

Abstract
The control of mRNA translation and degradation are critical for proper gene expression. A key regulator of both translation and degradation is Dhh1p, which is a DEAD-box protein, and functions both to repress translation and enhance decapping. We describe the crystal structure of the N- and C-terminal truncated Dhh1p (tDhh1p) determined at 2.1 Å resolution. This reveals that, like other DEAD-box proteins, tDhh1p contains two RecA-like domains, although with a unique arrangement. In contrast to eIF4A and mjDEAD, in which no motif interactions exist, in Dhh1p, motif V interacts with motif I and the Q-motif, thereby linking the two domains together. Electrostatic potential mapping combined with mutagenesis reveals that motifs I, V, and VI are involved in RNA binding. In addition, trypsin digestion of tDhh1p suggests that ATP binding enhances an RNA-induced conformational change. Interestingly, some mutations located in the conserved motifs and at the interface between the two Dhh1 domains confer dominant negative phenotypes in vivo and disrupt the conformational switch in vitro. This suggests that this conformational change is required in Dhh1 function and identifies key residues involved in that transition.
Keywords: protein crystallography, mRNA decay, mRNA decapping, RNA helicase, DEAD-box proteins, translation repression
INTRODUCTION
The regulation of mRNA decay plays an important role in control of gene expression. Studies performed primarily in yeast and human have identified two general mRNA decay pathways (Parker and Song 2004). The first step for both pathways is the shortening of the poly(A) tail. Following deadenylation, the deadenylated mRNAs can be degraded 3′ to 5′ by the cytoplasmic exosome. More commonly, the 5′ cap structure is removed by the Dcp1p/Dcp2p complex (decapping), allowing exonucleolytic 5′ to 3′ decay by Xrn1p. Decapping is a key step in the predominant mRNA decay pathway because it induces degradation of the mRNA, and thus it is subject to numerous control inputs (Coller and Parker 2004).
A key step in the decapping of eukaryotic mRNAs is the exiting of translation and the assembly of an mRNP state that targets the mRNA for decapping. Recently, Dhh1p, a member of the DEAD-box protein family, and Pat1p, an 88-kDa protein of unknown biochemical function, have been found to function in moving mRNAs from translation to the nontranslating pool of mRNAs, which are concentrated in specific subcellular sites of mRNA decapping and degradation referred to as P-bodies (J. Coller and R. Parker, in prep.). Dhh1p and Pat1p are also known to increase the rate of decapping of mRNAs (Bonnerot et al. 2000; Bouveret et al. 2000; Tharun et al. 2000; Coller et al. 2001; Fischer and Weis 2002). Thus, Dhh1p and Pat1p function as both activators of decapping and repressors of translation and are critical proteins modulating and connecting the transition from translation to mRNA degradation.
The Dhh1 protein is of particular interest because it is a member of a highly conserved class of DEAD-box proteins and has been implicated in a wide range of biological phenomena. For example, and consistent with the findings that Dhh1p functions as a translation repressor, Dhh1p homologs in Drosophila (Me31b), Caenorhabditis elegans (cgh-1), and Xenopus (Xp54) are involved in the storage of maternal mRNAs in a translationally repressed state (Ladomery et al. 1997; Minshall et al. 2001; Nakamura et al. 2001; Navarro et al. 2001; Minshall and Standart 2004). In addition, the Dhh1p homolog in Schizosaccharomyces pombe, Ste13, is required for meiosis (Maekawa et al. 1994). Finally, the Dhh1p homolog in mammals (RCK/p54) has been implicated in certain tumor types (Akao et al. 1995; Nakagawa et al. 1999). Interestingly, overexpression of RCK/p54 or Xp54 can rescue the loss of Dhh1p in yeast, demonstrating that the underlying biochemical function of Dhh1p is conserved (Tseng-Rogenski et al. 2003; Westmoreland et al. 2003). Thus, understanding Dhh1p function is likely to provide understanding in several biological contexts.
Proteins of the DEAD-box superfamily are widely distributed in nature and required for virtually all aspects of RNA metabolism. However, their precise contribution to most of these processes is not known (Tanner and Linder 2001; Rocak and Linder 2004). Sequence analysis has revealed that DEAD-box proteins contain eight to nine conserved sequence motifs including the eponymous DEAD motif and the newly identified Q-motif (Gorbalenya and Koonin 1993; de La Cruz et al. 1999; Hall and Matson 1999; Tanner et al. 2003). These motifs are believed to be involved in binding an NTP, generally ATP, and using the energy of ATP hydrolysis to unwind RNA duplexes and/or disrupt protein–RNA or protein–protein interactions (Jankowsky et al. 2001; Cordin et al. 2004; Rocak and Linder 2004). Consistent with this notion, the DEAD-box protein Ded1p has been shown to catalyze protein displacement independently of its ability to unwind RNA duplex, implying that DEAD-box proteins can function on a wide range of ribonucleoprotein substrates in addition to their duplex unwinding activity (Fairman et al. 2004).
Many of the DEAD-box proteins that have been studied in vitro have been shown to be RNA-dependent ATPases, contributing to the general model that these proteins function to rearrange RNA or RNP structure (Rocak and Linder 2004). Although most DEAD-box proteins are thought to possess ATP-dependent RNA helicase activity, it can be difficult to demonstrate a helicase activity for many of the DEAD-box proteins, presumably due to stringent substrate specificities or missing cofactors/helper proteins. Consistent with this observation, the helicase activity of Dhh1p has not been demonstrated in vitro so far. However, given its strong similarity to members of this protein family, and the fact that mutations in conserved motifs disrupt function in vivo (see below), it is highly likely that Dhh1p’s mode of function is similar to other DEAD-box proteins and involves coupling ATP hydrolysis to a conformational change in an RNA or RNP.
Despite the large number of DEAD-box proteins from bacteria to human and their universal roles in so many processes, high-resolution structures remain limited to three DEAD proteins, yeast eIF4A, a prototype minimal DEAD-box protein (Benz et al. 1999; Johnson and McKay 1999; Caruthers et al. 2000); mjDEAD from Methanococcus janaschii (Story et al. 2001); and the N-terminal domain of BstDEAD from Bacillus stearothermophilus (Carmel and Matthews 2004). Recently, the crystal structure of UAP56, a DExD/H-box protein involved in pre-mRNA splicing and mRNA export, revealed a unique spatial arrangement of the two conserved helicase domains (Shi et al. 2004). Here, we described the crystal structure of Dhh1p with truncations of the N- and C-terminal extensions from Saccharomyces cerevisiae. The structure shows that, like other DEAD-box proteins, Dhh1p contains two α/β domains, each with a RecA-like topology, although with a quite different domain arrangement. Structural comparison with other DEAD-box proteins showed that the unique domain arrangement leads to the novel motif interactions clustered in the cleft between two domains. Electrostatic potential mapping and the analysis of mutant proteins revealed a prominent channel that is involved in RNA binding. This putative RNA-binding surface spans the N-terminal and C-terminal domain and appears to be enlarged by a “closed” structural arrangement of Dhh1p. Interestingly, some mutations located in the conserved motifs and at the interface between the two Dhh1 domains confer dominant negative phenotypes in vivo and disrupt the conformational switch in vitro. This suggests that this conformational change is required in Dhh1 function and identifies key residues involved in that transition.
RESULTS AND DISCUSSION
Structure determination
Full-length Dhh1p from Saccharomyces cerevisiae was initially expressed and purified for crystallization. However, purified full-length Dhh1p underwent substantial proteolysis. Compared with its homologs in other organisms, and related DEAD-box proteins, Dhh1p contains extensions of ~50 amino acids at both its N and C terminals, which flank the conserved core domain (Fig. 1). Given these extensions, we expressed and purified to homogeneity an N- and C-terminal truncated yeast Dhh1p, which retains the entire helicase domain (residues 30–425; tDhh1p). tDhh1p was crystallized in space group P21 with one molecule per asymmetric unit. For phasing, multiwavelength anomalous dispersion (MAD) data were measured from a bromide (Br) derivative that yielded an easily interpretable electron density map at a resolution of 2.1 Å, allowing 98% of the model to be built automatically. Two regions of the polypeptide chain, residues 30–45 and residues 423–425, are not visible in the electron density map and are assumed to be disordered. Statistics of structure determination and refinement are summarized in Table 1 (see Materials and Methods).
FIGURE 1.

Sequence alignment of S. cerevisiae Dhh1p, Homo sapiens Rck/p54, S. cerevisiae eIF4A, and M. janaschii mjDEAD. The secondary structures of tDhh1p are shown. Invariant residues are shown in white letters, similar residues in red, and others in black. Residues mutated for in vivo and in vitro assays are indicated by *.
TABLE 1.
Data collection, phase determination, and refinement statistics
| Br MAD data | |||
| λ1 (peak) | λ2 (edge) | λ3 (remote) | |
| Wavelength (Å) | 0.9195 | 0.9198 | 0.9134 |
| Resolution (Å) | 2.1 | 2.1 | 2.1 |
| Unique reflections (N) | 23,845 | 23,855 | 23,816 |
| Completeness (%)a | 99.5 (98.7) | 99.5 (99.2) | 99.2 (98.5) |
| Redundancy | 3.6 | 3.7 | 3.6 |
| I/σ (I) | 12.8 (6.8) | 12.8 (7.2) | 11.6 (5.5) |
| Rmergea (%) | 4.0 (10.6) | 3.8 (9.3) | 4.4 (12.8) |
| Number of sites | 9 | ||
| Figure of merit | |||
| Before density modification | 0.45 | ||
| After density modification | 0.82 |
| Refinement statistics | |
| Values in parentheses indicate the specific values in the highest resolution shell (2.25–2.1 Å). | |
| aRmerge = ∑| Ij-<I>| ∑Ij, where Ij is the intensity of an individual reflection, and <I> is the average intensity of that reflection. | |
| bRcryst = ∑ || Fo| – |Fc||/ sum; |Fc|, where Fo denotes the observed structure factor amplitude and Fc denotes the structure factor amplitude calculated from the model. | |
| cRfree is as for Rcryst but calculated with 5.0% of randomly chosen reflections omitted from the refinement. | |
| Resolution range (Å) | 20–2.1 |
| Reflection used | 22,719 |
| Rcrystb (%) | 20.0 (20.3) |
| Rfreec (%) | 23.4 (23.6) |
| Nonhydrogen atoms | |
| Protein (N) | 3009 |
| Waters (N) | 253 |
| R.m.s deviations | |
| Bond length (Å) | 0.009 |
| Bond angle (°) | 1.09 |
Overall structure description
The polypeptide chain of tDhh1p is folded into two α/β domains, each with a RecA-like topology as observed in other DEAD-box proteins (Fig. 2). The N-terminal domain (residues 46–247) is composed of a pararllel seven-stranded β sheet flanked by four helices (α1–4) on one side and five helices on the other (α5–9). The N-terminal domain is connected to the C-terminal domain by a short stretch of peptide (residues 247–253). The C-terminal domain (residues 253–422) is also a parallel α/β structure but with three helices (α10–12) on one side and two helices (α13–14) on the other, surrounding the central seven-stranded β sheet. Consistent with their basic RecA-like topology, the N- and C-terminal domains are similar to each other. Superposition of these two domains gives an r.m.s.d. for 81 equivalent Cα atoms of 1.5 Å. The N-terminal domain contains sequence motifs associated with ATP binding and hydrolysis, while the C-terminal domain contains motifs associated with RNA binding. The N-terminal domain interacts with the C-terminal domain through the previously identified conserved motifs and helix α12 from the C-terminal domain, thereby creating a large channel between two domains (Fig. 2). Such a marked structural feature has not been seen in other DEAD-box structures and may have important functional implications for Dhh1p (see below).
FIGURE 2.

Structure of tDhh1p. (A) Ribbon diagram of tDhh1p with the N- and C-terminal domains colored in pale blue and light gray, respectively. The coloring scheme for conserved motifs is as follows: Q-motif in red, I in green, Ia in yellow, Ib in purple, II in orange, III in blue, IV in cyan, the α-12 helix in green-yellow, V in navy, and VI in magenta. The secondary structural elements are labeled. (B) The side view of tDhh1p. The molecule is rotated 90° along a vertical axis relative to the view in A. Figures 2, 3, and 4 were generated using MOLSCRIPT (Kraulis 1991).
Structural comparison
Comparison of the tDhh1p structure with the structures of eIF4A, mjDEAD, and UAP56 reveals three significant points. First, as pointed out earlier, the common feature of the DEAD-box proteins is that the conserved region of these proteins is composed of only two α/β domains (Caruthers et al. 2000; Story et al. 2001). Comparison of tDhh1p with eIF4A and mjDEAD shows that the individual domain structures are similar among these proteins. Superposition of the N-terminal domain of tDhh1p with those of eIF4A, mjDEAD, and UAP56 gives an r.m.s.d. of 1.0 Å, 1.3 Å, and 1.1 Å, respectively. When the C-terminal domain is superimposed, the r.m.s.d. values for tDhh1p versus eIF4A, tDhh1p versus mjDEAD, and tDhh1p versus UAP56 are 1.3 Å, 1.2 Å, and 1.1 Å, respectively. Thus, the overall structure of each domain is conserved between different DEAD-box proteins.
A second point is that the relative orientation of the N- and C-terminal domains in these DEAD-box proteins is significantly different. Yeast eIF4A is a “dumbbell” structure, having two domains connected to each other by an extended linker. mjDEAD is a compact molecule with two domains connected by a short linker. tDhh1p and UAP56 are more compact molecules with the two domains connected to each other by extensive interactions (see below). The orientation of the N- and C-terminal domains of Dhh1p is strikingly different in comparison to the other SF2 helicase structures. When the two N-terminal domains are superimposed, the C-terminal domain of eIF4A is rotated 50° with respect to the C-terminal domain of tDhh1p (Fig. 3A), whereas the corresponding rotation involving mjDEAD is 30° (Fig. 3B). Similarly, when the N-terminal domain of tDhh1p is superimposed with that of UAP56, the orientation of the C-terminal domain differs by 40° (data not shown).
FIGURE 3.

Comparison of tDhh1p with eIF4A and mjDEAD. (A) Superposition of the N-terminal domain of tDhh1p with that of eIF4A. tDhh1p is shown in light gray and eIF4A in green. The view of tDhh1p is as in Figure 2A. (B) Superposition of the N-terminal domain of tDhh1p with that of mjDEAD. mjDEAD is shown in magenta. Others are the same as in A. (C) Stereo view of the superposition of the N-terminal domain of tDhh1p with those of eIF4A and mjDEAD. tDhh1p is shown in light gray, eIF4A in light green, and mjDEAD in light pink. The view and the coloring scheme for motifs are as in Figure 2. (D) Stereo view of the superposition of the C-terminal domain of tDhh1p with those of eIF4A and mjDEAD. The color coding is as in C and Figure 2.
Finally, in addition to the differences in their relative domain orientations, there are local structural changes as well. In eIF4A, the N terminus forms a short antiparallel β strand that interacts with β1 and extends the seven-stranded β sheet to eight strands. The corresponding region (residues 30–46) in tDhh1p is disordered. mjDEAD lacks a C-terminal region (residues 410–419 in Dhh1p), which has different conformations in tDhh1p and eIF4A. Helix α5 and residues 199–205, which is a region immediately downstream of the DEAD motif, showed large positional shifts between tDhh1p and eIF4A and mjDEAD. The conformation of the loop connecting the α10 helix and β-9 strand in the C-terminal domain of tDhh1p has changed dramatically compared with that of the corresponding regions in both eIF4A and mjDEAD. Residues 90–95, 340–352, and 371–382, which respectively correspond to motifs I, V, and VI, showed larger conformational changes that may have functional implications (see below).
Location of the conserved sequence motifs
Dhh1p contains nine conserved sequence motifs as observed in other DEAD-box proteins (Figs. 1, 2). Structural and functional studies of several DNA and RNA helicases have suggested the general roles for some of these motifs (Tanner and Linder 2001; Caruthers and McKay 2002). Motif I, or the Walker A motif (Walker et al. 1982), forms a loop structure (P loop) that accommodates the phosphate groups of ATP. In tDhh1p structure, the P loop adopts an “open” conformation (Fig. 3C) similar to that observed in mjDEAD, but opposed to the “closed conformation” in the structures of eIF4A and BstDEAD without bound ligand (Johnson and McKay 1999; Story et al. 2001; Carmel and Matthews 2004). Such an open conformation of motif I would facilitate ATP binding. Motifs Ia and Ib have been suggested to be involved in binding of RNA substrates (Caruthers and McKay 2002), but their precise functional roles are not fully understood. The conformations of these two motifs in Dhh1p are very similar to those observed in eIF4A and mjDEAD (Fig. 3C).
Motif II is also called the Walker B, or DEAD motif (Walker et al. 1982). Structural, mutational, and biochemical studies indicated that motif II forms interactions with the β- and γ-phosphates of ATP through Mg2+ and is required for ATP hydrolysis (Schmid and Linder 1991; Pause and Sonenberg 1992, 1993; Benz et al. 1999; Caruthers and McKay 2002). In tDhh1p, the conformation of the DEAD motif is very similar to that in eIF4A or mjDEAD. However, a region immediately downstream of the DEAD motif, residues 199–205, shows large conformational changes with a positional shift of up to 8 Å for the equivalent Cα atom between tDhh1p, and eIF4A and mjDEAD (Fig. 3C). Whether this difference is idiosyncratic to Dhh1p or reflects a different basic conformation of the DEAD-box protein fold remains to be seen.
Motifs III and IV in tDhh1p are very similar structurally to those in mjDEAD and eIF4A (Fig. 3C,D). Motifs III and VI are proposed to link ATP binding and hydrolysis with conformational changes required for helicase activity (Caruthers and McKay 2002). Similar to mjDEAD and eIF4A, the SAT motif in tDhh1p interacts with the final aspartic acid of the DEAD motif, providing a conserved connection between motifs II and III. Motif IV is involved in single-strand DNA (ssDNA) binding in other helicases including PcrA, Rep, and hepatitis C virus (HCV) (Kim et al. 1998; Caruthers and McKay 2002). It is suggested that two Arg residues in motif IV of eIF4A might interact with single-strand RNA (Caruthers and McKay 2002).
Motif V in tDhh1p adopts a strikingly different conformation from those in mjDEAD and eIF4A (Fig. 3D). Superposition of motif V (residues 340–352) with those of mjDEAD and eIF4A gives an r.m.s.d. for 13 Cα atoms of 3.0 Å and 3.2 Å, respectively. The conformation of motif V is similar to that of UAP56 without bound ADP, although motif V is disordered in the structure of UAP56 with bound ADP (Shi et al. 2004). The conformation of motif V in tDhh1p allows it to interact with motif I and the Q-motif, thus linking the N- and C-terminal domains together (see below). This observation is similar to the findings that motif V in the DNA helicases PcrA and Rep bridges the contacts between domains 1A and 2A through interactions with motif II (Korolev et al. 1997; Velankar et al. 1999). This raises the possibility that motif V might play a general role in this class of proteins in bringing the two domains together. Interestingly, motif V also interacts with the DNA substrate in PcrA and Rep, suggesting this motif can have multiple roles and might couple substrate binding to domain interactions.
Structural studies on DNA helicases indicated that motif VI is involved in contacts with the γ phosphate of ATP (Caruthers and McKay 2002). Mutational and biochemical studies suggested that motif VI in eIF4A is required for RNA binding and ATP hydrolysis (Pause et al. 1993). In tDhh1p, the conformation of motif VI is dramatically different from that in both mjDEAD and UAP56, while the same region is disordered in eIF4A (Fig. 3D). In mjDEAD, the Q-motif, motifs I and II in the N-terminal domain, and motif VI in the C-terminal domain, form part of the ATP-binding site between these domains (Story et al. 2001). The second Arg residue in motif VI (HRxGRxGR) of DEAD-box proteins is proposed to perform a similar function as the “arginine finger” in G-proteins by sensing the γ-phosphate of the ATP (Caruthers and McKay 2002). Although it seems unlikely that motif VI of tDhh1p could make contact with the γ phosphate of ATP, it does interact with motif V (see below). Such interaction may push motif V forward for contacting motifs in the N-terminal domain.
The Q-motif is a newly identified motif and has been suggested to be involved in regulation of ATP binding by modulating the state of the P loop (Tanner et al. 2003). More recently, it has been shown that the Q-motif regulated RNA binding and helicase activity (Cordin et al. 2004). In tDhh1p, the Q-motif interacts extensively with the α12 helix and motif V from the C terminal, thus contributing part of the interactions between the N- and C-terminal domains (see below). Furthermore, structural comparison shows that the conformation of this motif in tDhh1p is similar to that in UAP56 (data not shown) but different from those in mjDEAD and eIF4A, with deviations of the equivalent Cα atoms being as large as 2.0 Å (Fig. 3C).
Interactions of the conserved motifs
An important issue in understanding the DEAD-box proteins is to understand how the conserved motifs function to couple ATP hydrolysis to perform work. Thus, a goal in gaining insight into helicase function is to understand the structural and dynamic interactions of the conserved motifs, particularly between the two different domains of the proteins. The possibility of dynamic interactions is suggested by multiple lines of evidence indicating that at least some of the DEAD-box proteins undergo conformational changes in response to substrate and/or ATP binding. For example, ATP and RNA binding have been found to change the protease digestion pattern in eIF4A and Dhh1p (see below; Lorsch and Herschlag 1998b). Moreover, the structures of PcrA and Rep (Korolev et al. 1997; Velankar et al. 1999) showed that these helicases contain four domains including two α/β domains (domains 1A and 2A), which are tightly packed against each other and correspond to the N- and C-terminal domains observed in DEAD-box proteins. Furthermore, the cleft between the two α/β domains closes upon ATP/DNA binding. Interestingly, the canonical helicase motifs in PcrA and Rep are close to each other and are clustered in the cleft between the two α/β domains, with motif V of domain 2A interacting with motif II in domain 1A, suggesting that the conserved motifs modulate the interaction between the two domains (Korolev et al. 1997; Velankar et al. 1999). Consistent with this possibility, in the structure of the related HCV helicase, all eight conserved motifs are found in a cleft between the two α/β domains (Kim et al. 1998).
In the eIF4A and mjDEAD structures, there are few interactions between the conserved motifs within different protein domains. The distant locations of the N- and C-terminal domains in the crystal structure of eIF4A prevent interactions between the two domains (Caruthers et al. 2000). However, it has been suggested that ATP and/or RNA binding would induce a dramatic conformational change in eIF4A, bringing the two domains as well as the conserved motifs closer together (Caruthers et al. 2000). Although mjDEAD has a compact two-domain structure, the two domains are in an open conformation and no motif interactions have been observed (Story et al. 2001).
In tDhh1p, the N- and C-terminal domains share an extensive interface similar to the two α/β domains in PcrA and Rep (Korolev et al. 1997; Velankar et al. 1999). The interaction between the N- and C-terminal domains in tDhh1p buries a pairwise accessible surface area of 1601.5 Å2. Close inspection of the tDhh1p structure shows several of the conserved motifs are clustered in the cleft between the N- and C-terminal domains, with motif V and the α12 helix in the C-terminal domain and motif I and the Q-motif in the N-terminal domain mediating the interdomain interactions (Fig. 1). Specifically, as shown in Figure 4, the backbone carbonyl oxygen atoms of both Leu342 and Thr344 in motif V are hydrogen-bonded to the NZ atom of Lys91 in motif I, while the side chain of Arg345 in motif V stacks against that of Arg89 in motif I. The side chain and the main-chain carbonyl oxygen of Leu343 in motif V make Van der Waals (VDW) contacts with the main-chain carbonyl oxygen of Gly93 and the side chain of Thr94 in motif I, respectively. Furthermore, the side chains of Leu343 and Thr344 make extensive hydrophobic interactions with Pro71 in the Q-motif. Helix α12 is situated in close proximity with the Q-motif and the putative ATP-binding pocket, thereby interacting with both the Q-motif and motif I via multiple contacts. Specifically, residues Arg330 and Phe326 in the α12 helix form a salt bridge and stack against Glu74 and Pro71 in the Q-motif, respectively, while His327 in the α12 helix stacks against the methylene group of Lys68 in the Q-motif. Moreover, residues Gln319, Gln320, Asn323, and His327 in helix α12 contact residues Gly95, Phe66, Ser70, and Pro69 in the Q-motif, respectively, through multiple VDW interactions. Interestingly, helix α12 also contacts the Q-motif in the structure of UAP56, suggesting this interaction will be common to multiple members of the SF2 family (Shi et al. 2004).
FIGURE 4.

Stereo view of interactions of the conserved motifs. Motifs Q, I, V, and VI and the α12 helix are colored as in Figure 2. Residues involved in the interactions are labeled and shown in stick models. Hydrogen bonds are shown as broken lines.
Apart from the interactions with motifs I and Q, motif V also interacts with motif VI and the α12 helix. For example, the main-chain carbonyl oxygen of Ile347 is hydrogen-bonded to the NH2 group of Arg376 in motif VI, while Gln350 interacts with R376 in motif VI. Inspection of the interaction between motif V and the α12 helix revealed that Ile349 makes extensive hydrophobic interactions with Phe326 and Phe329 of the α12 helix, and Asp341 forms a salt bridge with residue Arg322 in the α12 helix. An additional contact is made by the side chain of residue Leu343 of motif V stacking against the methylene group of Arg322 in helix α12.
The interactions between the conserved motifs are unique features of tDhh1p and correlate with the novel domain arrangement and conformational changes in motifs I, V, and VI in the tDhh1p structure. This observation is similar to the findings that motif V in the DNA helicases PcrA and Rep bridges the contacts between domains 1A and 2A through interactions with motif II (Korolev et al. 1997; Velenkar et al. 1999). Similar but weak interactions between motifs V and I have been observed in UAP56 (Shi et al. 2004). This raises the possibility that motif V of the SF2 protein family might play a general role in bringing the ATPase domain and the RNA-binding domain into close proximity. This would facilitate the coupling of ATP binding and hydrolysis with the RNA-specific functions. However, it is currently unclear if the unique features of the tDhh1p structure represent specific aspects of Dhh1p function, or represent a more general conformation that many DEAD-box proteins adopt at some stage of their conformational changes. This will be resolved as more structures of other DEAD-box proteins, and Dhh1p in different conformations, are reported.
Identification of residues required for RNA binding
DEAD-box proteins are generally capable of RNA binding. To identify potential surfaces for RNA binding on Dhh1p, we mapped the electrostatic potential on the molecular surface of tDhh1p. This analysis revealed a prominent positively charged patch on the molecular surface of tDhh1p (Fig. 5A). Part of the patch occupies the cleft between the N- and C-terminal domains, with the rest of the patch running across the C-terminal domain. Motifs I, V, and VI contribute amino acid side chains forming this patch. Strikingly, the residues that form this positive surface are highly conserved among Dhh1p homologs, suggesting that this region might represent a conserved RNA-binding pocket in these family members. To test whether this region is involved in RNA binding and is functionally important, we mutated several of the amino acids in this region to alanines (Fig. 5B) and examined their effect on RNA binding in vitro and protein function in vivo. Although Dhh1p has been reported to affect decapping directly in vitro (Fischer and Weis 2002), we did not examine the effects of these mutants on decapping in vitro, because we have been unable to document any direct role of Dhh1p on decapping in vitro despite repeated attempts (C. Decker and R. Parker, unpubl.).
FIGURE 5.

In vitro RNA-binding assay. (A) Solvent accessible and electrostatic potential of tDhh1p viewed as in Figure 2A showing a prominent RNA-binding channel. Residues located in the putative RNA-binding channel and Asp195 and Glu196 are labeled. (B) Schematic diagram showing the position of the various point mutants in Dhh1p. (C) Chart of the two RNA substrates: dsRNA-I and ss/dsRNA-II. (D) Effects of mutations of tDhh1p on RNA binding in vitro. BSA serves as a negative control. (E) Quantitation of RNA binding to wild-type and mutant tDhh1p proteins using three different RNA substrates. The percentage of RNA-binding activity of each mutant relative to that of wild-type Dhh1p is shown.
To examine the RNA binding in vitro, the mutant tDhh1p proteins were expressed in Escherichia coli and purified. The yield for each mutant protein was comparable to that of the wild-type protein. Moreover, CD spectroscopy gave the same spectra for wild type and mutant tDhh1p proteins. These observations indicate that these mutations had no significant effect on the general folding or stability of the Dhh1 protein. Wild-type and mutant tDhh1p proteins were examined for their binding, in the presence or absence of ATP, to a single-stranded poly(U) RNA oligo (12mer), a hairpin double-stranded RNA (dsRNA-I), and a hairpin dsRNA with the 11-base 5′-single-stranded tail (ss/dsRNA-II) (Fig. 5C). In the absence of ATP, wild-type tDhh1p binds to all three RNA substrates, and the addition of ATP to the RNA-binding buffer had no effect on RNA binding (data not shown). This observation implies that RNA binding to Dhh1p is not stimulated by ATP binding, at least under these biochemical conditions. The ability of Dhh1p to bind RNA independent of ATP is similar to the interaction of eIF4A with RNA, which can also occur independent of ATP, although ATP can enhance the eIF4A interaction with RNA (Lorsch and Herschlag 1998a).
An important result is that several changes in the putative RNA-binding channel significantly reduced RNA binding compared with the wild-type protein. Specifically, Ala mutations of Arg370 in motif VI, and Arg89 or Lys91 or both in motif I, drastically reduced binding to all three RNA substrates (Fig. 5D,E). Mutation of these residues to Ala would weaken the positively charged potential on the molecular surface of tDhh1p, thereby potentially reducing the binding of RNA. We also observed that mutations of Asp195 and Glu196 in the DEAD motif to Ala (D195A and E196A) increased RNA binding compared with the wild-type protein (Fig. 5D,E). These residues are located in the vicinity of the RNA-binding channel, and mutation of these negatively charged residues to Ala might thereby facilitate the RNA binding by reducing charge–charge repulsion. These results demonstrate that this region of Dhh1p is required for RNA binding. The simplest interpretation is that this positively charged region directly interacts with the RNA substrates. However, until a co-crystal of Dhh1p with RNA bound is analyzed, we cannot rule out the possibility that these mutations indirectly affect RNA binding by influencing conformational changes in the protein.
We also examined how mutations in motif V affected RNA binding. Individual substitutions of Arg345 and Gly346 by Ala (R345A and G346A) did not lead to substantial defects in RNA binding (Fig. 5D,E). However, altering Arg345 and Gly346 together (R345A G346A) dramatically decreased RNA binding (Fig. 5D,E). These results indicate that defects in motif V can affect RNA binding. Because multiple changes are required, we speculate that this effect may be caused by the combination of changes in the RNA-binding channel and conformational changes in Dhh1p.
To determine how these biochemical defects in tDhh1p are related to in vivo function, the tDhh1p variant proteins were examined in yeast for their effects on cell growth and mRNA turnover in both a wild-type and dhh1Δ strain. In these experiments, the variant Dhh1p are expressed in yeast under the control of their own promoter from single-copy plasmids. We observed that single mutants of D195A, E196A, and R370A and double mutants of R345A and G346A (R345A G346A), and R89A and K91A (R89A K91A), which all altered RNA binding in vitro (Fig. 5D,E), failed to complement the temperature sensitivity of the dhh1Δ strain (Fig. 6A). For R345A G346A double mutant, the loss of the ability to complement a dhh1Δ mutant was not specific to either Arg345 or Gly346, as similar results were seen with both R345A and G346A (Fig. 6A).
FIGURE 6.

In vivo mutagenesis analysis. (A) The growth of yeast cells expressing various Dhh1p mutants in either a dhh1Δ strain or wild-type (WT) background at 30°C or 37°C. (B) The effect of the various Dhh1p mutants on the steady-state accumulation of the EDC1 mRNA in a dhh1Δ strain or WT background. (C) Relative abundance of the EDC1 mRNA for the various Dhh1p mutants to that of the wild-type Dhh1p in a dhh1Δ strain or WT background.
The growth phenotypes of the Dhh1 mutant in vivo directly correlated with defects in mRNA turnover, which were determined by examining the levels of the EDC1 mRNA, which is sensitive to the loss of Dhh1p (Muhlrad and Parker 2005). Here we observed that mutants of D195A, E196A, and R370A and double mutants of R345A and G346A (R345A G346A), and R89A and K91A (R89A K91A), led to the accumulation of EDC1 mRNA in a dhh1Δ strain (Fig. 6B,C). Interestingly, individual substitution of Arg89 and Lys91 to Ala seems to be partially defective in both RNA binding and growth phenotype, and double mutation of these two residues (R89A K91A) is more extreme due to the additive effects of the singles (Figs. 5, 6). In combination, these results indicated a strong correlation between the in vitro defects in RNA binding and function of Dhh1p in vivo. The simplest interpretation of these observations is that RNA binding is required for Dhh1p function.
We also observed that the double mutants of R345A and G346A (R345A G346A), and R89A and K91A (R89A K91A), caused a dominant negative phenotype and caused death of wild-type cells at 37°C (Fig. 6A). For the combination of R345A and G346A, the dominant negative effects are solely due to G346A, as R345A did not by itself show a dominant negative phenotype. The dominant negative phenotype of R89A and K91A requires both mutations, as single mutants of both R89A and K91A have no effect on Dhh1p activity in wild-type cells, both by growth and RNA decay analysis (Fig. 6). Consistent with the dominant negative growth phenotype, double mutants of R345A and G346A (R345A G346A), and R89A and K91A (R89A K91A), and G346A also led to an increase in EDC1 mRNA levels even in a wild-type strain (Fig. 6B,C). Since G346A confers a dominant negative phenotype, yet still binds RNA, and mutants R89A, K91A, and R370A fail to bind RNA and are not dominant negative, the dominant negative phenotype is not simply due to the inability to bind RNA. Thus, we interpret these results to indicate that the alterations seen in G346A and R89A K91A double mutant alter the function of Dhh1p by locking the protein in a dominant negative conformation. Both of these changes would affect the structure of the interface between the two domains in tDhh1p. For example, the presence of an alanine at position 346 would be expected to interfere with motif VI moving close to and interacting with motif V. This steric interference would be expected to prevent Dhh1p from adopting the closed conformation seen in the structure. Similarly, substitutions of both Arg89 and Lys91 for Ala would reduce the specific contacts between the ATPase and RNA-binding domains of Dhh1p.
Conformational changes in Dhh1p
DEAD-box proteins are proposed to use the energy from ATP hydrolysis to rearrange inter- or intramolecular RNA structures or disrupt RNA–protein interactions (Tanner and Linder 2001; Rocak and Linder 2004). In tDhh1p, the interactions of motif V with motif I, the Q-motif, links the C-terminal domain to the N-terminal domain, thereby providing a mechanism for coupling ATP binding and hydrolysis with RNA-specific activities, probably through conformational changes. To test whether Dhh1p undergoes conformational changes upon ATP and/or RNA binding, we analyzed the trypsin cleavage pattern of tDhh1p in the presence of ATP and /or RNA.
Evidence that Dhh1p undergoes a conformational change was that the cleavage of tDhh1p by trypsin was slowed in the presence of ATP, whereas RNA alone had little, if any, effect on the cleavage of tDhh1p (Fig. 7). This implies that tDhh1p probably undergoes a significant conformational change upon ATP binding rather than RNA binding. In addition, ADP and AMP–PNP failed to confer trypsin resistance (Fig. 7). In the presence of both ATP and RNA, Dhh1p conferred strong trypsin resistance to such an extent that very little tDhh1p was cleaved even after 1 h of incubation with trypsin (Fig. 7). These observations provide evidence for conformational changes in tDhh1p and indicate that the most protease-resistant conformation forms in the presence of both ATP and RNA.
FIGURE 7.

Analysis of limited trypsin digestion. SDS-PAGE analysis of time courses for trypsin cleavage of wild-type tDhh1p (A) or the R345A G346A mutant (B) in the absence of ligands or presence of saturating concentrations of ATP, AMP-PNP, ADP, and 12mer poly(U) or their combinations. The position of the undigested tDhh1p is indicated with an arrow.
A reasonable hypothesis is that the protease-resistant form of Dhh1p is most likely a compact structure similar to the structure we observe. This predicts that alterations to Dhh1p that would prevent the closed conformation might prevent protease resistance in the presence of ATP and RNA. To examine this prediction, we examined the protease sensitivity of the tDhh1p with the R345A and G346A mutations, which would be predicted to disrupt the close packing of the two domains due to the substitution of G346 with the larger alanine residue. We observed that this mutant protein conferred much weaker trypsin resistance compared with the wild-type protein with added RNA and ATP (Fig. 7). This suggests that the mutant protein is unable to efficiently form the protected conformation of Dhh1p and has implications for the function of Dhh1p (see below).
Concluding remarks
The crystal structure of tDhh1p revealed that tDhh1p is generally similar to eIF4A, mjDEAD, and UAP56. However, in Dhh1p, the two domains are arranged in a unique manner and some of the conserved motifs show different structures than seen in earlier structures. One striking structural feature of tDhh1p is that motif V interacts with motif I and the Q-motif, thereby connecting the two domains. Since at least some mutations altering this interaction (R89A K91A and G346A) cause both loss-of-function phenotypes and confer dominant negative phenotypes, this interaction appears to be functionally important. Moreover, because motif V also comprises and interacts with motif VI to create the RNA-binding site, motif V therefore may function in connecting the ATP-binding site and the RNA-binding site together. This is analogous, although the molecular details differ, to the finding that motif V “bridges” the ATP-binding site and the oligo-nucleotide-binding site in DNA helicases, suggesting that motif V may play a general role in this class of proteins in coordinating domain interactions affected by ATP and substrate binding. Consistent with this view, in the structure of UAP56, the two helicase domains also contact each other through interactions between motifs V and I, and helix K (helix α12 in Dhh1p) and the Q-motif (Shi et al. 2004), but these interactions are not as extensive as those observed in tDhh1p.
Analysis of the electrostatic potential on the molecular surface of tDhh1p, combined with mutagenesis, identified a likely RNA-binding region, which is primarily composed of motifs I, V, and VI. An interesting feature of this binding surface is that it comprises residues from both the N- and C-terminal domains of Dhh1p. This fact dictates that the RNA-binding surface will be different dependent on the orientation of the two domains. Given this, we hypothesize that the nucleotide effects on RNA binding in DEAD-box proteins might generally be that ATP binding generates a close spatial arrangement of the two domains that forms an RNA-binding surface from residues in each domain. Future experiments to examine a Dhh1p-RNA co-crystal should shed light on this issue.
Evidence that Dhh1p undergoes a conformational change has come from examination of the trypsin cleavage pattern in the presence of nucleotide and/or RNA. This process is likely to occur in two steps, but probably without an obligate order in a manner similar to what has been seen with eIF4A (Lorsch and Hershlag 1998b). In one step, ATP binding would induce a conformational change of tDhh1p. In a second step, RNA binding, coupled with ATP binding, would induce a further conformational change. Such conformational changes are facilitated by the interdomain flexibility of DEAD-box proteins. In tDhh1p, the open conformation of motif I may facilitate ATP binding. This would lead to large-scale movements in the protein aided by specific interactions between motif V, and motif I and the Q-motif, which bring the ATP binding and RNA-binding domains into close proximity. Such domain rearrangement may also create the optimal RNA-binding surface by driving conformational transitions in the motif VI region, thereby exposing the Arg residues in motif VI to the RNA-binding channel. In support of this notion, mutations of any one of the three Arg residues in motif VI drastically reduce eIF4A binding to RNA (Pause et al. 1993). Taken together, these results suggest a model of Dhh1p function involving conformational changes, ATP binding, and hydrolysis, which facilitate a cycle of interactions between the conserved motifs and alter the nature of the RNA-binding surface.
Additional evidence that a conformational change in Dhh1p is required for function has come from the analysis of mutations at the interface between the two domains. Most strikingly, mutations of either G346 or K91A and R89A lead to loss of function and dominant negative phenotypes. These residues are located at the junction between the two domains (Fig. 8A) and would be predicted to block the formation of the closed structure due to steric hindrance (in the case of G346A), or to weaken the closed conformation by loss of interactions between the two domains (in the case of the K91A R89A mutant). Consistent with this, a Dhh1 variant containing the G346A proteins is defective in entering the protease-resistant conformation. Interestingly, this Gly residue in motif V is highly conserved in SF2 helicases. In contrast, the residues analogous to Lys89 and Arg91 are not generally conserved in SF2 family members and, for example, have been replaced by glutamine at these positions in eIF4A (Fig. 8B). Given this, we suggest that the G346 residue may be a critical residue in allowing the adoption of the closed conformation due to its small size in SF2 family members in general, although the molecular details of the interface will differ between different proteins.
FIGURE 8.
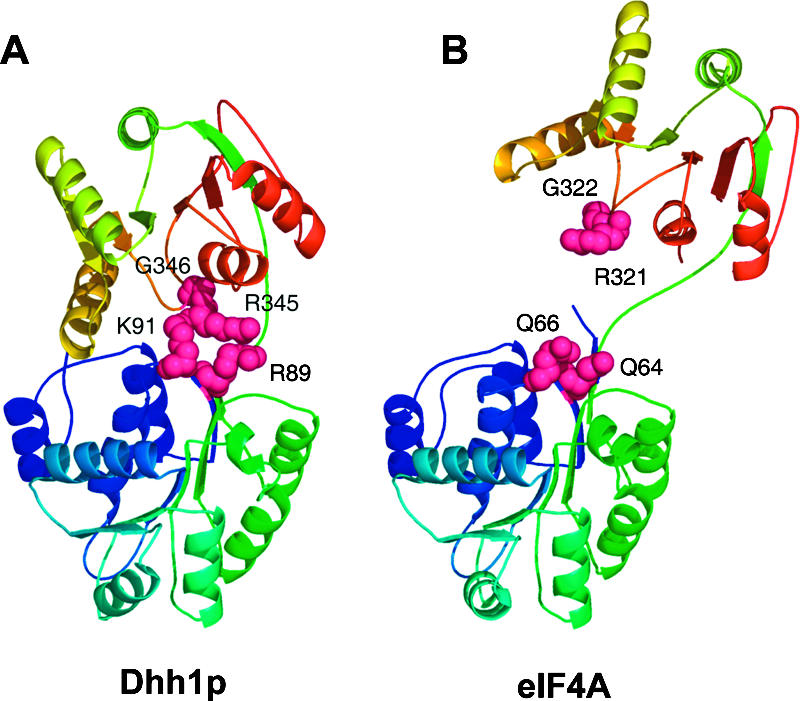
Comparison of Dhh1p to eIF4A. (A) The positions of Lys89, Arg91, Arg345, and Gly346. These residues span the junction between the N-terminal and C-terminal domains and appear to mediate the interaction between these two domains. (B) By comparison, the relative position of these residues in the eIF4A structure. Here Dhh1p R89 and K91 are replaced by Q64 and Q66 in eIF4A, respectively, while G346 is conserved in eIF4A at position 322.
MATERIALS AND METHODS
Protein expression and purification
The cDNA encoding the yeast Dhh1p core region (tDhh1p, residues 30–425) was cloned into pGEX-6P-1 vector (Amersham) and expressed as GST-fusion proteins in E. coli BL-21 Ril cells. Cells were resuspended in a lysis buffer (20 mM MES, pH 6.0, 500 mM NaCl, 2 mM DTT, 2 mM bendazole, 1 mM EDTA, 0.1 mM PMSF, 1 mg/mL lysozyme) for 30 min and lysed by sonication. The clarified cell lysate was loaded onto a glutathione-Sepharose 4B column (Amersham). GST-fusion protein was eluted by glutathione and cleaved by PreScission protease (Amersham) overnight at 4°C. After desalting, cleaved protein was loaded on a glutathione-Sepharose 4B column again and further purified by MonoS and Superdex-75 gel filtration columns (Amersham). Fractions containing tDhh1p were pooled and concentrated to ~10 mg/mL for crystallization.
Crystallization, data collection, and structure determination
Crystals were grown at 20°C using the hanging drops vapor diffusion method from a buffer containing 20% PEG400, 300 mM KBr, and 50 mM MES, pH 6.0. For data collection, crystals were transferred to the mother liquor containing 35% PEG400 and fast frozen in liquid nitrogen. Crystals belong to space group P21 with cell parameters a = 48.21 Å, b = 80.41 Å, c = 54.82 Å, α = 90°, β = 100.57°, γ = 90°, with one molecule per asymmetric unit. MAD data from a Br-derivative were collected at ID29, ESRF (Grenoble, France) and processed with the CCP4 package (CCP4 1994).
Br− sites were located by using the program SOLVE (Terwilliger and Berendzen 1999). Refinement of the heavy atom sites and phasing were carried out using SHARP (De la Fortelle and Bricogne 1997). After density modification and solvent flattening, 98% of the final model was built automatically by ARP/wARP (Perrakis et al. 1999). The rest of the model was built manually with the program O (Jones et al. 1991). Crystallographic refinement was performed with the programs CNS (Brunger et al. 1998) and REFMAC5 (Murshudov et al. 1997). Statistics for data collection and refinement are summarized in Table 1. The coordinates and structure-factor amplitudes for tDhh1p have been deposited in the Protein Data Bank with accession codes 1S2M.
CD spectroscopy
CD spectra were measured on a Jasco J-810 spectrapolarimeter at a resolution of 0.1 nm, with a bandwidth 2 nm. Five spectra were averaged for each derivative. For far-UV spectroscopy (200–250 nm), various protein samples were diluted in 50 mM NaCl, pH 6.0, at a concentration of 4 μM (derived from UV spectroscopy) using a cell of 0.1-cm path length. Mean residue ellipticity was calculated according to the CD measurement manual.
Mutagenesis and in vivo mRNA turnover assays
Alleles of Dhh1p used in both the in vitro and in vivo analyses were created using Quicksite-mutagenesis (Stratagene). Specific point mutants were generated and verified by sequence analysis. Complementation and dominant negative growth assays were performed by transforming each plasmid into either a wild-type (yRP841) or dhh1Δ deletion (yRP1561) (Coller et al. 2001) strain. Cells were then patched onto selective media and grown at either 30°C or 37°C.
RNA analysis was performed as previously described (Coller et al. 2001). Briefly, cells were transformed with plasmids expressing the various Dhh1p alleles or controls, and then grown in selective media until reaching mid-log phase. At this point, cells were harvested, RNA was extracted, and the EDC1 mRNA was visualized by Northern analysis using radiolabeled DNA oligonucleotide oRP1236 as a probe. The SCR1 RNA was probed as a loading control using radiolabeled DNA oligonucleotide oRP100.
Limited proteolysis
Reaction was performed in a buffer containing 20 mM Tris, 25 mM NaCl, 2 mM MgCl2, and 2 mM CaCl2, pH 7.0. Twenty micromoles per liter of the wild-type and mutant tDhh1p proteins and 10 μM trypsin were used for each reaction. The concentrations of ligands used were 40 μM poly(U) and 2 mM ATP, AMP-PNP, or ADP. Reactions were started by the addition of protease and stopped at various times from 5 min to 60 min by addition of standard protein sample buffer containing 100 mM NaOH, and the reaction solutions were then immediately boiled for 10 min. After being neutralized by the addition of 1N HCl, the samples were then analyzed by SDS-PAGE gels.
In vitro RNA-binding assay
Equal amounts (15 μg) of BSA, wild-type, and mutant tDhh1p proteins were dot-blotted on PVDF membrane. The membrane was blocked in buffer A (25 mM NaCl, 10 mM MgCl2, 10 mM HEPES, 0.1 mM EDTA, 1 mM DTT, 3% BSA, pH 7.0) for 1 h at room temperature. 12mer poly (U), dsRNA-I, and ss/dsRNA-II were purchased from Dharmacon. The latter two RNA substrates were annealed by boiling for 10 min at 95°C and then slowly cooling down to room temperature. RNA substrates were labeled with T4 kinase (Invitrogen) in the presence of [γ-32P]ATP (Amersham). Ten picomoles of 32P-labeled RNA substrates was added for binding assay in buffer B (50 mM NaCl, 10 mM MgCl2, 10 mM HEPES, 0.1 mM EDTA, 1 mM DTT, 1.5% BSA, pH 7.0). After being incubated in buffer B for 2 h, the membrane was washed with 20 mL buffer B three times and quantified using a phosphoimager (Molecular Dynamics). BSA protein served as a negative control.
Acknowledgments
We thank the beamline scientists at ID29 (ESRF, France) for assistance and access to synchrotron radiation facilities. In addition, we thank Drs. Nancy Standart, Nicola Minshall, and Carolyn Decker for helpful discussion. This work is financially supported by the Agency for Science, Technology and Research (A* Star) in Singapore (H.S.) and by the Howard Hughes Medical Institute (R.P.).
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2920905.
REFERENCES
- Akao, Y., Marukawa, O., Morikawa, H., Nakao, K., Kamei, M., Hachiya, T., and Tsujimoto, Y. 1995. The rck/p54 candidate proto-oncogene product is a 54-kilodalton D-E-A-D box protein differentially expressed in human and mouse tissues. Cancer Res. 55: 3444–3449. [PubMed] [Google Scholar]
- Benz, J., Trachsel, H., and Baumann, U. 1999. Crystal structure of the ATPase domain of translation initiation factor 4A from Saccharomyces cerevisiae—The prototype of the DEAD box protein family. Structure Fold. Des. 7: 671–679. [DOI] [PubMed] [Google Scholar]
- Bonnerot, C., Boeck, R., and Lapeyre, B. 2000. The two proteins Pat1p (Mrt1p) and Spb8p interact in vivo, are required for mRNA decay, and are functionally linked to Pab1p. Mol. Cell. Biol. 20: 5939–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouveret, E., Rigaut, G., Shevchenko, A., Wilm, M., and Seraphin, B. 2000. A Sm-like protein complex that participates in mRNA degradation. EMBO J. 19: 1661–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Carmel, A.B. and Matthews, B.W. 2004. Crystal structure of the BstDEAD N-terminal domain: A novel DEAD protein from Bacillus stearothermophilus. RNA 10: 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruthers, J.M. and McKay, D.B. 2002. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 12: 123–133. [DOI] [PubMed] [Google Scholar]
- Caruthers, J.M., Johnson, E.R., and McKay, D.B. 2000. Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc. Natl. Acad. Sci. 97: 13080–13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project No. 4). 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Coller, J. and Parker, R. 2004. Eukaryotic mRNA decapping. Annu. Rev. Biochem. 73: 861–890. [DOI] [PubMed] [Google Scholar]
- Coller, J.M., Tucker, M., Sheth, U., Valencia-Sanchez, M.A., and Parker, R. 2001. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA 7: 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordin, O., Tanner, N.K., Doere, M., Linder, P., and Banroques, J. 2004. The newly discovered Q motif of DEAD-box RNA heli-cases regulates RNA-binding and helicase activity. EMBO J. 23: 2478–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Cruz, J., Kressler, D., and Linder, P. 1999. Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci. 24: 192–198. [DOI] [PubMed] [Google Scholar]
- De la Fortelle, E. and Bricogne, G. 1997. Maximum-likelihood heavy-atom parameter refinement for Multiple Isomorphous Replacement and Multiwavelength Anomalous Diffraction method. Methods Enzymol. 276: 472–494. [DOI] [PubMed] [Google Scholar]
- Fairman, M.E., Maroney, P.A., Wang, W., Bowers, H.A., Gollnick, P., Nilsen, T.W., and Jankowsky, E. 2004. Protein displacement by DExH/D “RNA helicases” without duplex unwinding. Science 304: 730–734. [DOI] [PubMed] [Google Scholar]
- Fischer, N. and Weis, K. 2002. The DEAD box protein Dhh1 stimulates the decapping enzyme Dcp1. EMBO J. 21: 2788–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya, A.E. and Koonin, E.V. 1993. Helicases: Amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struct. Biol. 3: 419–429. [Google Scholar]
- Hall, M.C. and Matson, S.W. 1999. Helicase motifs: The engine that powers DNA unwinding. Mol. Microbiol. 34: 867–877. [DOI] [PubMed] [Google Scholar]
- Jankowsky, E., Gross, C.H., Shuman, S., and Pyle, A.M. 2001. Active disruption of an RNA-protein interaction by a DExH/D RNA helicase. Science 291: 121–125. [DOI] [PubMed] [Google Scholar]
- Johnson, E.R. and McKay, D.B. 1999. Crystallographic structure of the amino terminal domain of yeast initiation factor 4A, a representative DEAD-box RNA helicase. RNA 5: 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47(Pt 2): 110–119. [DOI] [PubMed] [Google Scholar]
- Kim, J.L., Morgenstern, K.A., Griffith, J.P., Dwyer, M.D., Thomson, J.A., Murcko, M.A., Lin, C., and Caron, P.R. 1998. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: The crystal structure provides insights into the mode of unwinding. Structure 6: 89–100. [DOI] [PubMed] [Google Scholar]
- Korolev, S., Hsieh, J., Gauss, G.H., Lohman, T.M., and Waksman, G. 1997. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell 90: 635–647. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24: 946–950. [Google Scholar]
- Ladomery, M., Wade, E., and Sommerville, J. 1997. Xp54, the Xenopus homologue of human RNA helicase p54, is an integral component of stored mRNP particles in oocytes. Nucleic Acids Res. 25: 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorsch, J.R. and Herschlag, D. 1998a. The DEAD box protein eIF4A. 1. A minimal kinetic and thermodynamic framework reveals coupled binding of RNA and nucleotide. Biochemistry 37: 2180–2193. [DOI] [PubMed] [Google Scholar]
- ———. 1998b. The DEAD box protein eIF4A. 2. A cycle of nucleotide and RNA-dependent conformational changes. Biochemistry 37: 2194–2206. [DOI] [PubMed] [Google Scholar]
- Maekawa, H., Nakagawa, T., Uno, Y., Kitamura, K., and Shimoda, C. 1994. The ste13+ gene encoding a putative RNA helicase is essential for nitrogen starvation-induced G1 arrest and initiation of sexual development in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 244: 456–464. [DOI] [PubMed] [Google Scholar]
- Minshall, N. and Standart, N. 2004. The active form of Xp54 RNA helicase in translational repression is an RNA-mediated oligomer. Nucleic Acids Res. 32: 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall, N., Thom, G., and Standart, N. 2001. Conserved role of a DEAD-box helicase in translational masking. RNA 7: 1728–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad, D. and Parker, R. 2005. The yeast EDC1 mRNA undergoes deadenylation-independent decapping stimulated by Not2p, Not4p, and Not5p. EMBO J. 24: 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Nakagawa, Y., Morikawa, H., Hirata, I., Shiozaki, M., Matsumoto, A., Maemura, K., Nishikawa, T., Niki, M., Tanigawa, N., Ikegami, M., et al. 1999. Overexpression of rck/p54, a DEAD box protein, in human colorectal tumours. Br. J. Cancer 80: 914–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, A., Amikura, R., Hanyu, K., and Kobayashi, S. 2001. Me31B silences translation of oocyte-localizing RNAs through the formation of cytoplasmic RNP complex during Drosophila oogenesis. Development 128: 3233–3242. [DOI] [PubMed] [Google Scholar]
- Parker, R. and Song, H. 2004. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 11: 121–127. [DOI] [PubMed] [Google Scholar]
- Pause, A. and Sonenberg, N. 1992. Mutational analysis of a DEAD box RNA helicase: The mammalian translation initiation factor eIF-4A. EMBO J. 11: 2643–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1993. Helicases and RNA unwinding in translation. Curr. Opin. Struct. Biol. 3: 953–959. [Google Scholar]
- Pause, A., Methot, N., and Sonenberg, N. 1993. The HRIGRXXR region of the DEAD box RNA helicase eukaryotic translation initiation factor 4A is required for RNA binding and ATP hydrolysis. Mol. Cell. Biol. 13: 6789–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis, A., Morris, R., and Lamzin, V.S. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6: 458–463. [DOI] [PubMed] [Google Scholar]
- Rocak, S. and Linder, P. 2004. DEAD-box proteins: The driving forces behind RNA metabolism. Nat. Rev. Mol. Cell Biol. 5: 232–241. [DOI] [PubMed] [Google Scholar]
- Schmid, S.R. and Linder, P. 1991. Translation initiation factor 4A from Saccharomyces cerevisiae: Analysis of residues conserved in the D-E-A-D family of RNA helicases. Mol. Cell. Biol. 11: 3463–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, H., Cordin, O., Minder, C.M., Linder, P., and Xu, R.M. 2004. Crystal structure of the human ATP-dependent splicing and export factor UAP56. Proc. Natl. Acad. Sci. 101: 17628–17633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story, R.M., Li, H., and Abelson, J.N. 2001. Crystal structure of a DEAD box protein from the hyperthermophile Methanococcus jannaschii. Proc. Natl. Acad. Sci. 98: 1465–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner, N.K. and Linder, P. 2001. DExD/H box RNA helicases: From generic motors to specific dissociation functions. Mol. Cell 8: 251–262. [DOI] [PubMed] [Google Scholar]
- Tanner, N.K., Cordin, O., Banroques, J., Doere, M., and Linder, P. 2003. The Q motif: A newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol. Cell 11: 127–138. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55: 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharun, S., He, W., Mayes, A.E., Lennertz, P., Beggs, J.D., and Parker, R. 2000. Yeast Sm-like proteins function in mRNA decapping and decay. Nature 404: 515–518. [DOI] [PubMed] [Google Scholar]
- Tseng-Rogenski, S.S., Chong, J.L., Thomas, C.B., Enomoto, S., Berman, J., and Chang, T.H. 2003. Functional conservation of Dhh1p, a cytoplasmic DExD/H-box protein present in large complexes. Nucleic Acids Res. 31: 4995–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar, S.S., Soultanas, P., Dillingham, M.S., Subramanya, H.S., and Wigley, D.B. 1999. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 97: 75–84. [DOI] [PubMed] [Google Scholar]
- Walker, J.E., Saraste, M., Runswick, M.J., and Gay, N.J. 1982. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1: 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmoreland, T.J., Olson, J.A., Saito, W.Y., Huper, G., Marks, J.R., and Bennett, C.B. 2003. Dhh1 regulates the G1/S-checkpoint following DNA damage or BRCA1 expression in yeast. J. Surg. Res. 113: 62–73. [DOI] [PubMed] [Google Scholar]