


Abstract
All Group I intron ribozymes contain a conserved core region consisting of two helical domains, P4–P6 and P3–P7. Recent studies have demonstrated that the elements required for catalysis are concentrated in the P3–P7 domain. We carried out in vitro selection experiments by using three newly constructed libraries on a variant of the T4 td Group I ribozyme containing only a P3–P7 domain in its core. Selected variants with new peripheral elements at L7.1, L8 or L9 after nine cycles efficiently catalyzed the reversal reaction of the first step of self-splicing. The variants from this selection contained a short sequence complementary to the substrate RNA without exception. The most active variant, which was 3-fold more active than the parental wild-type ribozyme, was developed from the second selection by employing a clone from the first selection. The results show that the P3–P7 domain can stand as an independent catalytic module to which a variety of new domains for enhancing the activity of the ribozyme can be added.
INTRODUCTION
Group I intron RNAs are ribozymes that catalyze two consecutive trans-esterification reactions to excise themselves from the precursor RNAs and ligate the flanking exons together (1). They are composed of a universally conserved core region and subgroup-specific peripheral regions (Fig. 1A for T4 td subgroup IA ribozyme) (2–4). Deletion experiments have demonstrated that the peripheral regions are not essential for catalysis (2–5). However, deletions of the peripheral regions generally cause a reduction in catalytic efficiency, although the effect can be compensated for, usually by adding a high concentration of magnesium ions and/or spermidine (chemicals that stabilize RNA structures). Thus the peripheral regions have been thought to stabilize the structure of the conserved core, which is essential for catalysis.
Figure 1.

Secondary structure of the ribozymes. (A) Wild-type T4 td ribozyme. (B) M1 mutant ribozyme. (C) Three sub-libraries used for the selection: (left) sub-library 7.1; (middle) sub-library 8; (right) sub-library 9. The structure model is according to Cech et al. (35). Gray arrowheads point to 5′ splice sites. Tertiary interactions are indicated by gray lines.
The core region of Group I ribozymes consists of two helical domains, P3–P7 and P4–P6, which are connected via base triples (Fig. 1A) (2,4,6,7). Biochemical studies have indicated that the portion essential for catalysis resides in the P3–P7 domain, especially the P7 and the J8/7 regions, although analyses using nucleotide analogs showed a modest contribution of the P4–P6 domain on transition state stabilization in the trans-esterification reactions (8–16). A recent study using the T4 td intron demonstrated that a mutant ribozyme lacking both the P4–P6 domain and the base triples (M1 mutant, Fig. 1B) can perform the trans-esterification reactions (17). Analogous deletion experiments using the Tetrahymena intron have revealed that L4 and the distal part of P4 in the P4–P6 domain are required to perform the intact splicing reaction (18). These elements are involved in the mechanism of recognizing the substrate helix and switching the sequential two trans-esterification reactions.
These studies prompted us to design and attempt a selection experiment in which the P3–P7 domain is treated as an independent catalytic module, as in the case of the C-domain (catalytic domain) of bacterial RNase P RNA (19,20): P RNA consists of two domains, the C-domain and S-domain (specificity domain), but only the C-domain contributes to catalysis (19,21,22). As anticipated, we identified replacements for the P4–P6 domain that can enhance the activity of the M1 mutant from the random sequences inserted in the peripheral loops.
MATERIALS AND METHODS
Library construction
Three sub-libraries were constructed from a mutant T4 td Group I ribozyme lacking both the P4–P6 domain and the base triples (M1 mutant, Fig. 1B). Initially, 5′ and 3′ fragments of the M1 mutant ribozyme and a synthetic DNA with 40 nt of random sequence were amplified by PCR (2.4 ml scale) using ExTaq DNA polymerase (Takara Shuzo, Japan). Oligodeoxynucleotides were purchased from Hokkaido System Science (Japan) and used for the amplification of the random sequence: template DNA, 5′-CGTGGACAGG GTGAGCGAGGCACCN40GAGCCCTGTCCACGCTCGCT-CAC-3′ (where N stands for any nucleotide, restriction sites in italic); forward primer, 5′-CGTGGACAGGGTGAGCGAG-3′); reverse primer, 5′-GTGAGCGAGCGTGGACAGG-3′). The oligonucleotides for the 5′ fragments of sub-libraries 7.1, 8 and 9 were, respectively: td-Fw, 5′-TTCTAATACGA CTCACTATAGGACAACTACATGTCCGAGGGGTATA AGGTGACTTATACTTGTAATCTATC-3′ (T7 promoter in bold), and 7.1-BanI-Rv, 5′-CTCCCGGTGCCGATAGTCGT TAGATTAGATAGATTAC-3′; td-Fw and 8-BanI-Rv, 5′-CTTGTGGTGCCAAGTTGTCTATCGTTTCGAGTC-3′; td-Fw and 9-BanI-Rv, 5′-GCATGGGTGCCTGCAGAGCAGA CTATATCTCC-3′. The oligonucleotides for the 3′ fragments of sub-libraries 7.1, 8 and 9 were, respectively; 7.1-BanII-Fw, 5′-TATCCCGAGCCCGAGTAGGGTCAAGTGACTCG-3′, and td-Rv, 5′-CATTATGTTCAGATAAGGTCGTTAATC-3′; 8-BanII-Fw, 5′-CTTGCGAGCCCAAGTTGGAGATATA GTCTGC-3′, and td-Rv; 9-BanII-Fw, 5′-GCATGGAG CCCTGCAGCTGGATATAATTCC-3′, and td-Rv. The PCR products were digested with non-palindromic restriction enzymes, BanI and BanII, and then ligated using T4 DNA ligase (Takara Shuzo). The ligation products were purified by native PAGE. About 5 × 1012 molecules of each sub-library DNA were transcribed using T7 RNA polymerase to yield RNA pools (∼100 copies each). The transcripts of the sub-libraries were mixed and used for the in vitro selection.
In vitro selection
The in vitro selection was carried out essentially as described (23,24). After in vitro transcription, the template DNA was digested using RQ1 DNase (Promega). The RNA from the mix was purified by denaturing PAGE and precipitated with ethanol. The purified RNA was dissolved in water and denatured by incubation at 70°C for 5 min followed by 50°C for 5 min. The resulting RNA was folded in concentrated reaction buffer at 50°C for 5 min, followed by 37°C for 5 min. The first round of selection was performed in a reaction mixture containing 1 µM ribozyme, 2 µM substrate oligo RNA (5′-biotin-GCAUCAUAGCAUCCACAUCGAAAUC AGGU-3′; Dharmacom Research), 50 mM MgCl2, 5 mM spermidine and 50 mM HEPES–KOH, pH 7.5, at 37°C for 4 h. For the subsequent rounds, the incubation time and the concentrations of the substrate RNA, MgCl2 and spermidine were progressively decreased (Table 1). The reaction was quenched by precipitation with ethanol. The reacted, or biotinylated, ribozymes were captured on streptavidin MagneSphere paramagnetic particles (Promega) and reverse transcribed with a reverse primer (td-Rv) using ReverTra Ace (Toyobo, Japan). The resulting cDNA was eluted from the particles by degrading the RNA with 150 mM KOH and then neutralizing with 150 mM HCl. The cDNAs were selectively amplified by PCR using a selective primer (5′-CTTGAC GTCAGCCTGGAAAACCCTC-3′) and td-Rv. A nested PCR was carried out with the primers td-Fw and td-Rv, to regenerate the T7 promoter and the 5′ guanosine residue. After nine cycles of selection, the variants were cloned into pUC18 and sequenced.
Table 1. Conditions for in vitro selection.
| Round | MgCl2 (mM) | Spermidine (mM) | Oligo RNA (µM) | Time |
|---|---|---|---|---|
| 1 | 50 | 5 | 2 | 4 h |
| 2 | 50 | 5 | 2 | 2 h |
| 3 | 50 | 5 | 2 | 1 h |
| 4 | 20 | 5 | 2 | 12 h |
| 5 | 20 | 5 | 2 | 1 h |
| 6 | 10 | 2.5 | 2 | 12 h |
| 7 | 10 | 2.5 | 2 | 1 h |
| 8 | 10 | 2.5 | 1 | 12 h |
| 9 | 10 | 2.5 | 1 | 1 h |
| 10 | 20 | 2.5 | 0.5 | 3 min |
| 11 | 20 | 2.5 | 0.5 | 1 min |
Assay of catalytic efficiency
The catalytic efficiencies of individual clones were assayed by the reversal reaction of the first step of the self-splicing reaction (23). The plasmid DNA was amplified by PCR with primers td-Fw and td-Rv. For mutational analysis of the clone 2 ribozyme, the variants were constructed by employing overlap extension PCR (25). The PCR product was transcribed in vitro with [α-32P]GTP. The internally labeled ribozyme was purified by denaturing PAGE and precipitated with ethanol. The ribozyme (10 nM) was dissolved in a solution with 2.5 mM spermidine and 50 mM HEPES–KOH, pH 7.5, followed by denaturation at 65°C for 5 min and then incubation at 37°C for 5 min. The resulting mix was folded by adding 20 mM MgCl2, followed by incubation at 37°C for 10 min. The reaction was started by adding the substrate oligo RNA at 37°C and stopped by adding EDTA. Reaction products were separated by denaturing PAGE and the extent of the reaction was quantified using a BAS 2500 II instrument (Fuji Film, Japan). The data were plotted using Deltagraph 4.0 (Polaroid).
Enzymatic footprinting
Enzymatic footprinting experiments were carried out as described (26) with minor modifications. For RNase V1 footprinting, clone 2 RNA (10 ng/µl) was dissolved in a solution containing 100 mM KCl and 50 mM HEPES–KOH, pH 7.5, and denatured by incubation at 65°C for 5 min and then at 37°C for 5 min. The resulting mix was folded by adding 20 mM MgCl2, followed by incubation at 37°C for 10 min. The RNA was further incubated with or without the substrate oligo RNA (3 µM) at 37°C for 10 min. The resulting RNA mix was digested with 1 U/ml RNase V1 (Amersham Pharmacia Biotech) at 37°C for 30 min. The digestion was stopped by phenol extraction and the RNA was precipitated with ethanol. The cleavage sites were mapped by reverse transcription (ReverTra Ace) using a 32P-labeled primer (td-Rv), followed by denaturing PAGE. For RNase A footprinting, clone 2 RNA (10 ng/µl) was dissolved in a solution containing 2.5 mM spermidine and 50 mM HEPES–KOH, pH 7.5, denatured and folded as described above. The folded RNA was digested with 1–50 ng/ml RNase A (Sigma) at 37°C for 30 min, followed by mapping as described above.
Construction and selection of the partially randomized clone 2 library
5′ and 3′ fragments of the library were amplified separately from clone 2 DNA by PCR using KOD DNA polymerase (Toyobo). Primers td-Fw and N4-R (5′-GAAAAATTACAT CTCGCTNNNNGCCTGCAGAGCAGACTATATCTCC-3′, 5′ end phosphorylated by T4 polynucleotide kinase; Takara Shuzo) or N5-F (5′-GTTNNTCTGGGTGTGGACAGG NNNCCTGCAGCTGGATATAATTCC-3′, 5′ end phosphorylated as described above) and td-Rv were used for the amplification of 5′ or 3′ fragments, respectively. The PCR products were assembled by blunt-end ligation (T4 DNA ligase; Takara Shuzo). The ligated DNA was purified by native PAGE and transcribed using T7 RNA polymerase. In vitro selection was carried out as described above (Table 1, rounds 10 and 11).
RESULTS
In vitro selection
The P3–P7 domain appears to represent an independent catalytic module because the essential regions required for the catalysis of Group I ribozymes are concentrated there. Thus it seemed possible that introduction of artificial modules into the domain might enhance the catalytic ability of a corresponding ribozyme lacking both the P4–P6 domain and the base triples. We constructed ribozyme libraries based on the P3–P7 domain in order to test this hypothesis through selection experiments. The libraries which were used in the selection were derived from the M1 mutant ribozyme, a mutant T4 td ribozyme lacking both the P4–P6 domain and the base triples (17). Three sub-libraries were constructed because it was uncertain which site would be best suited to generate new structural elements (Fig. 1C). In each sub-library, 40 nt of random sequence was inserted into the peripheral loop L7.1, L8 or L9: these sites were chosen because they are not critical for catalysis or are not involved in the long-range interactions of the M1 mutant (Fig. 1B) and insertions at these sites should eliminate enrichment of the variants of the wild-type containing the P4–P6 domain. Each sub-library, containing ∼5 × 1012 independent molecules, was transcribed to yield the pools; ∼100 copies for each molecule. The three pools were mixed and the resulting mixture was used for the selection by employing the reversal reaction of the first step of the self-splicing reaction (Fig. 2) (23,24). During the selection, the selection stringency was progressively increased (Table 1).
Figure 2.

Scheme for the in vitro selection. The circled B on the substrate oligo RNA indicates the 5′ biotin moiety. SA indicates streptavidin beads. For details see Materials and Methods.
After nine cycles of selection, the mixed library exhibited considerably elevated activity (Fig. 3). The activity of the round 9 pool was about one-fourth that of the wild-type ribozyme at 5 mM magnesium ions with 0.5 µM substrate oligo RNA (Fig. 3). Ten clones were randomly selected from the pool (Table 2). Interestingly, five, two and three clones had the insertions in L7.1, L8 and L9, respectively, indicating that these sites are tolerant of insertions that contribute to enhancement of activity (note that no particular preference for insertion site was observed from the digestion pattern of the round 9 pool DNAs by restriction enzymes; data not shown). Seven clones had point mutations in the constant regions but the altered nucleotides were clustered among the peripheral residues not responsible for long-range interactions, suggesting that these mutations do not significantly contribute to enhancement of the activity. In fact, a clone 4 variant without the point mutation still exhibited distinct activity compared with the M1 mutant, demonstrating that the insertions at three sites are competent independently (Table 2). Clones 1–10 contained short sequences which are complementary to the upper region of the substrate RNA. The best selected clone, termed clone 2, was employed for further analysis.
Figure 3.

Progress of the activity during the in vitro selection. The reaction of each pool of RNA was carried out in the presence of 5 mM MgCl2 and 0.5 µM substrate oligo RNA for 10 min.
Table 2. Round 9 clones.
| Insertion position | Inserted sequencea (5′→3′) | Mutations in the constant regionb | Activityc (kobs, per h) | |
|---|---|---|---|---|
| 1 | P7.1 | GGCACCAGUAAGUGUGGAUUGUACAUGAAUUUCCAAAAAUAACUUCGAGCCC | A896G (L7.2) | 0.58 |
| 2 | P9 | GGCACCAAGCGAGAUGUAAUUUUUCGUUACUCUGGGUGUGGACAGGAGCCC | None | 5.94 |
| 3 | P7.1 | GGCACCAGCUCUAUGAGAGUGGCCUUACGGAUGUGGAUAGAUAACCGGGAGCCC | None | 0.68 |
| 4 | P8 | GGCACCAGAAGUAAUUUCUAUGAUUUUGGCGUUUAUUUCGAUGGAUGGAGCCC | A897G (L7.2) | 3.72 (1.69)d |
| 5 | P8 | GGCACCUAUAAGUUCCAAAAACUCUUUUCCUUCUCUGAUGUGGUUGAGCCC | ΔA977 (L9.1) | 0.29 |
| 6 | P9 | GGCACCGCCCUUGAGAAAUUUGCUAGGGUCGUGUGAUGUGGUCACGAGCCC | None | 1.34 |
| 7 | P9 | GGCACCCGAUAAGAGGUAGCAUAGUCGAUGUGUGGAAGUGAUAUCAAGAGCCC | A928G (L8) | 2.16 |
| 8 | P7.1 | GGCACCGACUCCGUUCGGAGCGGUUCUGCGUGGAGAUGACAUUUAGGAGCCC | ΔG958 (L9) | 1.05 |
| 9 | P7.1 | GGCACCAUUUCGAUAGGUAAGGGAUGACCCUGUGGGUAGCAUUUGCGAGCCC | U34C (J2/3) ΔG891 (P7.2) | 1.52 |
| 10 | P7.1 | GGCACCAAACGAUAUCGUACGUGGAAAUGUUGCUCCUGAUAUACAAUGAGCCC | A918G (P8) | 0.67 |
| M1 mutant | 6.35 × 10–3 |
aThe restriction sites are shown in italic. Bold letters indicate the sequences complementary to the 5′ leader of the substrate RNA.
bNumbering of the sequence is according to Belfort et al. (36).
cActivity (kobs) was determined in the presence of 5 µM substrate RNA.
dThe activity of the clone 4 derivative which lacks the point mutation in the constant region.
Structural analysis of the clone 2 ribozyme
The clone 2 ribozyme had the insertion in the L9 region. Although its kcat value is lower than that of the wild-type ribozyme, its secondary rate constant (kcat/Km value) was slightly better than the wild-type because of its higher affinity for the substrate RNA (Table 3). One possible secondary structure of the insertion sequence of the clone 2 ribozyme was predicted as a branched structure with two stem–loops, P9a–L9a and P9b–L9b, using the Mfold program (Fig. 4A) (27,28).
Table 3. Kinetic parameters.
| T4 td ribozyme variants | kcata (per h) | Km (µM) | kcat/Km (per h/µM) |
|---|---|---|---|
| Wild-type | 24.61 | 5.53 | 4.45 |
| M1 mutant | NAb | NAb | 1.27 × 10–3 |
| Clone 2 | 6.44 | 1.29 | 4.99 |
| Clone 2.1 | 5.01 | 0.40 | 12.50 |
aIt was not determined whether kcat values reflect the rate of the chemical step.
bNot available. Because of its high Km value (>50 µM), kcat and Km values of the M1 mutant were undetermined.
Figure 4.

(A) Mutational analysis of the clone 2 ribozyme. Mutations are highlighted in boxes. ΔP9a-L9a indicates the deletion of P9a–L9a. Values show the activity of each mutant relative to the clone 2 ribozyme. The activity was determined with 5 µM substrate RNA. (B) Base pairings between L9b and the 5′ leader sequence of the substrate RNA: (top) base pairings between the clone 2 ribozyme and the original substrate RNA; (middle) the mutant substrate RNA; (bottom) base pairings between the L9b mutant ribozyme and the mutant substrate RNA. Mutations are highlighted in boxes. Values show the activity of each mutant relative to the clone 2 ribozyme. The activity was determined with 5 µM substrate RNA.
We carried out mutational analysis of the clone 2 ribozyme to see whether it possesses the predicted structure and also to identify the structural elements important for its function (Fig. 4A). Deletion of P9a–L9a significantly reduced the activity by ∼7-fold. However, neither the mutation in P9a nor L9a, which is designed to maintain its original secondary or loop structure, respectively, showed significant effect on the catalytic activity, supporting the predicted structure of P9a–L9a.
The mutation in P9b that is designed to give an alternative form of the predicted secondary structure resulted in an ∼50% reduction in activity, suggesting that the sequence of this region is involved in the mechanism of enhancing the activity (Fig. 4A).
The mutation in L9b that is designed to form an alternative loop structure showed the most dramatic effect by reducing the activity by 40-fold. The sequence of L9b is complementary to the 5′ leader region of the substrate RNA (Fig. 4B, top), suggesting that this loop forms base pairs with the substrate RNA as mentioned above. To see whether the predicted base pairings exist, we prepared a new substrate RNA which has a mutation disrupting the possible complementarity (Fig. 4B, middle). The clone 2 ribozyme cleaved the mutant substrate with significantly reduced efficiency. However, the compensatory mutation at L9b increased the activity ∼10-fold over the clone 2 ribozyme (Fig. 4B, bottom), suggesting that the predicted base pairings are involved in recognizing the substrate.
The predicted structure of the insertion in the clone 2 ribozyme was investigated by enzymatic footprinting (Fig. 5A). Footprinting with RNase V1, a double-stranded structure-specific endonuclease, showed cleavages at the putative P9a and P9b, indicating that they form helices as predicted. The cleavage at L9b was observed only in the presence of the substrate RNA, indicating that the loop interacts with the substrate. Footprinting with RNase A, a pyrimidine-specific endonuclease, showed cleavages at L9b and L9a (Fig. 5A). The nucleotides joining P9/P9a and P9a/P9b, as well as most of the nucleotides in L9a, were, however, protected. The slight cleavage by RNase V1 and the protection from RNase A suggest that the L9a loop forms an unusual structure, although its primary sequence is not significant for its function, as described above (Fig. 4).
Figure 5.
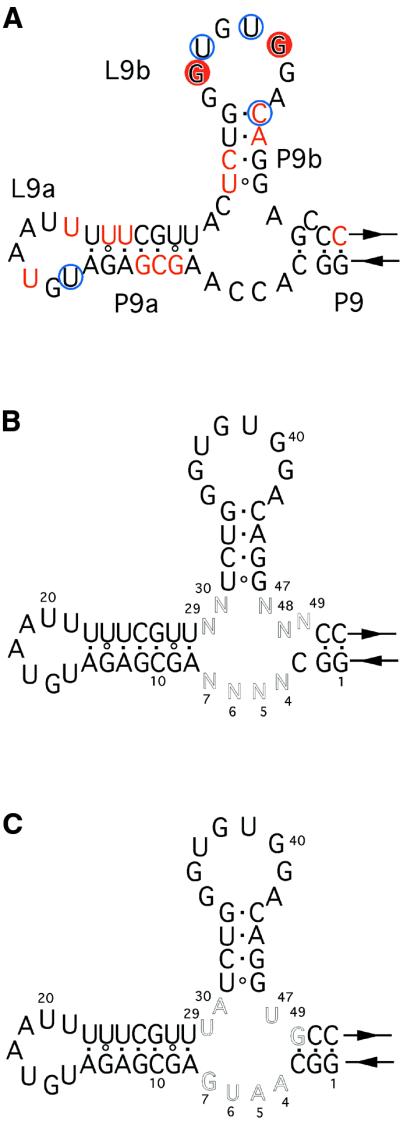
(A) Secondary structural model of the clone 2 ribozyme. The RNase V1 cleavage sites are indicated by red circles and the substrate- independent cleavage sites are shown by red letters. RNase A cleavage sites are shown by blue circles. (B) The partially randomized clone 2 library. (C) Secondary structure of the clone 2.1 ribozyme.
Secondary selection from the partially randomized clone 2 library
The results indicate that the L9b base-paired with the substrate RNA is fixed physically by other structural elements, such as the regions joining P9/P9a, P9a/P9b or P9/P9b. To investigate the role of the regions which presumably contribute to enhancing the activity of the clone 2 ribozyme, we constructed a partially randomized clone 2 library for the new selection experiment (Fig. 5B). In the library, containing 262 144 variants, the nine residues that include the corresponding regions and the 5′-AGC-3′ starting from the A nucleotide next to the 3′ end of P9b (N47–N49 in Fig. 5B) were randomized. The activity of the library was comparable to that of the clone 2 ribozyme after two rounds of in vitro selection (Table 1, rounds 10 and 11). Thus, the corresponding DNAs were cloned and sequenced. The selected clones were more active than the clone 2 ribozyme, although none possessed sequence analogous to that of the clone 2 ribozyme. They showed that the original sequences in regions of the clone 2 ribozyme are not optimal, suggesting that further evolution of the newly added peripheral domain is possible.
The consensus sequence from one class of the resulting highly active variants was 5′-GGCA4R5U6G7AGCGAGA UGUAAUUUUUCGUUY29A30UCUGGGUGUGGACAGG U47(N48)G49CC-3′ (where Y is C or U, R is A or G and the randomized positions are shown in bold) (Table 4). The most active ribozyme, termed clone 2.1, exhibited ∼3-fold higher activity than that of the wild-type T4 td ribozyme (Table 3 and Fig. 5C). The three residues at the 3′ end of P9b turned out to be 5′-U47G49-3′ in the clone 2.1 ribozyme, suggesting that the bulged C residue in P9 of the clone 2 ribozyme is likely deleterious. The upstream nucleotide, indicated by (N48) in the consensus, next to G49, was frequently deleted, suggesting that the region between P9 and P9b might prefer one rather than two bases, although it is also possible that the reverse transcriptase has a tendency to skip this position.
Table 4. Round 11 clones.
| Clone | Inserted sequencea (5′→3′) | Activityb |
|---|---|---|
| 2 | GGCACCAAGCGAGAUGUAAUUUUUCGUUACUCUGGGUGUGGACAGGAGCCC | 1.0 |
| 2.1 | GGCAAUGAGCGAGAUGUAAUUUUUCGUUUAUCUGGGUGUGGACAGGUGCC | 2.5 |
| 2.2 | GGCAGUGAGCGAGAUGUAAUUUUUCGUUCAUCUGGGUGUGGACAGGUUGCC | 2.3 |
| 2.3 | GGCUCUGAGCGAGAUGUAAUUUUUCGUUUCUCUGGGUGUGGACAGGUGCC | 1.4 |
| 2.4 | GGCAAGAAGCGAGAUGUAAUUUUUCGUUCGUCUGGGUGUGGACAGGUAGCC | 1.3 |
aThe randomized positions are shown in bold. Underlined letters indicate the sequences that coincide with the consensus sequence.
bThe activity relative to the clone 2 ribozyme was determined with 0.5 µM substrate RNA.
DISCUSSION
New peripheral elements were added to the three sites L7.1, L8 and L9 in a mutant T4 td Group I ribozyme lacking the P4–P6 domain together with the base triples from its catalytic core. The variants with the inserted elements at each site efficiently catalyzed the reversal reaction of the first step of self-splicing. From our experiments, no preference was observed for the insertion sites where new structural elements are attached, although this does not negate the possibility that one particular site is favored under certain conditions. The results may reflect the structural flexibility of the peripheral elements in Group I ribozymes (29,30) and explain why many different classes (subgroups IA–IE) of Group I ribozymes could have evolved.
The clone 2 ribozyme was unable to perform intact splicing (cis-splicing) (data not shown). This is presumably because the substrate recognition mechanism of the selected clone is different from that of the wild-type ribozyme. The clone 2 ribozyme has a sequence complementary to the 5′ leader sequence of the substrate RNA in its L9b loop. Our data indicate that the loop forms base pairs with the substrate RNA. However, the base pairings by themselves are insufficient because elimination of P9a–L9a from the clone 2 ribozyme significantly reduced the activity. Thus, the results indicate that this region might be involved in fixing base pairings with the substrate RNA and/or in long-range interactions with other regions of the ribozyme.
The selected clones from the round 9 pool share similar complementary sequences without exception (Table 2, letters in bold), suggesting that the base pairings with the substrate RNA strongly contribute to enhancing activity in the newly obtained ribozymes. If this is the case, the selected ribozymes cannot employ other RNAs as substrates. However, this potential problem can be technically overcome by altering the sequence of the substrate RNA during selection or physically protecting the 5′ portion of the substrate from the ribozyme.
Clone 2 was the most active variant of the round 9 clones. A modified clone 2 containing partially randomized sequences was subjected to the second selection. Unexpectedly, the selection generated another class of ribozymes with significantly improved activities, emphasizing the importance of extra mutagenic steps (typically carried out by error-prone PCR) to produce superior variants.
The selection experiment resulted in a ribozyme (clone 2.1 ribozyme) that was 3-fold more active than the wild-type ribozyme. However, its first rate constant (kcat value) was lower than that of the wild-type ribozyme. This is probably due to the selection conditions, such as a long reaction time and low substrate concentration. If so, other conditions may be useful in enriching for ribozymes with high kcat values.
Our results show that new artificial domains can be easily added to the P3–P7 domain, which serves as an independent catalytic module. This class of artificial modular evolution (20,31–34) may generate a variety of ribozymes with new properties.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Tsutomu Suzuki for the gift of the expression system of T7 RNA polymerase and Dr Kazuei Igarashi for the gift of RNase V1. We also thank Dr Ruth T. Yu and the members of the Inoue laboratory for critical reading of and comments on the manuscript. This work was supported by Grants-in-Aid for Scientific Research on Priority Areas (T.I.) and Encouragement of Young Scientists (Y.I.) from the Ministry of Education, Science, Sports and Culture, Japan.
REFERENCES
- 1.Cech T.R. (1990) Self-splicing of group I introns. Annu. Rev. Biochem., 59, 543–568. [DOI] [PubMed] [Google Scholar]
- 2.Jaeger L., Michel,F. and Westhof,E. (1996) The structure of group I ribozymes. In Eckstein,F. and Lilley,D.M.J. (eds), Catalytic RNA. Springer-Verlag, Berlin, Germany, pp. 33–51.
- 3.Beaudry A.A. and Joyce,G.F. (1990) Minimum secondary structure requirements for catalytic activity of a self-splicing group I intron. Biochemistry, 29, 6534–6539. [DOI] [PubMed] [Google Scholar]
- 4.Cech T.R. and Golden,B.L. (1998) Building a catalytic active site using only RNA. In Gesteland,R.F., Cech,T.R. and Atkins,J.F. (eds), The RNA World, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 321–349.
- 5.Lehnert V., Jaeger,L., Michel,F. and Westhof,E. (1996) New loop-loop tertiary interactions in self-splicing introns of subgroup IC and ID: a complete 3D model of the Tetrahymena thermophila ribozyme. Chem. Biol., 3, 993–1009. [DOI] [PubMed] [Google Scholar]
- 6.Michel F. and Westhof,E. (1990) Modeling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J. Mol. Biol., 216, 585–610. [DOI] [PubMed] [Google Scholar]
- 7.Golden B.L., Gooding,A.R., Podell,E.R. and Cech,T.R. (1998) A preorganized active site in the crystal structure of the Tetrahymena ribozyme. Science, 282, 259–263. [DOI] [PubMed] [Google Scholar]
- 8.Strobel S.A. and Ortoleva-Donnelly,L. (1999) A hydrogen-bonding triad stabilizes the chemical transition state of a group I ribozyme. Chem. Biol., 6, 153–165. [DOI] [PubMed] [Google Scholar]
- 9.Waring R.B. (1989) Identification of phosphate groups important to self-splicing of the Tetrahymena rRNA intron as determined by phosphorothioate substitution. Nucleic Acids Res., 17, 10281–10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christian E.L. and Yarus,M. (1992) Analysis of the role of phosphate oxygens in the group I intron from Tetrahymena. J. Mol. Biol., 228, 743–758. [DOI] [PubMed] [Google Scholar]
- 11.Christian E.L. and Yarus,M. (1993) Metal coordination sites that contribute to structure and catalysis in the group I intron from Tetrahymena. Biochemistry, 32, 4475–4480. [DOI] [PubMed] [Google Scholar]
- 12.Streicher B., Westhof,E. and Schroeder,R. (1996) The environment of two metal ions surrounding the splice site of a group I intron. EMBO J., 15, 2556–2564. [PMC free article] [PubMed] [Google Scholar]
- 13.Berens C., Streicher,B., Schroeder,R. and Hillen,W. (1998) Visualizing metal-ion-binding sites in group I introns by iron(II)-mediated Fenton reactions. Chem. Biol., 5, 163–175. [DOI] [PubMed] [Google Scholar]
- 14.Ortoleva-Donnelly L., Szewczak,A.A., Gutell,R.R. and Strobel,S.A. (1998) The chemical basis of adenosine conservation throughout the Tetrahymena ribozyme. RNA, 4, 498–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pichler A., Berens,C. and Schroeder,R. (2000) Localization of metal ion binding sites in Group I intron RNA. In Krupp,G. and Gaur,R.K. (eds), Ribozyme Biochemistry and Biotechnology. Eaton Publishing, Westborough, MA, pp. 7–25.
- 16.Szewczak A.A., Kosek,A.B., Piccirilli,J.A. and Strobel,S.A. (2002) Identification of an active site ligand for a Group I ribozyme catalytic metal ion. Biochemistry, 41, 516–525. [DOI] [PubMed] [Google Scholar]
- 17.Ikawa Y., Shiraishi,H. and Inoue,T. (2000) Minimal catalytic domain of a group I self-splicing intron RNA. Nature Struct. Biol., 7, 1032–1035. [DOI] [PubMed] [Google Scholar]
- 18.Ikawa Y., Yoshioka,W., Ohki,Y., Shiraishi,H. and Inoue,T. (2001) Self-splicing of the Tetrahymena group I ribozyme without conserved base-triples. Genes Cells, 6, 411–420. [DOI] [PubMed] [Google Scholar]
- 19.Loria A. and Pan,T. (1996) Domain structure of the ribozyme from eubacterial ribonuclease P. RNA 2, 551–563. [PMC free article] [PubMed] [Google Scholar]
- 20.Mobley E.M. and Pan,T. (1999) Design and isolation of ribozyme–substrate pairs using RNase P-based ribozymes containing altered substrate binding sites. Nucleic Acids Res., 27, 4298–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frank D.N. and Pace,N.R. (1998) Ribonuclease P: unity and diversity in a tRNA processing ribozyme. Annu. Rev. Biochem., 67, 153–180. [DOI] [PubMed] [Google Scholar]
- 22.Altman S. and Kirsebom,L. (1999) Ribonuclease P. In Gesteland,R.F., Cech,T.R. and Atkins,J.F. (eds), The RNA World, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 351–378.
- 23.Green R. and Szostak,W.R. (1992) Selection of a ribozyme that functions as a superior template in a self-copying reaction. Science, 258, 1910–1915. [DOI] [PubMed] [Google Scholar]
- 24.Green R., Ellington,A.D. and Szostak,W.R. (1990) In vitro genetic analysis of the Tetrahymena self-splicing intron. Nature, 347, 406–408. [DOI] [PubMed] [Google Scholar]
- 25.Pogulis R.J., Vallejo,A.N. and Pease,L.R. (1996) In vitro recombination and mutagenesis by overlap extension PCR. Methods Mol. Biol., 57, 167–176. [DOI] [PubMed] [Google Scholar]
- 26.Knapp G. (1989) Enzymatic approaches to probing of RNA secondary and tertiary structure. Methods Enzymol., 180, 192–212. [DOI] [PubMed] [Google Scholar]
- 27.Zuker M., Mathews,D.H. and Turner,D.H. (1999) Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide. In Barciszewski,J. and Clark,B.F.C. (eds), RNA Biochemistry and Biotechnology. Kluwer Academic, Boston, MA, pp. 11–43.
- 28.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- 29.Williams K.P., Imahori,H., Fujimoto,D.N. and Inoue,T. (1994) Selection of novel forms of a functional domain within the Tetrahymena ribozyme. Nucleic Acids Res., 22, 2003–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikawa Y., Shiraishi,H. and Inoue,T. (1996) Characterization of the newly constructed domains that replace P5abc within the Tetrahymena ribozyme. FEBS Lett., 394, 5–8. [DOI] [PubMed] [Google Scholar]
- 31.Tuschl T., Sharp,P.A. and Bartel,D.P. (1998) Selection in vitro of novel ribozymes from a partially randomized U2 and U6 snRNA library. EMBO J., 17, 2637–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnston W.K., Unrau,P.J., Lawrence,M.S., Glasner,M.E. and Bartel,D.P. (2001) RNA-catalyzed RNA polymerization: accurate and general RNA-templated primer extension. Science, 292, 1319–1325. [DOI] [PubMed] [Google Scholar]
- 33.Robertson M.P. and Ellington,A.D. (2001) In vitro selection of nucleoprotein enzymes. Nat. Biotechnol., 19, 650–655. [DOI] [PubMed] [Google Scholar]
- 34.Koizumi M., Soukup,G.A., Kerr,J.N.Q. and Breaker,R.R. (1999) Allosteric selection of ribozymes that respond to the second messengers cGMP and cAMP. Nature Struct. Biol., 6, 1062–1071. [DOI] [PubMed] [Google Scholar]
- 35.Cech T.R., Damberger,S.H. and Gutell,R.R. (1994) Representation of the secondary and tertiary structure of group I introns. Nature Struct. Biol., 1, 265–266. [DOI] [PubMed] [Google Scholar]
- 36.Belfort M., Chandry,P.S. and Pedersen-Lane,J. (1987) Genetic delineation of functional components of the group I intron in the phage T4 td gene. Cold Spring Harbor Symp. Quant. Biol., 52, 181–192. [DOI] [PubMed] [Google Scholar]