

Abstract
The signal recognition particle (SRP) mediates membrane targeting of translating ribosomes displaying a signal-anchor sequence. In Escherichia coli, SRP consists of 4.5S RNA and a protein, Ffh, that recognizes the signal peptide emerging from the ribosome and the SRP receptor at the membrane, FtsY. In the present work, we studied the interactions between the NG and M domains in Ffh and their rearrangements upon complex formation with 4.5S RNA and/or FtsY. In free Ffh, the NG and M domains are facing one another in an orientation that allows cross-linking between positions 231 in the G domain and 377 in the M domain. There are binding interactions between the two domains, as the isolated domains form a strong complex. The interdomain contacts are disrupted upon binding of Ffh to 4.5S RNA, consuming a part of the total binding energy of 4.5S RNA-Ffh association that is roughly equivalent to the free energy of domain binding to each other. In the SRP particle, the NG domain binds to 4.5S RNA in a region adjacent to the binding site of the M domain. Ffh binding to FtsY also requires a reorientation of NG and M domains. These results suggest that in free Ffh, the binding sites for 4.5S RNA and FtsY are occluded by strong domain–domain interactions which must be disrupted for the formation of SRP or the Ffh-FtsY complex.
Keywords: protein targeting, translation, protein export, bimane cross-linking, fluorescence resonance energy transfer
INTRODUCTION
The signal recognition particle (SRP) is a ribonucleoprotein that targets ribosomes translating secretory or inner-membrane proteins to the translocation pore located in the membrane of the endoplasmic reticulum in eukaryotes or the plasma membrane in prokaryotes (Keenan et al. 2001; Nagai et al. 2003; Doudna and Batey 2004). The SRP recognizes and binds to hydrophobic signal sequences as they emerge from ribosome nascent-chain complexes (RNC). The RNC-SRP complex is directed to the membrane by the interaction with the membrane-associated SRP receptor, and the RNC is transferred to the translocation pore through which the nascent protein passes during further elongation. In Escherichia coli, the SRP consists of 4.5S RNA (114 nucleotides) and Ffh, a 48-kDa protein. Ffh is a GTPase that consists of three domains, the N-terminal N domain, the G domain, comprising the GTP binding site, and the C-terminal M domain where the RNA-binding site is located (Bernstein et al. 1989; Poritz et al. 1990). The N domain packs against the G domain to form a contiguous unit called the NG domain. The bacterial SRP receptor is FtsY, a membrane-associated 52-kDa protein that has an acidic N-terminal A domain followed by an NG domain (Gill and Salmond 1990). The NG domains of Ffh and FtsY are homologous in sequence, and the crystal structures of the NG domains of Ffh from Thermus aquaticus and of FtsY from E. coli revealed a high degree of structural similarity (Freymann et al. 1997; Montoya et al. 1997). Complex formation between Ffh and FtsY involves the symmetric association of their G domains, as shown by the recent crystal structures of the complex of the two NG domains (Egea et al. 2004; Focia et al. 2004).
The structure of the SRP is not known. Two crystal structures of free Ffh are available (Keenan et al. 1998; Rosendal et al. 2003). In the crystal structure of Ffh from T. aquaticus, there were three molecules of Ffh in the asymmetric unit, and the linkers between G and M domains were disordered (Keenan et al. 1998). Based on that crystal structure, there are three possible arrangements of NG and M domains, two of which (A/A, i.e., A chain for both NG and M domains; or B/A, B chain for NG and A chain for M) are compatible with the length of the linker. In both arrangements, the NG and M domains are found sufficiently close to each other to make intramolecular contacts. In a recent crystal structure of the homologous SRP54 from Sulfolobus solfataricus alone and in complex with domain IV of SRP RNA, determined at a resolution of 4 Å, the linker could be traced, and the protein was present in a conformation in which the NG domain was detached from the M domain and which appeared to be stabilized by a hydrophobic interaction between N and M domains (Rosendal et al. 2003).
The binding site of Ffh on 4.5S RNA was mapped by genetic analysis (Wood et al. 1992), as well as by chemical and enzymatic footprinting (Lentzen et al. 1996). The RNA-binding site of Ffh was localized within the M domain (Römisch et al. 1989), and the crystal structure of the complex of the RNA-binding fragment of the M domain of E. coli Ffh with a 49-nt fragment of 4.5S RNA comprising domain IV was determined (Batey et al. 2000). The crystal structure revealed contacts between the M domain and 4.5S RNA that explained the footprints of Ffh in internal loops A and B of 4.5S RNA. Internal loops C and D and stem d of 4.5S RNA, where Ffh footprints were observed as well, were not included in the core structure (Batey et al. 2000), suggesting that those footprints (Lentzen et al. 1996) were due to protection by the NG domain or conformational changes in 4.5S RNA upon binding of the M domain. In the cryo-EM structure of the eukaryotic SRP homolog, the contact between the NG and M domains is limited to a potential connection trough the flexible loop of the M domain to the NG interface, and there is no contact between the NG domain and 4.5S RNA (Halic et al. 2004), and the domain arrangement of Ffh is different from either crystal structure (Keenan et al. 1998; Rosendal et al. 2003).
The structure of a complex of the NG domains of Ffh and FtsY revealed a symmetric heterodimer featuring a composite active site that contains two molecules of GDPNP (Egea et al. 2004; Focia et al. 2004). The arrangement of the two NG domains in the structure imposed constrains on the possible positions of the M domain of Ffh. Probing the structure of the Ffh-FtsY complex by cross-linking and mass spectroscopy (Chu et al. 2004) suggested that the solution structure of the Ffh-FtsY complex is in good agreement with the crystal structure (Egea et al. 2004; Focia et al. 2004) and that the M domain is positioned in close proximity to the Ffh-FtsY interface in the complex. Furthermore, the cross-linking studies suggested that in the Ffh-FtsY complex the M domain assumes a position different from that suggested by Ffh crystal structures (Keenan et al. 1998; Rosendal et al. 2003).
The aim of the present work was to clarify whether the NG domain interacts with the M domain or 4.5S RNA in free Ffh, SRP, and in the complex with FtsY. The arrangement of NG and M domains was studied by cross-linking and fluorescence resonance energy transfer (FRET) techniques. The binding of the NG domain to 4.5S RNA and the conformational rearrangements taking place upon SRP formation were monitored by chemical footprinting and fluorescence measurements. Finally, the interaction between Ffh and FtsY was studied by fluorescence using a variant of Ffh in which the relative mobility of NG and M domains was restricted by a cross-link.
RESULTS
Complex formation between isolated NG and M domains of Ffh
The interaction between the domains of Ffh was examined using the isolated NG domain (residues 1–295; 33 kDa) and M domain (positions 297–453; 17 kDa), monitoring fluorescence resonance energy transfer (FRET) between fluorophores attached to a single cysteine residue in either domain. BODIPY FL (Bpy) at position 406 of the M domain was used as donor [Ffh-M (Bpy406) ] and Alexa 546 (Alx) at position 84 of the NG domain as acceptor [Ffh-NG (Alx84) ]. FRET was monitored by the decrease of the fluorescence lifetime of Bpy (Table 1). With Ffh-M (Bpy406) alone, the lifetime was 5.2 nsec, and the lifetime decreased to 4.7 nsec upon addition of unlabeled NG domain. When Ffh-NG (Alx84) was added, the lifetime of Ffh-M (Bpy406) dropped to 0.90 nsec, indicating FRET at an efficiency of 81% (Fig. 1B; Table 1). Analogous results were obtained by monitoring the steady-state fluorescence emission of Ffh-M (Bpy406) : the emission decreased to 74% upon addition of unlabeled NG domain and to 21% when the Alx-labeled NG domain was added, yielding a FRET efficiency of 72% (Table 1). Distances between donor and acceptor of 38 ± 6 Å and 41 ± 8 Å, respectively, were calculated from the changes in the lifetime and fluorescence intensity of the Bpy donor, comparing the values measured in the presence of unlabeled and acceptor-labeled NG domain (Materials and Methods). The indicated distance ranges take into account the uncertainty about the orientation of the fluorophores, as assessed by depolarization measurements (Materials and Methods), and the standard deviations of the lifetime and fluorescence measurements (Table 1). Within these ranges, the results of the lifetime and steady-state fluorescence measurements coincide. About the same distance was derived from FRET measurements in native Ffh (I. Buskiewicz, unpubl.), supporting the view that the arrangement of NG and M domains in the complex of the isolated domains and in full-length Ffh is the same.
TABLE 1.
Complex formation between NG and M domains monitored by FRET
| Ligand | Ligand | Lifetimea (nsec) | Rel. fluorescenceb |
| Ffh-M (Bpy406) | — | 5.2 | 1.0 |
| “ | Ffh-NG | 4.7 | 0.74 |
| “ | Ffh-NG (Alx84) | 0.9c | 0.21 |
a SD ± 0.1 nsec.
b SD ± 2%.
c Average lifetime, see Materials and Methods.
FIGURE 1.

Interaction of isolated NG and M domains monitored by fluorescence. (A) Positions of fluorescence labels. NG and M domains are shown in the A/A arrangement (Keenan et al. 1998) ; cysteine residues that were used for attaching fluorescence labels are indicated. (B) FRET measurements. Fluorescence decay curves were measured for Ffh-M (Bpy406) (fluorescence donor) alone (1), in the presence of unlabeled NG domain (2), or in the presence of Ffh-NG (Alx84) (acceptor) (3) ; the time response curve of the excitation pulse is also indicated (4). Fluorescence lifetimes derived from the decay curves are summarized in Table 1. (C) Fluorescence titrations. The fluorescence emission of Ffh-NG (OG84) upon addition of unlabeled Ffh-M (○) or of Ffh-M (Bpy406) upon addition of unlabeled Ffh-NG (•) was monitored. The amplitudes of the fluorescence changes (11% increase and 26% decrease for OG84 and Bpy406, respectively) were set to 1.0.
The binding affinity between NG and M domains was determined by equilibrium titration, monitoring the fluorescence change of Ffh-M (Bpy406) caused by binding of the unlabeled NG domain or of Ffh-NG (Alx84) by binding of the unlabeled M domain (Fig. 1C). Similar Kd values of 90 ± 20 and 40 ± 10 nM, respectively, were obtained by the two measurements. For calculation of the free binding energy, we used an average Kd value of 60 ± 30 nM, corresponding to a free binding energy of ΔG° = −9.8 kcal/mol (Table 2).
TABLE 2.
Summary of binding affinities
| Ligand | Ligand | Kd (M) | ΔG° (kcal/mol) |
| Ffh-NG | Ffh-M | (6.0 ± 0.3) ×10−8 | −9.8 ± 0.5 |
| Ffh | 4.5S RNA | (5.2 ± 0.5) ×10−11 | −14.0 ± 1.3 |
| “ | 61 mer | (3.3 ± 0.3) ×10−11 | −14.3 ± 1.3 |
| Ffh-M | 4.5S RNA | (3.3 ± 0.2) ×10−11 | − 14.3 ± 0.9 |
| “ | 61 mer | (2.8 ± 0.3) ×10−11 | −14.4 ± 1.5 |
| Ffh-NG | 4.5S RNA | (5.6 ± 1.2) ×10−7 | −8.5 ± 1.8 |
| “ | 61 mer | (6.2 ± 0.9) ×10−7 | −8.4 ± 1.2 |
| “ | 49 mer | (2.0 ± 0.5) ×10−5 | −6.4 ± 1.6 |
Binding of the isolated NG domain of Ffh to 4.5S RNA
Binding of full-length Ffh protein (48 kDa) to 4.5S RNA (114 nt; 35 kDa) can be studied by band shift in nondenaturing gel electrophoresis (Lentzen et al. 1994; Kusters et al. 1995; Jagath et al. 2001). Under the same conditions, the isolated NG domain of Ffh (33 kDa) did not cause a significant shift of 4.5S RNA (data not shown), suggesting that the complex—if formed—was of low stability. However, using gel filtration on Superdex 75, a complex of 4.5S RNA and the NG domain, both present at 5 μM concentration, could be detected (Fig. 2, trace 1). At lower concentrations of protein (0.5 μM) and RNA (0.1 μM), part of the RNA remained unbound (trace 2), indicating a Kd of the complex of ~0.5 μM.
FIGURE 2.

Complex formation between the NG domain of Ffh and 4.5S RNA. The isolated NG domain (Materials and Methods) and 4.5S RNA were mixed in 100 μL of buffer A at 5 μM concentration each (1) or 0.5 μM and 0.1 μM (2), respectively, and separated on a Superdex 75 gel filtration column in buffer A at 25°C. Absorbance at 260 nm is plotted in arbitrary units.
Chemical footprinting reveals separate binding sites for M and NG domains on 4.5S RNA
According to previous chemical footprinting results, the binding site of Ffh on 4.5S RNA extends from internal loop A through internal loop C and the adjacent stem (Fig. 3A). The M-domain footprints in internal loops A and B (DMS modification; Fig. 3B) are consistent with the contacts revealed by the crystal structure of the complex of an M-domain fragment of Ffh and a 49 mer construct of 4.5S RNA comprising loops A and B (Batey et al. 2000, 2001). Using kethoxal modification, a strong footprint at position G27 was observed for both full-length Ffh, as described earlier (Lentzen et al. 1996), and the isolated NG domain (Fig. 3C). The isolated M domain caused no protection in that region of 4.5S RNA. These results suggest that the NG domain binds to 4.5S RNA in the region of internal loop C and the adjacent stem; alternatively, the change of the reactivity against kethoxal could be caused indirectly by changing the conformation at this region. Thus, the footprint of full-length Ffh appears to be composed of the footprints of the M and NG domains, indicating that in the complex of 4.5S RNA with full-length Ffh, both M domain and NG domain are bound to the RNA.
FIGURE 3.

Footprinting of full-length Ffh, NG domain, and M domain on 4.5S RNA. (A) Predicted secondary structure of 4.5S RNA. Encircled nucleotides (A,C) were found reactive toward dimethyl sulfate, the boxed nucleotide (G) toward kethoxal. Strong and weak reactivities are indicated in black and gray, respectively. Protection is indicated by small circles or a square. The binding site of Ffh, as derived from previous footprinting results (Lentzen et al. 1996), is delineated and the approximate binding sites of M and NG domains are indicated. (B) DMS modification. C,G, sequencing lanes. (C) Kethoxal modification. A,C,G,T, sequencing lanes. The positions of nucleotides protected from modification are indicated in the sequences to the right.
Affinities of Ffh, M domain, and NG domain for 4.5S RNA
The binding affinities of Ffh and the isolated M and NG domains to 4.5S RNA were determined by titration experiments. In addition to full-length 4.5S RNA, two fragments of 4.5S RNA were used, a 61mer (nt 21–81), comprising internal loops A through C, and a 49 mer that comprised internal loops A and B and was closed by an extended double-stranded stem replacing loop C (Batey et al. 2000). 5′-32P-labeled RNAs were used at 5 pM concentration, and RNA-protein complexes were isolated by nitrocellulose filtration. Titration of 4.5S RNA with full-length Ffh yielded a Kd of 52 pM (Fig. 4A), close to the value previously obtained at similar buffer conditions (Batey et al. 2001; Batey and Doudna 2002). At different buffer conditions and in the presence of detergent, we previously obtained significantly weaker binding (Jagath et al. 2001). A similar Kd value of 33 pM was obtained for Ffh binding to the 61mer fragment of 4.5S RNA (Table 2). The isolated M domain exhibited the same affinity, ~30 pM, of binding to 4.5S RNA (Fig. 4A) and the 61mer (Table 2). The titration of 4.5S RNA with the isolated NG domain of Ffh yielded a much higher Kd value of 0.7 μM (Fig. 4B) ; the yield of complex was reduced to 60%, indicating some dissociation of the complex during isolation. No complex between the NG domain and the 49mer fragment of 4.5S RNA was observed by nitrocellulose filtration, suggesting that the complex did not form or was too unstable to be detected by filtration. Complex formation was specific, as the addition of tRNA or 5S rRNA in 100-fold excess had no influence on the titration (data not shown).
FIGURE 4.

Quantitation of Ffh and Ffh domains binding to 4.5S RNA. (A) Binding of Ffh and M domain. Binding was measured by nitrocellulose filtration with 32P-labeled 4.5S RNA (5 pM). Ffh (•), M domain (○). (B) Binding of NG domain. 32P-labeled 4.5S RNA (▪) or 49mer (⋄) were present at 80 nM concentration. (C) Fluorescence titrations with NG domain. 4.5S RNA (▾), 61mer (▿), and 49mer (⋄) were labeled with Alexa 555 (Alx) at the 3′ end; titrations, monitoring the fluorescence emission of the Alx probe, were performed and evaluated as described in Materials and Methods. Standard deviations of individual points are indicated by error bars (three or more independent measurements). The respective fits are indicated by continuous lines; Kd values obtained from the fits are summarized in Table 2.
Binding of the NG domain to 4.5S RNA and 4.5S RNA fragments was also monitored by fluorescence titrations, i.e., at true equilibrium conditions. RNAs were labeled by introducing the fluorescent probe Alexa 555 at the 3′ end (Materials and Methods). Fluorescence titrations yielded Kd values of about 0.6 μM for NG binding to full-length 4.5S RNA and the 61mer fragment (Fig. 4C), in keeping with the results from nitrocellulose filtration. Binding of the NG domain to the 49 mer fragment was very weak, Kd=20 μM, indicating that the 49mer fragment does not comprise the (complete) binding site of the NG domain. Practically no signal change was observed with an even shorter fragment of 39 nt comprising internal loops A and B closed by a short double helix (data not shown).
Binding of 4.5S RNA to the NG domain labeled at position 84 with Oregon Green (OG), Ffh-NG (OG84), increased the fluorescence by 15%. Fluorescence titrations yielded Kd values of 0.6 ± 0.1 μM for binding of 4.5S RNA and the 61mer fragment to the labeled NG domain (Table 2).
Cross-linking G and M domains of Ffh abrogates 4.5S RNA binding
In order to verify whether NG and M domains of Ffh must come apart to allow complex formation with 4.5S RNA, we studied the effect of introducing a chemical cross-link between G and M domain. As a cross-linker, we used dibromobimane (dBrB), which specifically reacts with thiol groups and can cross-link pairs of thiol groups within 3–6 Å of one another (Mornet et al. 1985). The cross-linking reaction can be monitored by fluorescence, because dBrB becomes fluorescent when the two bromine atoms, which are strong quenchers of fluorescence, are released during the reaction with thiol groups. Control reactions were performed with monobromobimane (mBrB), which becomes fluorescent upon coupling to a single thiol group. Cysteine residues were engineered into two positions of Ffh, 231 and 377, that appeared sufficiently close in the A/A arrangement of NG and M domains in the crystal structure of T. aquaticus Ffh (Keenan et al. 1998) (Fig. 5A). The reaction of the double mutant Ffh (C231/377) with dBrB was measured after a 2-h incubation (Fig. 5B). Control reactions of the respective single mutants Ffh (C231) and Ffh (C377) with mBrB reached comparable fluorescence levels. This strongly indicates that the cross-linked cysteine residues, 231 and 377, were close to one another in the native form of the double mutant. Another double mutant, Ffh (C17/344), did not yield a fluorescent product with dBrB, i.e., no cross-link, consistent with the large distance between those two cysteine residues in the tertiary structure of Ffh, independently of the configuration of domains. Mutant Ffh lacking any cysteine, Ffh (S406), did not react with dBrB. Ffh (C231/377) bound to 4.5S RNA formed hardly any cross-linked product upon reaction with dBrB. This indicates that in the complex, the two cysteine residues at positions 231 and 377 are too far apart to formthe cross-link, suggesting a separation of M and G domains upon Ffh binding to 4.5S RNA. The electrophoretic mobility of bimane–cross-linked Ffh was the same as that of free Ffh (Fig. 5B, inset), which demonstrates that the cross-linked protein was monomeric, i.e., that the cross-link was strictly intramolecular.
FIGURE 5.

Bimane cross-linking of G and M domains of Ffh. (A) Model of bimane crosslinked to Ffh (C231/377). The structure of T. aquaticus Ffh in the A/A orientation was used (Keenan et al. 1998) (PDF acc. no. 2FFH) in which residues 228 and 363, in E. coli Ffh corresponding to residues 231 and 377, respectively, were replaced with cysteines using RasMol software. (B) Reaction of dibromobimane (dBrB) and monobromobimane (mBrB) with cysteine mutants of Ffh as monitored by bimane fluorescence after 120-min incubation. (Inset) SDS gel showing the amounts of Ffh protein in the samples. (C) Ffh binding to 4.5S RNA. The formation of SRP was monitored by band shift in a 7% nondenaturing polyacrylamide gel; 4.5S RNA was stained with ethidium bromide. Lane 1, 4.5S RNA; 2, + Ffh; 3, + Ffh (C231/377) ; 4, + Ffh (C231/377) cross-linked with dBrB; 5, Ffh (C231) reacted with mBrB; 6, Ffh (C377) reacted with mBrB; 7, Ffh (C17/344) reacted with dBrB.
The inhibition of dBrB cross-linking in the Ffh (C231/377) -4.5S RNA complex suggested that the arrangement of M and NG domains was changed upon formation of SRP. Therefore, cross-linked Ffh was examined with respect to 4.5S RNA binding, using the gel shift assay (Valent et al. 1995; Jagath et al. 2001) (Fig. 5C). According to this assay, bimane–cross-linked Ffh was no longer able to bind 4.5S RNA, whereas Ffh modified with bimane at single positions was as active as unmodified Ffh. This result strongly indicates that binding of Ffh to 4.5SRNA requires a rearrangement of M and G domains.
Cross-linking G and M domains of Ffh impairs FtsY binding
Finally, we examined the interaction of bimane–crosslinked Ffh with the SRP receptor, FtsY, monitoring the fluorescence of a single tryptophan residue in mutant FtsY (Trp342) (Jagath et al. 2000). Binding of wild-type Ffh (data not shown) (Jagath et al. 2000) or mBrB-modified Ffh (C231/377) to FtsY caused a twofold fluorescence increase of Trp342 and a blue-shift of the emission spectrum by ~10 nm (Fig. 6A). With bimane–cross-linked Ffh, the fluorescence increase of Trp342 was less, ~50%, and there was no spectral shift. Fluorescence titrations revealed an affinity of wild-type Ffh for FtsY of ~60 nM (Fig. 6B). The affinity was not affected by the attachment of bimane from mBrB to Ffh (C231/377), whereas cross-linking of Ffh (C231/377) with dBrB diminished the affinity ~20-fold to 1.3 μM.
FIGURE 6.

Inhibition of FtsY binding to Ffh by bimane cross-linking. (A) Fluorescence emission spectra of FtsY (Trp342). 1, FtsY (Trp342), 0.5 μM; 2, + Ffh (C231/377) cross-linked with dBrB, 6 μM; 3, + Ffh (C231/377) reacted with mBrB, 3 μM. 4, Ffh (C231/377) crosslinked with dBrB in the absence of FtsY. (B) Fluorescence titrations. Increasing amounts of Ffh (○), Ffh (C231/377) reacted with mBrB (•), or Ffh (C231/377) cross-linked with dBrB (▿) were added to FtsY (Trp342) (0.01 μM).
DISCUSSION
The domain arrangement of bacterial Ffh is not known, and the crystal and cryo-EM structures solved thus far show different relative orientations of NG and M domains (Keenan et al. 1998; Rosendal et al. 2003; Halic et al. 2004). The present data show that in free Ffh the NG domain binds to the M domain with high affinity (60 nM), implying an extended interaction area between the two domains. In the arrangements reported for S. sulfolobus Ffh (Rosendal et al. 2003) and for canine SRP54 on the ribosome (Halic et al. 2004), or in the B/A domain arrangement in T. aquaticus Ffh (Keenan et al. 1998), the areas of interaction between NG and M domains are too small to account for the observed high affinity of the interaction. In contrast, in the A/A domain arrangement in the crystal structure of Ffh from T. aquaticus (Keenan et al. 1998), the M domain forms extensive contacts with both N and G domains, which would account for the observed affinity of the complex of NG and M domains. Furthermore, the observation that cysteine residues at positions 231 (G domain) and 377 (M domain) were readily cross-linked by dibromobimane strongly supports the A/A arrangement, as the two residues are within cross-linking distance (5 Å ) in the A/A configuration only, and far apart (> 50 Å ) in all other configurations (Keenan et al. 1998). Additional support for the A/A configuration of free Ffh comes from FRET measurements between fluorophores placed at various positions in the NG and M domains (I. Buskiewicz, unpubl.). Thus, bacterial Ffh in solution apparently is present in a compact conformation with NG and M domains tightly bound to one another. On the other hand, the domains appear to separate upon Ffh binding to 4.5S RNA (see below), and may separate further at some later stage in the SRP cycle.
The present results reveal that Ffh interacts with 4.5S RNA through both M and NG domains. While the M domain-RNA interaction previously was characterized at atomic resolution (Batey et al. 2000), not much is known about the interaction of the NG domain. The results from chemical footprinting suggest that the interaction involves the region around G27 of 4.5S RNA (present paper), explaining the part of the Ffh footprint that extends beyond the footprint due to binding of the M domain (Lentzen et al. 1996). Modeling 4.5S RNA into the crystal structure of free Ffh in the A/A configuration on the basis of the M domain-RNA crystal structure (Batey et al. 2000) indicates extensive steric clashes between the RNA and the NG domain. In order to avoid clashes and keep the NG domain in binding distance to the RNA, the NG and M domains have to be separated by a moderate movement of NG away from the M domain. In keeping with such a movement, Ffh in which the A/A configuration of G and M domains was fixed by introducing a bimane cross-link was no longer active in binding to 4.5S RNA, and, conversely, in Ffh bound to 4.5S RNA the cross-link was no longer formed. The conformation of Ffh in the complex with 4.5S RNA is not known. It is not excluded that it assumes the unfolded conformation observed for archaeal SRP54 and its complex with domain IV RNA (Rosendal et al. 2003), although results from FRET measurements appear to indicate a different conformation (I. Buskiewicz, unpubl.).
The isolated NG domain of Ffh binds to 4.5S RNA with a free energy of binding of ~8.5 kcal/mol (Table 2). The affinity of the isolated M domain is much higher, 14.3 kcal/mol. Were the RNA-binding affinities of the isolated domains simply additive, full-length Ffh would be expected to bind with a Kd close to 10−18 M. However, it binds with a Kd of ~50 pM (5 × 10−11 M), comparable to that of the M domain alone, indicating that part of the total binding energy, roughly equivalent to the free energy of NG domain binding, is consumed by a conformational change required for complex formation. The observation that cross-linking the M and G domains, i.e., fixing them in the A/A conformation, abrogates 4.5S RNA binding indicates that Ffh must change its conformation in order to accommodate the RNA. In fact, FRET measurements show that M and NG domains come apart upon binding the RNA by widening the cleft between M and G domain (I. Buskiewicz, unpubl.). The isolated domains bind strongly to one another, the binding energy amounting to 9.8 kcal/mol (Table 2). This value is close to the free-energy difference between full-length Ffh and the isolated NG and M domains. Assuming that in intact Ffh the two domains interact in the same way, this indicates that the major part of the conformational work during complex formation consists in the separation of the domains, although additional, less energy-consuming rearrangements are possible. In fact, fluorescence and chemical footprinting data indicate that Ffh binding induces a global conformational change in the RNA (Lentzen et al. 1994, 1996).
Cross-linking the G and M domains of Ffh also impaired complex formation with the SRP receptor, FtsY, lowering the binding affinity 20-fold. This can be explained on the basis of the crystal structures of free Ffh in the A/A configuration and of the complex of the NG domains of both Ffh and FtsY (Keenan et al. 1998; Egea et al. 2004; Focia et al. 2004). When the two structures are aligned on the G domain of Ffh, the M domain of Ffh and the NG domain of FtsY to a large part occupy the same space. Thus, in order to allow Ffh-FtsY complex formation, M and NG domains of Ffh must move away from each other, and the cross-link apparently interferes with that movement. Interestingly, the binding of 4.5S RNA to Ffh greatly accelerates complex formation with FtsY (Peluso et al. 2000). The present results suggest that the acceleration is due to the domain rearrangement in Ffh that is induced by binding of 4.5S RNA and makes the interaction interface on the NG domain of Ffh more easily accessible for FtsY. FtsY binding to SRP is impaired by base changes in the tetraloop and the adjacent stem of 4.5S RNA (Jagath et al. 2001), indicating that a conformational change of the RNA is involved in ternary complex formation as well.
In conclusion, the present results show that conformational changes of Ffh, notably a change of the arrangement of NG and M domains, are necessary for the binding of Ffh to 4.5S RNA to form SRP and for the binding of Ffh to the SRP receptor, FtsY. One may speculate that the modulation of these changes, e.g., by SRP binding to ribosomes displaying a signal sequence at the peptide exit, contributes to the interplay between SRP, FtsY, and trigger factor on the ribosome (Buskiewicz et al. 2004) and, thereby, to the timing of events during ribosome targeting to the membrane. Studying the dynamics of the interactions between translating ribosomes, SRP, and the SRP receptor will be one major objective of research in the future.
MATERIALS AND METHODS
Buffers and materials
Buffer A: 25 mM HEPES (pH 7.5), 70 mM ammonium acetate, 30 mM potassium acetate, 7 mM magnesium acetate. Alexa Fluor 546 maleimide, Alexa Fluor 555 maleimide and hydrazide, Oregon Green 488 maleimide, BODIPY-Fl iodoacetamide, and mono- and dibromobimane were from Molecular Probes. Ni-NTA agarose was from QIAGEN. All other chemicals were obtained from Sigma or Merck.
Preparation of 4.5S RNA
Full-size 4.5S RNA and 61mer was prepared by T7 RNA polymerase transcription. The respective templates were amplified by Pfu polymerase using two primers, one coding for the region of the T7-RNA polymerase promoter and a second coding for the end of the RNA, and used in transcription reaction without further purification. In vitro transcription was carried out in 5 mL of 40 mM Tris-HCl (pH 7.5), 1 mM spermidine, 10 mM DTT, 0.05% Tween-20, 8 mM MgCl2 containing 1 mM GMP, 2 mM of ATP, GTP, CTP, and UTP each, 5 μg/mL of amplified DNA template, 1600 units/mL of T7 RNA polymerase (Fermentas), and 500 U/mL of RNase inhibitor (Fermentas) for 4 h at 37°C. RNA was purified by ion exchange chromatography on MonoQ using a linear gradient of 0–1 M LiCl in 10 mM Bis-Tris (pH 6.0), 10 mM MgCl2, 1 mM EDTA. The 49-nt fragment of 4.5S RNA comprising nt 32–74 of 4.5S RNA and additional three base pairs at the ends of the molecule (Batey et al. 2000) was purchased from Dharmacon.
Plasmid construction, protein expression, and purification
The constructs coding for the NG (residues 1–295) or M (residues 297–453 or 357–453) domains of Ffh were prepared by PCR mutagenesis and cloned into pET-16 and pET-24 (Novagen), respectively. The NG domain construct contained an N-terminal tag of 10 histidines and nine additional amino acids; the M domain constructs contained a C-terminal six-histidine tag. To control the functional activity of the recombinant domains, the NG and M domains were also prepared from full-length Ffh by V8 digestion (Zheng and Gierasch 1997) and tested for 4.5S RNA binding by fluorescence titrations similar to those shown in Figure 4. No differences in binding to 4.5S RNA were found when recombinant domains and native domains produced by protease cleavage were compared (data not shown). The properties of the longer (residues 297–453) and shorter (residues 357–453) M domain constructs with respect to binding to 4.5S RNA and NG domain were identical (data not shown).
To construct Ffh mutants with single and double cysteine substitutions, the single cysteine residue present at position 406 of native Ffh was substituted with serine. Ffh mutants with double cysteine replacements at positions 231/377 or 17/344, and single cysteine replacements at positions 84, 231, and 377 were generated by PCR mutagenesis by the QuickChange method using Pfu polymerase (Promega). Mutations were generated in plasmid pET24-Ffh coding for Ffh extended by six histidines at the C terminus. Mutations were confirmed by DNA sequencing. Introducing mutations and fluorescence dyes at the above positions did not affect the binding of Ffh to 4.5S RNA, FtsY, or ribosomes (data not shown).
Ffh mutants were expressed in E. coli BL21 (DE3) pLysS cells and purified on Ni-NTA agarose under nondenaturing conditions. Ten g of cell pellet were resuspended in 40 mL of 20 mMHEPES (pH 7.5), 300 mM NaCl, 0.1 mM EDTA, 0.1 mM Pefablock SC, and 10 mM 2-mercaptoethanol, and the cells were opened by sonification (Branson Sonifier, duty cycle 50%, output 4, 3× for 5 min on ice). The extract was centrifuged at 20,000g for 30 min. The supernatant was incubated with 5 mL of Ni-NTA agarose equilibrated with 20 mM HEPES (pH 7.5), 300 mM KCl, and 10 mM 2-mercaptoethanol on ice for 60 min under shaking. The resin was washed with 150 mL of 20 mM HEPES (pH 7.5), 1 M KCl, 10 mM imidazole, and 10 mM 2-mercaptoethanol, and Ffh was eluted with 20 mL of 20mM HEPES (pH 7.5), 300mM KCl, 250mM imidazole, and 20% glycerol. The proteins were further purified by gel filtration on a Superdex 75 column (Pharmacia) in buffer A with 10% glycerol. Proteins were concentrated by ultrafiltration using 30-kDa (NG domain, full-size Ffh) or 5-kDa (M domain) membranes (Vivaspin) at 4°C. The purity of proteins was > 95% according to SDS-PAGE. FtsY (Trp342) was expressed in E. coli BL21 (DE3) pLysS cells and purified as described (Jagath et al. 2000).
Fluorescence labeling
Labeling of full-size Ffh and Ffh domains containing single cysteine residues with OG 488-maleimide, Alx 546-maleimide, or Bpy-iodoacetamide was carried out by incubating with a fivefold excess of dye over protein for 5 h on ice. Free dye was removed by gel filtration through Sephadex G-25.
For 3′-end labeling, 4.5S RNA (100 A260 units/mL) was oxidized by incubation in 0.1 M sodium acetate (pH 5.3), 5 mM KIO4 for 30 min at 0°C in the dark. The reaction was stopped by adding ethylene glycol to a concentration of 10 mM and incubating further for 5 min at 0°C. After ethanol precipitation, RNA was dissolved in 0.1 M sodium acetate (pH 5.3), and reacted with a threefold excess of Alx 555 hydrazide for 5 h at 20°C in the dark. Free dye was removed by phenol extraction, RNA precipitated with ethanol, and finally dissolved in 20 mM HEPES (pH 7.5), 7 mM magnesium acetate, 0.5 mM EDTA, and 100 mM NH4Cl. The extent of labeling was ~90%, based on absorption measurements. Labeled 4.5S RNA was separated from unlabeled by FPLC on MonoQ using a gradient from 0.5 to 0.8 M LiCl in 20 mM HEPES (pH 7.5), 7 mM magnesium acetate, 0.5 mM EDTA, and 100 mM NH4Cl. Fractions containing 4.5S RNA were pooled, and the RNA was precipitated with ethanol.
Dibromobimane cross-linking
The homobifunctional cross-linker 4,6-bis (bromomethyl) -3,7- dimethyl-1,5-diazabicyclo [3.3.0]octa-3,6-diene-2,8-dione (dibromobimane (dBrB) ) was used to introduce a cross-link between cysteines 231 and 377 in Ffh (C231/377). dBrB has two equivalent bromomethyl groups that can cross-link a thiol pair located within 3–6 Å of each other (Mornet et al. 1985). dBrB is nonfluorescent in solution but becomes fluorescent when both of its alkylating groups have reacted (Kosower and Kosower 1987; Ue 1987).
Double mutants Ffh (C231/377) and Ffh (C17/344) were treated with a 1.1-fold molar excess of dBrB. Samples were withdrawn over time, then purified from unbound cross-linker by gel filtration through Sephadex G-25, and bimane fluorescence was measured (excitation 390 nm; emission 470 nm). Single cysteine mutants Ffh (C231) or Ffh (C377) were labeled with a fivefold excess of mBrB and treated in the same way as the double mutant. Proteins were purified by FPLC on MonoQ using a gradient of 0.25–0.30 M KCl in 20 mM HEPES (pH 7.5) ; fractions containing bimane-labeled proteins were identified by monitoring fluorescence as above. Samples were analyzed by SDS-PAGE.
Gel shift and gel filtration assays
SRP complex formation wasmonitored by nondenaturing 7% PAGE (Jagath et al. 2001) in 50 mM Tris-acetate (pH 6.5), 75 mM ammonium acetate, 10 mM magnesium acetate, and 1 mM EDTA at 4°C. SRP was prepared by incubating Ffh (2 μM) with 4.5S RNA (1 μM) in buffer A in the presence of 0.2-mM GDPNP for 20 min at 20°C and loaded on the gel. GDPNP was present in the upper tank of the electrophoresis chamber as well; the running buffer was exchanged every 30 min. Gels were stained with ethidium bromide and with Coomassie.
Complex formation between 4.5S RNA and the NG domain of Ffh was monitored by gel filtration on Superdex 75 (two tandem columns Pharmacia HR30; 1.6 cm×30 cm). The samples were applied in 100 μL and the column developed with 20 mM HEPES (pH 7.5), 70 mM NH4Cl, 30 mM KCl, 7 mM MgCl2 at 0.5 mL/min. The position of RNA was monitored by absorbance at 260 nm in a flow-through monitor.
Chemical probing
To form the complex, 2 μM 4.5S RNA was incubated for 15 min at 25°C in buffer A in the presence of 6 μM full-length Ffh, 6 μM M domain, or 6 μM NG domain. Kethoxal modification of 4.5S RNA complexes was performed in 50 μL of buffer A containing 15 mg/mL kethoxal for 30 min at 25°C. The reaction was stopped by the addition of 24 μL of 250 mM potassium borate, 150 mM sodium acetate, pH 7.0, and the complexes were precipitated with ethanol. DMS modification was carried out in 50 μL of buffer A containing 0.5%DMS for 10 min at 25°C. The reaction was stopped by addition of 24 μL of 1 M Tris-HCl, pH 7.5, 1 M 2-mercaptoethanol, 100 mM EDTA. Pellets were dissolved in 200 μL of 0.3 M sodium acetate, pH 7.0, 0.5% SDS, 5mMEDTA, and RNA was purified by phenol extraction and ethanol precipitation. Two oligodeoxynucleotide primers complementary to nt 94–105 and 39–52 of 4.5S RNA were used for primer extension that was carried out as described (Lentzen et al. 1996).
Filter binding assay
Ffh binding to 4.5S RNA and variants was measured by nitrocellulose filtration in buffer A containing 1 mM DTT, 0.1 mg/mL bovine serum albumin, and 0.1 mg/mL poly (U). 5′-32P-labeled 4.5S RNA, 61mer, or 49mer (5 pM; 1.6×109 dpm/pmol) were incubated with varying amounts of Ffh for 10 min at 25°C. The mixtures were filtrated through nitrocellulose filters (0.45 μm; Sartorius), filters were washed with 4 mL of reaction buffer, and bound RNA was determined by liquid scintillation counting. Titration curves were evaluated by nonlinear fitting. At saturating protein concentrations, 85%–90% of RNA was found in complexes with full-length Ffh or the M domain; in the presence of saturating amounts of the NG domain, ~60% of the RNA was recovered in the complex.
Fluorescence titrations
Steady-state fluorescence was measured in a PTI QuantaMaster C-61/2000 T-Format scanning spectrofluorometer. All titrations were carried out in buffer A containing 10% (v/v) glycerol at 20°C. Binding of the NG domain to M domain was monitored by fluorescence change of Ffh-NG (OG84) or Ffh-M (Bpy406). Initial concentrations of labeled NG or M domains were 0.013–0.06 μM. Binding of 4.5S RNA and derivatives to the NG domain was monitored by the fluorescence change of Ffh-NG (OG84) or Alx 555 attached to the 3′ end of 4.5S RNA (4.5S RNA (Alx114) ). Excitation and emission wavelengths were 475 and 517 nm (OG) or 535 and 565 nm (Alx), respectively. The initial concentration of the Ffh-NG (OG84) was 0.052 μM in 4.5S RNA and 61mer titrations, and 0.49 μM in the 49mer titration. When Alx-labeled 4.5S RNA derivatives were used, the initial concentrations were 0.041 μM (full-length 4.5S RNA), 0.048 μM ( 61mer), and 0.51 μM (49mer). Binding of Ffh to FtsY was monitored by the fluorescence of the single Trp residue in FtsY (Trp342). After correction for dilution, the data were evaluated by nonlinear fitting to a quadratic equation describing ligand binding to one site using Table Curve software (Jandel Scientific).
FRET measurements
FRET was monitored by steady-state fluorescence or fluorescence lifetime measurements. Fluorescence of Bpy FL (excitation 490 nm, emission 530 nm) was measured on a PTI QuantaMaster C-61/2000 scanning spectrofluorometer. Time-domain lifetime measurements were carried out using a fluorescence lifetime spectrometer FluoTime 100 (PicoQuanT). Excitation pulses (440 nm, 10 MHz, 60 psec width) were generated by a laser diode system (PTD 800B with LDH PC 440, PicoQuanT). To exclude scattered light, a 500-nm liquid cut-off filter (CrO4 2−/Cr2O7 2−, 0.3 M, basic pH) was used in the emission channel. Fluorescence decay measurements were carried out in buffer A containing 10% (v/v) glycerol at room temperature.
Data analysis was performed using multiexponential fluorescence decay fitting software FluoFit v. 3.2.0 (PicoQuanT). For Ffh-M (Bpy406) alone or in the presence of unlabeled NG domain, single-exponential decay curves were obtained, yielding lifetimes of 5.2 and 4.7 nsec, respectively. In the presence of Ffh-NG (Alx84), a two-exponential decay curve with lifetimes of 1.38 and 0.46 nsec and respective amplitudes of 48% and 52% was obtained, yielding an average lifetime of 0.9 nsec, which was used for calculating the FRET efficiency.
Distances between donor and acceptor, R, were estimated from FRET efficiencies, E, as calculated from the decrease of the fluorescence intensity or the lifetime of the donor according to the following equations (Lakowicz 1999) :
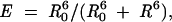 |
where R0 is the distance at which the FRET efficiency is 50%;
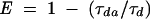 |
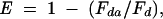 |
where τda, Fda, and τd, Fd are the lifetimes (τ) and fluorescence intensities (F) of the donor measured in the presence and in the absence of acceptor, respectively. The value for R0 was calculated using the equation:
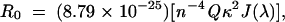 |
where n is the refractive index (n = 1.4 was used), Q the quantum yield of the donor in the absence of acceptor, κ2 the orientation factor, and J (λ) the overlap integral between donor emission and acceptor absorption. Quantum yields were determined by comparing the integrated emission spectra of protein-bound dyes to that of a known standard as described (Lakowicz 1999) ; the quantum yield of Bpy FL attached to Ffh was 0.90. Distance ranges were calculated from the ranges of κ2 values as determined from anisotropy measurements (Lakowicz et al. 1988) :
 |
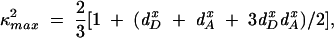 |
where di x = (ri /r0) 1/2 are depolarization factors, and ri and r0 are the measured limiting and fundamental anisotropies of donor (D) and acceptor (A), respectively.
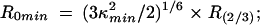 |
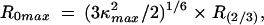 |
where R (2/3) is the distance calculated assuming κ2 = 2/3. The calculated R0 value was 48 ± 2 Å for Bpy FL and Alx 546 attached to protein.
Acknowledgments
Time-resolved measurements were performed in the Center for Fluorescence Spectroscopy, University of Maryland at Baltimore, School of Medicine, MD, USA. We thank Ignacy Gryczynski for help with the time-resolved fluorescence measurements and discussions, and Carmen Schillings, Astrid Böhm, SimoneMöbitz, and Petra Striebeck for expert technical assistance. The work was supported by the Deutsche Forschungsgemeinschaft, the Alfried Krupp von Bohlen und Halbach-Stiftung, and the Fonds der Chemischen Industrie.
Abbreviations
Alx, Alexa Fluor
Bpy, BODIPY Fl
FRET, fluorescence resonance energy transfer
mBrB and dBrB, mono- and dibromobimane
OG, Oregon Green
RNC, ribosome nascent-chain complex
SRP, signal recognition particle.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.7242305.
REFERENCES
- Batey, R.T. and Doudna, J.A. 2002. Structural and energetic analysis of metal ions essential to SRP signal recognition domain assembly. Biochemistry 41: 11703–11710. [DOI] [PubMed] [Google Scholar]
- Batey, R.T., Rambo, R.P., Lucast, L., Rha, B., and Doudna, J.A. 2000. Crystal structure of the ribonucleoprotein core of the signal recognition particle. Science 287: 1232–1239. [DOI] [PubMed] [Google Scholar]
- Batey, R.T., Sagar, M.B., and Doudna, J.A. 2001. Structural and energetic analysis of RNA recognition by a universally conserved protein from the signal recognition particle. J. Mol. Biol. 307: 229–246. [DOI] [PubMed] [Google Scholar]
- Bernstein, H.D., Poritz, M.A., Strub, K., Hoben, P.J., Brenner, S., and Walter, P. 1989. Model for signal sequence recognition from amino-acid sequence of 54K subunit of signal recognition particle. Nature 340: 482–486. [DOI] [PubMed] [Google Scholar]
- Buskiewicz, I., Deuerling, E., Gu, S.Q., Jockel, J., Rodnina, M.V., Bukau, B., and Wintermeyer, W. 2004. Trigger factor binds to ribosome-signal-recognition particle (SRP) complexes and is excluded by binding of the SRP receptor. Proc. Natl. Acad. Sci. 101: 7902–7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, F., Shan, S., Moustakas, D.T., Alber, F., Egea, P., Stroud, R.M., Walter, P., and Burlingame, A.L. 2004. Unraveling the interface of signal recognition particle and its receptor using chemical crosslinking and mass spectrometry. Proc. Natl. Acad. Sci. 101: 16454–16459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna, J.A. and Batey, R.T. 2004. Structural insights into the signal recognition particle. Annu. Rev. BioChem. 73: 539–557. [DOI] [PubMed] [Google Scholar]
- Egea, P.F., Shan, S.O., Napetschnig, J., Savage, D.F., Walter, P., and Stroud, R.M. 2004. Substrate twinning activates the signal recognition particle and its receptor. Nature 427: 215–221. [DOI] [PubMed] [Google Scholar]
- Focia, P.J., Shepotinovskaya, I.V., Seidler, J.A., and Freymann, D.M. 2004. Heterodimeric GTPase core of the SRP targeting complex. Science 303: 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freymann, D.M., Keenan, R.J., Stroud, R.M., and Walter, P. 1997. Structure of the conserved GTPase domain of the signal recognition particle. Nature 385: 361–364. [DOI] [PubMed] [Google Scholar]
- Gill, D.R. and Salmond, G.P. 1990. The identification of the Escherichia coli ftsY gene product: An unusual protein. Mol. Microbiol. 4: 575–583. [DOI] [PubMed] [Google Scholar]
- Halic, M., Becker, T., Pool, M.R., Spahn, C.M., Grassucci, R.A., Frank, J., and Beckmann, R. 2004. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature 427: 808–814. [DOI] [PubMed] [Google Scholar]
- Jagath, J.R., Rodnina, M.V., and Wintermeyer, W. 2000. Conformational changes in the bacterial SRP receptor FtsY upon binding of guanine nucleotides and SRP. J. Mol. Biol. 295: 745–753. [DOI] [PubMed] [Google Scholar]
- Jagath, J.R., Matassova, N.B., de Leeuw, E., Warnecke, J.M., Lentzen, G., Rodnina, M.V., Luirink, J., and Wintermeyer, W. 2001. Important role of the tetraloop region of 4.5S RNA in SRP binding to its receptor FtsY. RNA 7: 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan, R.J., Freymann, D.M., Walter, P., and Stroud, R.M. 1998. Crystal structure of the signal sequence binding subunit of the signal recognition particle. Cell 94: 181–191. [DOI] [PubMed] [Google Scholar]
- Keenan, R.J., Freymann, D.M., Stroud, R.M., and Walter, P. 2001. The signal recognition particle. Annu. Rev. BioChem. 70: 755–775. [DOI] [PubMed] [Google Scholar]
- Kosower, N.S. and Kosower, E.M. 1987. Thiol labeling with bromobimanes. Methods Enzymol. 143: 76–84. [DOI] [PubMed] [Google Scholar]
- Kusters, R., Lentzen, G., Eppens, E., van Geel, A., van der Weijden, C.C., Wintermeyer, W., and Luirink, J. 1995. The functioning of the SRP receptor FtsY in protein-targeting in E. coli is correlated with its ability to bind and hydrolyse GTP. FEBS Lett. 372: 253–258. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy, pp. 305–309. Kluwer Academic, New York.
- Lakowicz, J.R., Gryczynski, I., Cheung, H.C., Wang, C.K., Johnson, M.L., and Joshi, N. 1988. Distance distributions in proteins recovered by using frequency-domain fluorometry. Applications to troponin I and its complex with troponin C. Biochemistry 27: 9149–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentzen, G., Dobberstein, B., and Wintermeyer, W. 1994. Formation of SRP-like particle induces a conformational change in E. coli 4.5S RNA. FEBS Lett. 348: 233–238. [DOI] [PubMed] [Google Scholar]
- Lentzen, G., Moine, H., Ehresmann, C., Ehresmann, B., and Wintermeyer, W. 1996. Structure of 4.5S RNA in the signal recognition particle of Escherichia coli as studied by enzymatic and chemical probing. RNA 2: 244–253. [PMC free article] [PubMed] [Google Scholar]
- Montoya, G., Svensson, C., Luirink, J., and Sinning, I. 1997. Crystal structure of the NG domain from the signal-recognition particle receptor FtsY. Nature 385: 365–368. [DOI] [PubMed] [Google Scholar]
- Mornet, D., Ue, K., and Morales, M.F. 1985. Stabilization of a primary loop in myosin subfragment 1 with a fluorescent crosslinker. Proc. Natl. Acad. Sci. 82: 1658–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai, K., Oubridge, C., Kuglstatter, A., Menichelli, E., Isel, C., and Jovine, L. 2003. Structure, function and evolution of the signal recognition particle. EMBO J. 22: 3479–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluso, P., Herschlag, D., Nock, S., Freymann, D.M., Johnson, A.E., and Walter, P. 2000. Role of 4.5S RNA in assembly of the bacterial signal recognition particle with its receptor. Science 288: 1640–1643. [DOI] [PubMed] [Google Scholar]
- Poritz, M.A., Bernstein, H.D., Strub, K., Zopf, D., Wilhelm, H., and Walter, P. 1990. An E. coli ribonucleoprotein containing 4.5S RNA resembles mammalian signal recognition particle. Science 250: 1111–1117. [DOI] [PubMed] [Google Scholar]
- Römisch, K., Webb, J., Herz, J., Prehn, S., Frank, R., Vingron, M., and Dobberstein, B. 1989. Homology of 54K protein of signal-recognition particle, docking protein and two E. coli proteins with putative GTP-binding domains. Nature 340: 478–482. [DOI] [PubMed] [Google Scholar]
- Rosendal, K.R., Wild, K., Montoya, G., and Sinning, I. 2003. Crystal structure of the complete core of archaeal signal recognition particle and implications for interdomain communication. Proc. Natl. Acad. Sci. 100: 14701–14706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ue, K. 1987. Intramolecular cross-linking of myosin subfragment 1 with bimane. Biochemistry 26: 1889–1894. [DOI] [PubMed] [Google Scholar]
- Valent, Q.A., Kendall, D.A., High, S., Kusters, R., Oudega, B., and Luirink, J. 1995. Early events in preprotein recognition in E. coli: interaction of SRP and trigger factor with nascent polypeptides. EMBO J. 14: 5494–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, H., Luirink, J., and Tollervey, D. 1992. Evolutionary conserved nucleotides within the E. coli 4.5S RNA are required for association with P48 in vitro and for optimal function in vivo. Nucleic Acids Res. 20: 5919–5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, N. and Gierasch, L.M. 1997. Domain interactions in E. coli SRP: Stabilization of M domain by RNA is required for effective signal sequence modulation of NG domain. Mol. Cell 1: 79–87. [DOI] [PubMed] [Google Scholar]