, the H2′ to following aromatic carbon (C6 or C8) distance is very small (<2.5 Å) and uniform. In this particular alignment system, the observed RDCs sampled a broad absolute range from −84 to +80 Hz. The H2′ to C6 or C8 RDCs, while small, may still be significant relative to the measurement error when the degree of alignment is this large. Consequently, the uniform and small distances seen for many helical H2′ to C6 or C8 distances may explain why it is difficult to fit this class of RDCs. This odd behavior of the H2′ RDCs may not have been noticed or reported previously because most degrees of alignment tend to be smaller, with magnitudes of Da values ∼20 Hz.
Structure analysis
Structural analysis to extract helical parameters and evaluate the distortion induced by the bulge was executed with the program CURVES (Lavery and Sklenar 1988). A break was introduced at the site of the J6 internal bulge region, nucleotides 266–267 and 28–291, to allow calculation of interhelical deviations between P6a and P6b. The bulged nucleotide in P6a, C262, was omitted from the analysis.
Acknowledgments
This work was supported by a National Institutes of Health (NIH)–National Cancer Institute (NCI) award to G. Varani, and a NIH Ruth L. Kirschstein National Research Service Award (NRSA) to T. Leeper. Spectrometer time and the assistance of Nancy Isern at the William R. Wiley Environmental Molecular Sciences Laboratory (EMSL), Richland, Washington, were greatly appreciated.
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.7222505.
REFERENCES
- Aboul-ela, F., Karn, J., and Varani, G. 1995. The structure of the human immunodeficiency virus type-1 Tar RNA reveals principles of RNA recognition by Tat protein. J. Mol. Biol. 253: 313–332. [DOI] [PubMed] [Google Scholar]
- Al-Hashimi, H.M., Gosser, Y., Gorin, A., Hu, W., Majumdar, A., and Patel, D.J. 2002. Concerted motions in Hiv-1 Tar RNA may allow access to bound state conformations: RNA dynamics from Nmr residual dipolar couplings. J. Mol. Biol. 315: 95–102. [DOI] [PubMed] [Google Scholar]
- Allain, F.H.-T. and Varani, G. 1995. Structure of the P1 helix from group I self splicing introns. J. Mol. Biol. 250: 333–353. [DOI] [PubMed] [Google Scholar]
- Andersson, P., Weigelt, J., and Otting, G. 1998. Spin-state selection filters for the measurement of heteronuclear one-bond coupling constants. J. Biomol. NMR 12: 435–441. [DOI] [PubMed] [Google Scholar]
- Antal, M., Boros, E., Solymosy, F., and Kiss, T. 2002. Analysis of the structure of human telomerase RNA in vivo. Nucleic Acids Res. 30: 912–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J.L. and Greider, C.W. 2003. Determinants in mammalian telomerase RNA that mediate enzyme processivity and cross-species incompatibility. EMBO J. 22: 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2004. Telomerase RNA structure and function: Implications for dyskeratosis congenita. Trends Biochem. Sci. 29: 183–192. [DOI] [PubMed] [Google Scholar]
- Chen, J.L., Blasco, M.A., and Greider, C.W. 2000. Secondary structure of vertebrate telomerase RNA. Cell 100: 503–514. [DOI] [PubMed] [Google Scholar]
- Chen, J.L., Opperman, K.K., and Greider, C.W. 2002. A critical stem-loop structure in the Cr4-Cr5 domain of mammalian telomerase RNA. Nucleic Acids Res. 30: 592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore, G.M. and Kuszewski, J. 2003. Improving the accuracy of NMR structures of RNA by means of conformational database potentials of mean force as assessed by complete dipolar coupling cross-validation. J. Am. Chem. Soc. 125: 1518–1525. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Gronenborn, A.M., and Bax, A. 1998a. A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J. Magn. Reson. 133: 216–221. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Gronenborn, A.M., and Tjandra, N. 1998b. Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J. Magn. Reson. 131: 159–162. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NmrPipe: A multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Delange, T. 1994. Activation of telomerase in a human tumor. Proc. Natl. Acad. Sci. 91: 2882–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingley, A.J. and Grzesiek, S. 1998. Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide (2)J(Nn) couplings. J. Am. Chem. Soc. 120: 8293–8297. [Google Scholar]
- Freier, S.M., Kierzek, R., Jaeger, J.A., Sugimoto, N., Caruthers, M.H., Neilson, T., and Turner, D.H. 1986. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. 83: 9373–9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, D. and Collins, K. 2003. Distinct biogenesis pathways for human telomerase RNA and H/Aca small nucleolar RNAs. Mol. Cell 11: 1361–1372. [DOI] [PubMed] [Google Scholar]
- Hansen, M.R., Mueller, L., and Pardi, A. 1998. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling Interactions. Nat. Struct. Biol. 5: 1065–1074. [DOI] [PubMed] [Google Scholar]
- Jady, B.E., Bertrand, E., and Kiss, T. 2004. Human telomerase RNA and box H/Aca scarnas share a common cajal body-specific localization signal. J. Cell Biol. 164: 647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelland, L.R. 2000. Telomerase inhibitors: Targeting the vulnerable end of cancer? Anticancer Drugs 11: 503–513. [DOI] [PubMed] [Google Scholar]
- Kneller, D.G. and Kuntz, I.D. 1993. UCSF Sparky: An Nmr display, annotation and assignment tool. J. Cell. Biochem. 53: 254. [Google Scholar]
- Lai, C.K., Mitchell, J.R., and Collins, K. 2001. RNA binding domain of telomerase reverse transcriptase. Mol. Cell. Biol. 21: 990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery, R. and Sklenar, H. 1988. The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic-acids. J. Biomol. Struct. Dyn. 6: 63–91. [DOI] [PubMed] [Google Scholar]
- Leeper, T., Leulliot, N., and Varani, G. 2003. The solution structure of an essential stem-loop of human telomerase RNA. Nucleic Acids Res. 31: 2614–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingner, J. and Cech, T.R. 1998. Telomerase and chromosome end maintenance. Curr. Opin. Gen. Dev. 8: 226–232. [DOI] [PubMed] [Google Scholar]
- Lingner, J., Hughes, T.R., Shevchenko, A., Mann, M., Lundblad, V., and Cech, T.R. 1997. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276: 561–567. [DOI] [PubMed] [Google Scholar]
- Mason, D.X., Goneska, E., and Greider, C.W. 2003. Stem-loop IV of Tetrahymena telomerase RNA stimulates processivity in trans. Mol. Cell. Biol. 23: 5606–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElligott, R. and Wellinger, R.J. 1997. The Terminal DNA structure of mammalian chromosomes. EMBO J. 16: 3705–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, J.R. and Collins, K. 2000. Human telomerase action requires two independent interactions between telomerase RNA and telomerase reverse transcriptase. Mol. Cell 6: 361–371. [DOI] [PubMed] [Google Scholar]
- Mitchell, J.R., Cheng, J., and Collins, K. 1999. A Box H/Aca small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol. Cell. Biol. 19: 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty, T.J., Huard, S., Dupuis, S., and Autexier, C. 2002. Functional multimerization of human telomerase requires an RNA interaction domain in the N terminus of the catalytic subunit. Mol. Cell. Biol. 22: 1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty, T.J., Marie-Egyptienne, D.T., and Autexier, C. 2004. Functional organization of repeat addition processivity and DNA synthesis determinants in the human telomerase multimer. Mol. Cell. Biol. 24: 3720–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmichi, T., Nakano, S., Miyoshi, D., and Sugimoto, N. 2002. Long RNA dangling end has large energetic contribution to duplex stability. J. Am. Chem. Soc. 124: 10367–10372. [DOI] [PubMed] [Google Scholar]
- Pardi, A. and Nikonowicz, E.P. 1992. A simple procedure for resonance assignment of the sugar protons in 13c-labeled RNA. J. Am. Chem. Soc. 114: 9202–9203. [Google Scholar]
- Pervushin, K., Ono, A., Fernandez, C., Szyperski, T., Kainosho, M., and Wüthrich, K. 1998. Nmr scalar coupling across Watson-Crick base pair hydrogen bonds in DNA observed by transverse relaxation-optimized spectroscopy. Proc. Natl. Acad. Sci. 95: 14147–14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklénar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2: 661–665. [DOI] [PubMed] [Google Scholar]
- Pogacic, V., Dragon, F., and Filipowicz, W. 2000. Human H/Aca small nucleolar Rnps and telomerase share evolutionarily conserved proteins Nhp2 and Nop10. Mol. Cell. Biol. 20: 9028–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, S.R., Oubridge, C., Varani, G., and Nagai, K. 1998. Preparation of RNA–protein complexes for X-ray crystallography and NMR. RNA–protein interaction: Practical approach (ed. C. Smith), pp. 37–74. Oxford University Press, Oxford, UK.
- Sayle, R.A. and Milnerwhite, E.J. 1995. RASMOL: Biomolecular graphics for all. Trends Biochem. Sci. 20: 374–376. [DOI] [PubMed] [Google Scholar]
- Schwieters, C.D., Kuszewski, J.J., Tjandra, N., and Clore, G.M. 2003. The Xplor-Nih NMR molecular structure determination package. J. Magn. Reson. 160: 65–73. [DOI] [PubMed] [Google Scholar]
- Sperger, J.M. and Cech, T.R. 2001. A stem-loop of tetrahymena telomerase RNA distant from the template potentiates RNA folding and telomerase activity. Biochemistry 40: 7005–7016. [DOI] [PubMed] [Google Scholar]
- Tesmer, V.M., Ford, L.P., Holt, S.E., Frank, B.C., Yi, X., Aisner, D.L., Ouellette, M., Shay, J.W., and Wright, W.E. 1999. Two inactive fragments of the integral RNA cooperate to assemble active telomerase with the human protein catalytic subunit (Htert) in vitro. Mol. Cell. Biol. 19: 6207–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolman, J.R., Al-Hashimi, H.M., Kay, L.E., and Prestegard, J.H. 2001. Structural and dynamic analysis of residual dipolar coupling data for proteins. J. Am. Chem. Soc. 123: 1416–1424. [DOI] [PubMed] [Google Scholar]
- Varani, G., Aboul-ela, F., Allain, F.H.-T., and Gubser, C.C. 1995. Novel three-dimensional 1h-13c-31p triple resonance experiments for sequential backbone correlations in nucleic acids. J. Biomol. NMR 5: 315–320. [DOI] [PubMed] [Google Scholar]
- Varani, G., Aboul-ela, F., and Allain, F.H.-T. 1996. NMR investigations of RNA structure. Progr. NMR Spectr. 29: 51–127. [Google Scholar]
- Walberer, B.J., Cheng, A.C., and Frankel, A.D. 2003. Structural diversity and isomorphism of hydrogen-bonded base interactions in nucleic acids. J. Mol. Biol. 327: 767–780. [DOI] [PubMed] [Google Scholar]
- Xia, J., Peng, Y., Mian, I.S., and Lue, N.F. 2000. Identification of functionally important domains in the N-terminal region of telomerase reverse transcriptase. Mol. Cell. Biol. 20: 5196–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q., Throolin, R., Pitt, S.W., Serganov, A., and Al-Hashimi, H.M. 2003. Probing motions between equivalent RNA domains using magnetic field induced residual dipolar couplings: Accounting for correlations between motions and alignment. J. Am. Chem. Soc. 125: 10530–10531. [DOI] [PubMed] [Google Scholar]