

Abstract
The interaction between box C/D small nucleolar (sno)RNAs and the 15.5K protein nucleates snoRNP assembly. Many eukaryotic snoRNAs contain two potential binding sites for this protein, only one of which appears to be utilized in vivo. The binding site conforms to the consensus for a kink-turn motif. We have investigated the molecular basis for selection of one potential site over the other using in vitro mobility shift assays and nucleotide analog interference mapping of Xenopus U25 snoRNA and of a circularly permuted form. We find that preferential binding of human 15.5K is not dependent on the proximity of RNA ends, but instead appears to require a structural context beyond the kink-turn itself. Direct analysis of the energetic contributions to binding made by 18 functional groups within the kink-turn identified both backbone atoms and base functionalities as key for interaction. An intramolecular RNA–RNA contact via a 2′-hydroxyl may supercede a putative Type I A-minor interaction in stabilizing the RNA–protein complex.
Keywords: snoRNA, snoRNP, kink-turn, 15.5K protein, methylation guide
INTRODUCTION
Many stable cellular RNAs including ribosomal RNAs (rRNAs), small nuclear RNAs (snRNAs), and transfer RNAs (tRNAs) acquire covalent nucleotide modifications during their biogenesis. Small ribonucleoprotein (RNP) complexes found in the nucleolus or nucleoplasmic Cajal bodies (Henras et al. 2004) participate in the modification of eukaryotic rRNAs and snRNAs. Methylation and pseudouridylation of rRNA occur in the nucleolus at an early stage of ribosome biogenesis. Methylation of specific ribose 2′-hydroxyls is performed by small nucleolar RNPs (snoRNPs) containing well-defined conserved sequence elements termed boxes C and D (Kiss-László et al. 1996; Nicoloso et al. 1996; Tycowski et al. 1996). A separate class of snoRNPs, box H/ACA snoRNPs, isomerizes specific rRNA uridines to pseudouridines (for review, see Decatur and Fournier 2003).
Each box C/D snoRNP contains four proteins and a single small nucleolar RNA (snoRNA). These snoRNAs (Fig. 1) harbor two remarkably conserved sequence elements, box C (RUGAUGA) and box D (CUGA), near the paired 5′- and 3′-termini of the mature snoRNA (Decatur and Fournier 2003). Most snoRNAs also contain more degenerate internal copies of these elements, boxes C′ and D′ (Tycowski et al. 1996; Kiss-László et al. 1998). Box C/D snoRNAs confer site specificity on the functioning snoRNPs through one or two regions of extended sequence complementarity to the region(s) on the rRNA destined for methylation. These 10–20 nucleotide (nt) “guide” sequences reside immediately upstream of box D or box D′ (or in some cases, both) and form duplexes with the target RNA, directing methylation of the fifth nucleotide paired upstream of the 5′ end of box D or D′ (Decatur and Fournier 2003).
FIGURE 1.

Secondary structures of U25 and cpU25. Full sequences of Xenopus laevis U25 and cpU25 are shown. Boxes C, D, C′, and D′ are in bold. The 2-nt change disrupting the terminal stem (TS mutant) of cpU25 is shown in the box labeled TSM. CpU25 retains the numbering of U25. Nucleotides added to cpU25 to create the terminal stem are designated s-i through s-xiii, and nucleotides added to seal the U25 termini into a loop are labeled L-i through L-iii. The secondary structure of the box C/D kink-turn motif is shown.
The snoRNA also serves as an organizational scaffold for the four snoRNP proteins. Studies in Xenopus oocytes (Szewczak et al. 2002) and in mammalian cell extracts (Watkins et al. 2002) demonstrated that particle assembly is initiated by binding of the 15.5K protein to sequences in boxes C and D. Incorporation of the three additional snoRNP proteins depends on the snoRNA–15.5K interaction (Watkins et al. 2002). The two related proteins, Nop56 and Nop58, recognize boxes C′ and C, respectively, as demonstrated by in vivo UV crosslinking (Cahill et al. 2002). Fibrillarin, which crosslinks to boxes D, D′ and C′ in vivo (Cahill et al. 2002), functions as the snoRNP methyl transferase (Ochs et al. 1985; Tollervey et al. 1993; Niewmierzycka and Clarke 1999; Galardi et al. 2002).
The 15.5K protein is the only snoRNP protein with a structurally well-characterized RNA binding site. Biochemical studies (Watkins et al. 2000) and functional group analysis (Szewczak et al. 2002) are consistent with boxes C and D forming a kink-turn structure. Kink-turns were originally identified in the X-ray crystal structures of U4 snRNA bound to h15.5K (Nottrott et al. 1999; Vidovic et al. 2000) and in the large and small subunit rRNAs from Haloarcula marismortui (Klein et al. 2001). The kink-turn motif features a stem–bulge–stem structure (Fig. 1). Stem 1 contains Watson-Crick base pairs followed by a 3-nt bulge, whereas stem 2 includes noncanonical interactions, specifically two G–A pairs and, for box C/D snoRNAs, a highly conserved U–U pair (Watkins et al. 2000).
Many box C/D snoRNAs have the potential to form two kink-turns, one using boxes C and D and the other using boxes C′ and D′. However, boxes C′ and D′ do not adhere as strongly to the consensus sequences UGAUGA and CUGA as do boxes C and D (Kiss-László et al. 1998). Consequently, it has not been clear whether a second kink-turn structure forms in the functional snoRNP. In fact, a designed snoRNA containing perfect consensus sequences in boxes C′ and D′ was found to bind the 15.5K protein only at boxes C and D both in vivo and in vitro (Szewczak et al. 2002). Moreover, point mutations in box D and box D′ (CUGA to CUGC) had been shown to have differential effects on 15.5K binding to box C/D RNAs; mutation of the box D sequence, which disrupted one of the strongly conserved G–A pairs in the noncanonical stem of the kinkturn, abrogated protein binding (Watkins et al. 2000; Cahill et al. 2002) while the change in box D′ had little or no effect (Watkins et al. 2000; Szewczak et al. 2002). Why, if the appropriate sequences are present, does the protein recognize the box C/D elements in one region of the RNA (boxes C and D), but not another (C′ and D′)?
We selected U25, a box C/D snoRNA from Xenopus laevis, for a more detailed study of the snoRNA–15.5K interaction. To investigate the ability of box C′ and D′ sequences to form a well-defined RNA structure, we circularly permuted U25, and examined its interactions with recombinant human 15.5K (h15.5K) protein (Fig. 1, right). To further our understanding of kink-turn formation and recognition by 15.5K, we determined the energetic contributions of individual functional groups within the Xenopus U25 kink-turn motif to the RNA–protein interaction.
RESULTS
15.5K binds eukaryotic box C/D snoRNAs asymmetrically
Our original study of 15.5K binding (Szewczak et al. 2002) utilized a composite snoRNA containing a 5′-sequence extension fused to a typical box C/D snoRNA. In a gel mobility shift assay, recombinant h15.5K bound to the composite snoRNA with an apparent Kd of 130 nM, producing a single shifted complex (Szewczak et al. 2002). To confirm this interaction for a bona fide vertebrate snoRNA, in vitro transcribed Xenopus U25 (Fig. 1) was examined (Tycowski et al. 1996). At all protein concentrations tested, the interaction produced a single shifted complex (Fig. 2A) with a binding affinity of 73 nM. Apparent Kd values were derived from protein titrations with a minimum of eight concentrations, and the values were adjusted for the maximum fraction bound (see Materials and Methods). Because only a single RNA–protein complex is observed, this result is consistent with only boxes C and D being recognized in a eukaryotic snoRNP.
FIGURE 2.

cpU25 forms two complexes with h15.5K. (A) Uniformly [32P]-uridine-labeled U25 (3 nM) forms a single RNP complex when incubated with increasing amounts of h15.5K, while uniformly [32P]- uridine-labeled cpU25 (2.5 nM) shifts sequentially into two complexes, RNP1 and RNP2. (B) Uniformly [32P]-uridine-labeled TSM-cpU25 (7 nM) forms a single complex with h15.5K of different mobility than either RNP1 or RNP2 formed by cpU25 (7 nM).
Circularly permuted U25 forms two complexes with h15.5K
One clear difference between the C/D and C′/D′ elements is their location within snoRNA sequences. If proximity to the RNA termini facilitates 15.5K binding, then boxes C′ and D′ would not be selected as the protein binding site. This possibility was investigated using a circularly permuted version of U25, termed cpU25 (Fig. 1). The U25 terminal stem was sealed with a UUCG tetraloop, and a new stem was inserted between boxes C′ and D′. If proximity to the ends is the only determinant, cpU25 and the protein should form a single complex with h15.5K bound to boxes C′ and D′.
Figure 2A shows a comparison of the complexes formed by h15.5K with U25 and cpU25. An initial RNP complex formed (RNP 1) at protein concentrations comparable to those seen for h15.5K interacting with U25. At elevated protein concentrations, a second shifted complex (RNP 2) was observed for cpU25. Under similar conditions, the U25–h15.5K interaction produced a single shifted complex (RNP 1, cf. lanes 6 and 13). Formation of two shifted complexes is not consistent with complete relocalization of 15.5K binding proximal to the 5′ and 3′ termini of the molecule. These data instead suggest that cpU25 contains two 15.5K binding sites with different binding affinities.
To define further the region of the RNA recognized in RNP 2, we disrupted the terminal stem of cpU25 (TSM– cpU25, Fig. 1). If the cpU25 terminal stem is a binding determinant for RNP 2, then TSM–cpU25 should not form that complex. As shown in Figure 2B, TSM–cpU25 formed a single RNP complex with 15.5K over the full concentration range tested, in contrast to cpU25. Moreover, the 2-nt change resulted in a substantial change in the RNA electrophoretic mobility. Since mobility on nondenaturing PAGE is sensitive to changes in RNA conformation, the difference suggests that TSM–U25 cannot adopt the same global conformation as cpU25, consistent with loss of a structural element that may be recognized by h15.5K.
In the construction of cpU25, none of the U25 box elements were altered. Consequently, box C′ (RUGAGGA) differs from the consensus box C sequence (RUGAUGA). In a K-turn motif, this change would produce a G–U pair instead of the highly conserved U–U pair found in stem 2. To test whether this pairing is essential, cpU25 G45U was synthesized. As with cpU25, cpU25 G45U produced two shifts in an electrophoretic mobility shift assay (data not shown). RNP 2 appeared at lower protein concentration for the G45U mutant (L.B.W. Szewczak, S.J. DeGregorio, and J.A. Steitz, unpubl.), suggesting that U–U is preferred over a G–U pair. This result also points to RNP 2 arising from binding of h15.5K to boxes C′ and D′.
NAIM analysis of the U25–15.5K binding interaction
Nucleotide analog interference mapping (NAIM) was employed to identify precisely the binding site(s) for 15.5K on both U25 and cpU25 snoRNAs. In vitro transcribed RNAs containing nucleotide analogs were subjected to a binding selection by forming and excising gel shift bands RNP1 and RNP2 (Szewczak et al. 2002).
U25 snoRNA transcripts containing 5′-α-thiophosphate derivatives of adenosine (AαS), guanosine (GαS), cytidine (CαS), or uridine (UαS) parental analogs as well as inosine 5′-α-thiophosphate (InoαS) and purine 5′-α-thiophosphate (PurαS) were examined (Fig. 3A,C; Table 1). Interactions with ribose sugars were probed using transcripts containing 2′-deoxynucleotide analogs, dAαS, dGαS, dCαS, and dUαS (Fig. 3A,B). The selection for h15.5K binding involved incubating 5′-end labeled U25 transcripts with 30 nM h15.5K at 4°C for 1 h, and then separating bound from free RNAs on nondenaturing polyacrylamide gels (see Fig. 2). Following iodine treatment of the recovered, shifted RNAs and resolution of the resulting sequence ladders by denaturing gel electrophoresis (Strobel and Shetty 1997), sites of analog interference appear as gaps or bands of diminished intensity in selected (shifted) versus unselected lanes.
FIGURE 3.

h15.5K recognizes box C/D in U25, and both boxes C/D and C′/D′ in cpU25. (A) U25 transcripts containing AαS, dAαS, GαS, dGαS, CαS, or dCαS were selected by binding to h15.5K and the shifted RNAs were compared to unselected RNAs after treatment with I2. Substitution of dA at positions 5 and 9 impairs the RNA–protein interaction. dA18 did not cause interference reproducibly. (B) Interference values for dAαS, dGαS, and dCαS are plotted against nucleotide position for U25. Standard errors of the mean are shown for A (circles, n = 4), G (squares, n = 4), C (triangles, n = 2), and U (diamonds, n = 2). (C) Observed sites of interference, which are consistent with h15.5K binding to a kink-turn, are shown for U25 (left) and cpU25 (middle, RNP1 and right, RNP2) as white letters on black circles. Additional sites of interference are circled. Sites where removal of the 2′-hydroxyl caused interference are indicated with arrows. Sequences of the conserved box elements are in bold.
TABLE 1.
Interference effects for U25 binding to h15.5Ka
| Functional group | Kappa or phosphorothioate value | |
| Box C | ||
| A5 | 2′-OH | 5 ± 1 |
| U7 | pro RP-phosphate | 57 ± 19 |
| G8 | N2-amine | 17 ± 7 |
| A9 | N6-amine | 4 ± 1 |
| 2′-OH | 3 ± 0.6 | |
| Box D | ||
| U62 | pro RP-phosphate | 4 ± 1 |
| G63 | N2-amine | 33 ± 14 |
| 2′-OH | 2 ± 0.1 | |
| A64 | N6-amine | 10 ± 6 |
| 2′-OH | 3 ± 0.7 | |
| pro RP-phosphate | 2 ± 0.2 | |
| Other nucleotides | ||
| C4 | 2′-OH | 0.7 ± 0.1 |
| G65 | N2-amine | 5 ± 2 |
| 2′-OH | 1 ± 0.1 | |
| G66 | N2-amine | 2 ± 0.5 |
aNormalized kappa and phosphorothioate values represent averages of two to six independent experiments. Standard errors of the mean are shown.
2′-Hydroxyl groups and phosphate oxygens contribute to RNP formation
Extensive hydrogen bonding interactions involving ribose 2′-hydroxyls are key features in structural models for protein recognition of kink-turns (Vidovic et al. 2000; Klein et al. 2001; Moore et al. 2004). However, there are no biochemical data addressing the functional contributions of these contacts. Using NAIM, we found four sites where loss of the 2′-hydroxyl impaired protein association (arrows in Fig. 3C). In and near box C, 2′-deoxy substitutions at A5 and A9 were deleterious (Fig. 3A,C; Table 1), as were substitutions in box D at G63 and A64 (Fig. 3B,C; Table 1). The 2′-hydroxyl of A5 is particularly interesting, since the base in this position, the first nucleotide of the asymmetric bulge, is not phylogenetically conserved in box C/D snoRNAs. Thus, the backbone rather than the base appears to participate in an essential hydrogen bonding interaction.
Analysis of RNAs containing the parental α-thiophosphates (AαS, GαS, CαS, and UαS) provides information about the involvement of backbone oxygens in protein binding (Strobel and Shetty 1997; Batey et al. 2001; Szewczak et al. 2002). In agreement with previous results, three important pro-RP phosphate oxygens were identified: U7, U62, and A64 (Fig. 3C; Table 1; Szewczak et al. 2002). The phosphates of U7 in box C and U62 in box D correspond to U37 and U87 in the composite snoRNA, both of which were important for in vivo snoRNP assembly and in vitro h15.5K binding (Szewczak et al. 2002). The phosphate of A64 (A89 in the composite) contributed to in vitro binding as well.
U25 forms a kink-turn structure
The heart of the kink-turn motif is the set of sheared G–A pairs in the noncanonical stem (Fig. 1, stem 2). NAIM analysis identified the functional groups involved in their formation (Table 1). Deletion of the G8 N2 amine, the A64 N6 amine, the A9 N6 amine or the G63 N2 amine resulted in loss of each respective transcript from the proteinbound pool. Substitution of inosine for guanosine at positions G65 and G66, involved in the top two base pairs of U25’s terminal stem, also created sites of interference. The C4–G65 base pair has been proposed to participate in an A-minor tertiary interaction (Klein et al. 2001), and loss of the amine would potentially disrupt both the base pair and a tertiary contact (see Discussion). Destabilization of the C3–G66 pair may compromise the terminal stem, consistent with the observation that a stem of three contiguous base pairs is required for snoRNA processing Xenopus oocytes (Xia et al. 1997). In concert with the key phosphate positions identified, these results confirm that U25 forms a kink-turn structure from sequences in boxes C and D.
cpU25 retains the box C/D kink-turn
NAIM analysis of the cpU25–15.5K interaction involved examination of both RNP 1 and RNP 2. Transcripts containing the parental α-thiotriphosphates, InoαS and PurαS were recovered from shifted complexes formed with either 40 nM (RNP 1) or 1000 nM (RNP 2) h15.5K, and analyzed (see Materials and Methods).
As shown in Figure 3C and Table 2, the functional group changes in cpU25 that impaired formation of RNP 1 on cpU25 clustered in boxes C and D. Sulfur substitution of the pro-RP phosphates of U7 and U62 interfered, as did removal of each of the exocyclic amines of G8, A9, G63, and A64. A similar pattern of interference was observed for U25 (compare U25 and cpU25 RNP 1 in Fig. 3C), arguing that cpU25 forms a kink-turn using boxes C and D. Additionally, deletion of the exocyclic amines of Gs-v and As-vi (Fig. 1) reduced protein recognition. These nucleotides form two of the four base pairs in the “new” terminal stem, and may thus contribute to the global structure of the RNA.
TABLE 2.
Interference effects for cpU25 binding to h15.5Ka
| RNP 1 | RNP 2 | |||
| Functional group | Kappa or phosphorothioate value | Functional group | Kappa or phosphorothioate value | |
| Box C | ||||
| U7 | pro RP-phosphate | 63 ± 31 | pro RP-phosphate | 4 ± 0.5 |
| G8 | N2-amine | 12 ± 3 | ||
| A9 | N6-amine | 40 ± 24 | N6-amine | 5 ± 1 |
| Box D | ||||
| U62 | pro RP-phosphate | 3 ± 1 | ||
| G63 | N2-amine | 6 ± 2 | ||
| A64 | N6-amine | 12 ± 4 | N6-amine | 28 ± 18 |
| Box C′ | ||||
| U42 | pro RP-phosphate | 12 ± 8 | ||
| A44 | pro RP-phosphate | 12 ± 3 | ||
| Box D′ | ||||
| A35 | pro RP-phosphate | 8 ± 3 | ||
| Other nucleotides | ||||
| Gs-v | N2-amine | 3 ± 1 | ||
| As-vi | N6-amine | 4 ± 2 |
aNormalized phosphorothioate and kappa values represent averages of two to five independent experiments. Standard errors of the mean are shown.
RNAs recovered from RNP 2 containing cpU25 showed two regions of analog interference. A subset of the key functional groups required to form RNP 1 also contribute to RNP 2: the pro-RP phosphate of U7, and the amines of A9 and A64. It thus seems likely that RNP 2 retains the kink-turn structure in boxes C and D. G43 was obscured in the gels by pronounced background hydrolysis, so that the effect of substituting inosine at that position could not be quantified.
Three sites of α-thiophosphate interference unique to RNP 2 were detected. U42 and A44 lie within box C′, and A35 lies within box D′. The effects observed at U42 and A35 correspond to those seen in the kink-turns of U25, cpU25 RNP 1, and a composite snoRNA (Szewczak et al. 2002).
Together, the NAIM data indicate that moving the RNA termini did not result in relocalization of the h15.5K binding site. Instead, introducing a four base-pair terminal stem between boxes C′ and D′ led to formation of a second shifted complex (RNP2) while retaining association of 15.5K with boxes C and D (RNP 1).
A GST tag does not affect the RNA binding activity of 15.5K
To investigate further the molecular basis for kink-turn recognition by h15.5K, we examined the energetic contributions of individual functional groups to the binding interaction. A double membrane filter binding assay (Wong and Lohman 1993; Batey et al. 2001; Kuhn et al. 2002) was used for rapid determination of the effects of single functional group changes on the binding interaction between U25 and the 15.5K protein. In our hands, the untagged h15.5K protein was not retained on the nitrocellulose membrane (J.S. Gabrielsen, L.B.W. Szewczak, and J.A. Steitz, unpubl.), and so the glutathione S-transferase (GST)-tagged fusion protein was used. As shown in Figure 4A, the interaction between GST–15.5K and ligated U25 (wt box C ligated Kd = 41.9 nM, wt box D ligated Kd = 70 nM) was comparable to that for the binding of untagged h15.5K to transcribed U25 (Kd = 73.1 nM).
FIGURE 4.

U25 binds equivalently to h15.5K and GST-15.5K. (A) Uniformly [32P]-uridine-labeled U25 binds h15.5K (closed triangles) with an apparent Kd = 73 ± 9 nM in a gel mobility shift assay. Wild-type U25, produced by ligating box C (open circles) or box D (open squares) oligonucleotides, binds GST-15.5K in a filter binding assay with affinities of 42 ± 3 nM or 70 ± 7 nM, respectively. (B) GST-15.5K binds wild type (circles, Kd = 35.9 ± 2.0 nM) and A9dA (triangles, Kd = 57 ± 3 nM) RNAs equivalently in the filter-binding assay. A9Pur (squares, Kd > 330 ± 30 nM) and A5dA (diamonds, Kd > 4200 ± 500 nM) RNAs exhibit substantially weaker binding.
The U25–15.5K interaction depends on contacts in the 3-nt bulge
U25 RNAs containing site-specific functional group alterations were generated by DNA-mediated ligation of synthetic oligonucleotides with partial U25 transcripts (Moore and Sharp 1992). For substitutions in box C, 12-mer oligonucleotides were ligated onto a transcript containing U25 nt 13–75 (U25ΔC), and for box D substitutions a transcript of nt 1–60 (U25ΔD) was ligated to 15- or 8-mer oligonucleotides (see Materials and Methods). Eighteen atomic substitutions were examined within and bordering the kink-turn, chosen based on their identification in the NAIM experiments or because a group had been highlighted as important in structural models (Vidovic et al. 2000; Klein et al. 2001). In each filter-binding experiment, ligated mutant RNA was compared directly to ligated wild-type RNA (Fig. 4B). Changes in the free energy of binding (ΔΔG) were calculated relative to the wild type in each experiment, and values from independent experiments were averaged to produce the values shown in Figure 5.
FIGURE 5.

Ribose and phosphate atoms contribute to the U25-GST- 15.5K interaction, assessed by filter binding. The change in free energy of binding relative to wild-type ligated U25 (ΔΔG) is plotted for the individual functional groups measured. Error bars show the standard error of the mean for a minimum of three independent experiments. Asterisks indicate measurements that represent minima. Dashed lines, ΔΔG values > 0.2 kcal·mol−1 or < −0.2 kcal·mol−1 are considered significant.
Removing the 2′-hydroxyl of A5, the first unpaired nucleotide in the K-turn bulge, in the NAIM experiment produced a site of strong interference (Fig. 3A,B). Likewise, RNA bearing a deoxy substitution at A5 generated by site-specific ligation was severely impaired in binding. The binding curve was not saturable (Fig. 4B), and the resulting energetic penalty (Fig. 5, ΔΔG > 2.05 kcal·mol−1) is the minimum estimate for removal of the 2′-hydroxyl. We conclude that the proposed contact made by the 2′-OH of A5 to the N1 of A64 (Fig. 6A,B) is crucial for RNP formation (Vidovic et al. 2000; Klein et al. 2001).
FIGURE 6.

A tertiary contact promotes K-turn recognition. (A) The primary and secondary structure of the U25 kink-turn is shown. Two proposed tertiary contacts are indicated with dashed lines. (B) (Left) Tertiary contacts between the first bulged nucleotide and the first G-A pair in KT-#7 in the Haloarcula marismortui 50S subunit (Klein et al. 2001). Hydrogen bonds forming both secondary (dashed lines) and tertiary (bold dashed lines) interactions are shown. (Right) Comparable nucleotides from Xenopus U25 are shown. Functional groups with κ values > 2 and contributing > 0.2 kcal/mol to binding are marked with a gray circle outlined in black. (C) (Left) Type I A-minor interaction from the H. marismortui 50S ribosomal subunit (Nissen et al. 2001). (Right) Proposed A-minor interaction in box C/D snoRNAs formed by the second G-A pair and the first base pair in the canonical stem. Functional groups with K values > 2 are in gray circles, as in (A). Functional groups with κ values > 2, but ΔΔG < 0.2 kcal/mol are shown with black circles. Functional groups examined in the NAIM analysis, but showing no interference, are in black boxes in (B) and (C).
Previous NAIM studies in vivo and in vitro identified the pro-RP oxygen of the third phosphate in the bulge as essential for snoRNP assembly and h15.5K binding (Szewczak et al. 2002). However, as T7 RNA polymerase only accepts the SP α-thiotriphosphate isomer and thus produces RNA with exclusively RP sulfur substitutions, the effect of substitution at the pro-SP oxygen could not be determined using T7 transcripts. To examine both the pro-SP and pro-RP oxygens, a 13-mer synthetic oligonucleotide containing either the RP or the SP sulfur substitution at position 7 was synthesized (Dharmacon), purified by HPLC, and ligated onto U25ΔC transcripts as described (see Materials and Methods). The binding affinities of the single isomer-containing RNAs were determined in the filter binding assay (Fig. 5). Replacing the pro-RP oxygen of U7 with sulfur destabilized the complex by at least 0.9 kcal·mol−1. Substitution of the pro-SP oxygen did not affect binding.
The third bulged nucleotide (U7 in U25) juts out of the kinked structure in the U4 snRNA–15.5K X-ray structure and was modeled as forming an intramolecular RNA–RNA contact with the phosphate of the preceding nucleotide (A6 in U25) through its 2′-hydroxyl (Vidovic et al. 2000). This potential interaction could not be addressed by NAIM because of the large phosphorothioate effect at position U7 (see Fig. 3C; Table 1). Instead, U25 RNA bearing a single dU substitution at position U7 was generated. It binds GST–15.5K with the same affinity as the wild type, suggesting that the intramolecular contact is not essential for the stability of the RNA–protein interaction (Fig. 5). A6 did not show phosphorothioate interference in the NAIM experiment (Fig. 3A), consistent with this interpretation.
The G–A pairs energetically anchor the K-turn
The tandem G–A pairs found in virtually all box C/D snoRNAs form the core of the kink-turn motif. This central role is borne out energetically. Removing the exocyclic amines of the box C nucleotides G8 and A9 destabilizes the RNA–protein complex by 0.72 kcal·mol−1 and at least 1.9 kcal·mol−1, respectively (Figs. 4B, 5). Similarly, in box D, loss of the exocyclic amines of G63 and A64 cost 0.58 kcal·mol−1 and 0.86 kcal·mol−1 (Fig. 5).
However, the K-turn motif is knit together by more than just the base functional groups of these noncanonical pairs. Deletion of the 2′-hydroxyl of G63 impairs binding by 0.36 kcal/mol (Fig. 5). The corresponding position in the U4 snRNA–15.5K crystal structure (G43) is within hydrogen bonding distance of the preceding nucleotide’s phosphate (A44, A64 in the U25 sequence) in one of the two crystallographically independent complexes solved (Vidovic et al. 2000). A low level of phosphorothioate interference at A64 is consistent with this interaction (Fig. 3C; Table 1).
Although deoxy substitution at positions A9 (Fig. 4B) and A64 appeared as modest sites of interference in the NAIM experiments (Fig. 3; Table 1), they did not significantly affect the binding interaction when examined in the filter binding assay (Fig. 5). This difference may arise because of the two assay formats used: gel mobility shift with untagged protein versus filter binding with GST-tagged h15.5K.
Phosphorothioate substitution at A64 produced analog interference in experiments with both U25 (Fig. 3C; Table 1) and a composite snoRNA (Szewczak et al. 2002). A synthetic oligonucleotide corresponding to the last 8 nt of the U25 sequence with a sulfur substitution at the pro-RP-or the pro-SP oxygen at A64 was subjected to HPLC purification and used in ligation reactions (see Materials and Methods). No affect on GST–15.5K binding was observed (Fig. 5). This functional group is proposed to interact with the 2′-hydroxyl of the G63 equivalent in the U4–h15.5K structure. If an intramolecular RNA–RNA contact contributes energetically to a complex, deletion of either functional group should destabilize the binding (Szewczak et al. 1998). Since removal of the G63 2′-OH does incur an energetic penalty, but substitution of either nonbridging oxygen does not, the G63 2′-OH may be involved in a different stabilizing interaction, either within the RNA or with the protein as previously suggested (Vidovic et al. 2000).
A strongly conserved U–U pair sits above the G–A pairs in stem 2 of the K-turn. The pro-RP oxygen of U62 was identified as a site of sulfur interference in the NAIM experiment (Table 1). However, neither the RP-substitution nor the SP-substitution made a significant energetic contribution to GST–15.5K binding when fully substituted in ligated U25 (Fig. 5).
U25 interaction with GST–15.5K does not depend on a type I A-minor interaction
An A-minor interaction (Fig. 6A,C) was included in the initial modeling of the kink-turn motif structure based on examination of rRNA crystal structures (Klein et al. 2001; Nissen et al. 2001). There are four potential tertiary hydrogen bonds that can form in the type I A-minor interaction proposed for the K-turn (Fig. 6C, left). For U25 (Fig. 6C, right), the contacts would involve the 2′-OH of C4, the 2′-OH of A9, and both the exocyclic amine and 2′-OH of G65. Only the 2′- OH of A9 and the N2 of G65 were identified as sites of interference by NAIM (Table 1). However, energetic analysis of RNAs bearing single-site substitutions (A9dA, G65Ino, and G65dG) did not reveal a significant contribution for any of these functional groups to the U25–GST–15.5K interaction (Fig. 5), suggesting that the proposed A-minor interaction is not a key stabilizing feature of the RNA–protein complex.
DISCUSSION
To broaden our understanding of the box C/D snoRNP assembly, we examined the interaction between a box C/D snoRNA and the human 15.5K protein in detail. Recombinant h15.5K bound both Xenopus U25 and a circularly permuted RNA at the kink-turn motif formed by boxes C and D. The circularly permuted RNA produced two protein-dependent shifts on nondenaturing gels, suggesting the presence of a second binding site. Mutagenesis of the terminal stem of cpU25 demonstrated that the stem is required for production of the second shifted complex. NAIM analysis of U25 and cpU25 binding to h15.5K revealed that similar functional groups participate in the interaction in each RNA. Functional groups identified in the combinatorial experiments were assayed individually for their energetic contribution to the U25 RNA–protein interaction. In addition to atoms crucial for forming known kink-turn elements such as the tandem G–A pairs, several backbone atoms were found to be essential for maintaining strong affinity to 15.5K.
h15.5K interacts with a single binding site on eukaryotic box C/D snoRNAs
Electrophoretic mobility shift analysis and NAIM experiments confirmed that h15.5K interacts with a single binding site on Xenopus U25 RNA in vitro (Figs. 2A, 3). While this had been demonstrated previously for a composite snoRNA in vivo and in vitro (Szewczak et al. 2002), this work extends that observation to a Xenopus box C/D snoRNA transcript. Our results are in contrast to those on archaeal sRNPs, where each of the C/D motifs may be bound by L7Ae, the 15.5K homolog (Omer et al. 2002; Rashid et al. 2003; Tran et al. 2003).
Binding determinants outside the kink-turn motif
Many eukaryotic box C/D snoRNAs have the potential to form two kink-turn motifs (Kiss-László et al. 1998). Despite this possibility, we find that h15.5K associates only with the structure formed by boxes C and D. One obvious difference between the sets of conserved elements is proximity to the mature snoRNA termini. To test their geographic importance, we moved the 5′ and 3′ ends of the U25 RNA to fall between boxes C′ and D′, creating cpU25 (Fig. 1). If proximity to the ends is a key binding determinant, then cpU25 might relocalize to interact with h15.5K through boxes C′ and D′.
Unlike U25, cpU25 produced protein-dependent shifts with two different and distinct mobilities on nondenaturing polyacrylamide gels (Fig. 2). NAIM analysis of RNP 1 confirmed that the protein binds more strongly to boxes C and D, and produces a pattern consistent with recognition of a kink-turn (Fig. 3). These results suggest that the RNA– protein interaction does not require access to the RNA termini, supporting the recent demonstration of coupling between pre-mRNA splicing and box C/D snoRNP protein deposition in which the 15.5K protein binds while snoRNAs are still embedded within intron sequences (Hirose et al. 2003).
While circular permutation did not alter the initial protein binding site, it did create a second shifted complex. The second shift (RNP 2) occurred at roughly 10-fold higher protein concentration than the first (RNP 1), and was dependent on the presence of the new terminal stem (Fig. 2). Functional group analysis of RNP 2 (Fig. 3) demonstrated the continued contribution of the G–A pairs in boxes C/D to binding, as well as the nonbridging oxygen at the third position in the bulge, consistent with retention of the kink-turn. Two new sites of phosphorothioate interference at A35 and U42 match the pattern expected for recognition of a second kink-turn within the protein-bound RNA. Since disruption of the terminal stem eliminated the second complex, the stem appears to support kink-turn formation/recognition. Consistent with this model, eukaryotic box C/D snoRNAs require either a short terminal stem (Xia et al. 1997), or a longer external stem for their production in vivo (Villa et al. 1998; Darzacq and Kiss 2000). It may be that the protein recognizes the kink-turn presented in the context of a stem, but not the stem itself (Vidovic et al. 2000). Alternatively, the protein could recognize a pre-formed kink-turn, and the box C/D snoRNAs require the stem to initiate formation of the structure.
However, NAIM analysis of RNP 2 did not show the full interference pattern anticipated for a kink-turn in boxes C′/ D′ (Fig. 3). In particular, the functional groups that would interact to form the G–A pairs in the second motif were either not sites of interference or not informative in the assay (G43). Consequently, the data are also consistent with weaker association (Fig. 2) of the protein with an alternate RNA structure. Since several methylation guide RNAs do contain sequences predicted to form stem structures between boxes C′ and D′ analogous to stem I in cpU25 (Kiss-László et al. 1998; Tycowski et al. 1998), it remains to be determined if these RNAs assemble into functional RNPs containing one or two copies of the 15.5K protein.
Backbone atoms in the asymmetric loop make critical contacts
In the sequences of box C/D snoRNAs (Weinstein and Steitz 1999) and K-turns (D. Klein, pers. comm.), the identity of the first nucleotide in the asymmetric bulge is not conserved. Both NAIM studies (Fig. 3; Table 1) and energetic analyses (Figs. 4, 5) showed that the 2′-hydroxyl of A5 plays a substantial role in RNP formation. Although a base-specific hydrogen bond appears in KT-#7 from the Haloarcula LSU rRNA (Fig. 6A, left), it is not conserved among the eight rRNA kink-turns (Klein et al. 2001), whereas the contact between the 2′-hydroxyl and the N1 of the adenine in the first G–A pair is (D. Klein, pers. comm.). The same 2′-OH to N1 hydrogen bonding interaction appears in the U4–15.5K structure (Vidovic et al. 2000), and a recent study of the U4–h15.5K interaction using computational and biochemical approaches also supports a key role for this interstrand contact in RNA–protein recognition (Cojocaru et al. 2005).
The third nucleotide of the asymmetric bulge (U7) sits at the vertex of the kink-turn (Vidovic et al. 2000; Klein et al. 2001). In the U4–h15.5K structure, its base points away from the converging helices and is buried in the protein (Vidovic et al. 2000). As a consequence, its nonbridging oxygens and 2′-hydroxyl are available for hydrogen bonding. The pro-RP oxygen at this position contributes strongly to RNP formation. This atom is intolerant of sulfur substitution in U25 (Fig. 3C) as had been observed previously for a different RNA both in vitro and in vivo (Szewczak et al. 2002). The corresponding position in the U4–h15.5K structure interacts directly with the protein (Vidovic et al. 2000). However in that model, both nonbridging oxygens are within hydrogen bonding distance of functional groups on the protein (Vidovic et al. 2000). Energetic analysis of RNA containing either the pro-RP or the pro-SP sulfur substitution revealed that only the pro-Rp oxygen was sensitive to sulfur substitution (Fig. 5).
The 2′-hydroxyl of the extruded nucleotide (U7) is predicted to make a stabilizing hydrogen bond to the phosphate of the preceding nucleotide (Vidovic et al. 2000; Cojocaru et al. 2005). However, this functional group was not found to be an energetic factor in GST–15.5K binding (Fig. 5). A strong phosphorothioate effect at this nucleotide precludes comparison of this result with NAIM data (Fig. 3C).
The A-minor interaction is not an essential determinant of h15.5K protein binding
Two recent biochemical studies have highlighted the importance of forming an A-minor interaction (Nissen et al. 2001) in the tight folding of kink-turn-containing RNAs in the absence of protein. DMS footprinting of rRNA KT-38 placed within the Tetrahymena P4-P6 fragment showed that the N1 positions of the adenines corresponding to A64 and A9 (A939 and A1032) were protected in the compactly folded RNA (Matsumura et al. 2003). These results are consistent with a hydrogen bonding interaction between A64 and the 2′-hydroxyl of A5 (Fig. 6A,B) and a type I A-minor interaction between A9 and the uppermost base pair of stem 1 (C4–G65, Fig. 6A,C). A second study described a two-state folding equilibrium for KT-7 lodged between two extended duplexes (Goody et al. 2004), with the A-minor interaction serving as a key stabilizing element of the kink-turn. Both studies therefore suggest that a compact, fully folded kink-turn requires additional stabilization, either from mono- or divalent ions (Matsumura et al. 2003; Goody et al. 2004) or perhaps from bound proteins (Goody et al. 2004).
In the case of h15.5K and the box C/D motif, none of the functional groups that participate in the proposed A-minor interaction make substantial energetic contributions to the binding affinity in the filter binding assay (Figs. 4–6), suggesting that in this RNP the importance of these contacts is minimized. Since both the amine of G65 and the 2′-hydroxyl of A9 appeared as sites where functional group changes were deleterious in the NAIM experiments (Fig. 3, an assay closely related to those used to examine the RNA-only folding interactions), we conclude that the A-minor motif likely forms, but is not the linchpin for kink-turn formation. Instead, the data presented here implicate the 2′-OH to N1 interaction between A5 and A64 as the crucial interaction stabilizing the K-turn fold for protein recognition (Fig. 6).
MATERIALS AND METHODS
Oligonucleotides
DNA oligonucleotides
DNA oligonucleotides (W.M. Keck Facility, Yale University) were dissolved in 400 μL TE buffer, PCA extracted, ethanol precipitated, and used without additional purification.
ET7FU25: GGCGAATTCTAATACGACTCACTATAGGCCAATG ATGAGACCAATCACAGACC
BSRU25: CGCGGATCCCGGGCCTCAGAGATAGTAAGC
5′cpU25: GGCGAATTCTAATACGACTCACTATAGGTCAATGA GGACAGCTTACTATCTCTGAGGCTTCGGCCAATGATGAG ACC
3′cpU25: CGCGGATCCGGTCTCAGAACAGGTCTGTGATTGGT CTCATCATTGGCCGAAGCC
cpU25MF: CGACTCACTATAGCACAATGAGGACAG
cpU25MR: GCTGTCCTCATTGTGCTATAGTGAGTCG
U2513–34: GCGAAGCTTAATACGACTCACTATAGACCAATC ACAGACCTGTTCTG
U25C-SP: GGTCTGTGATTGGTCTCATCATTGGCC
U2541–60R: AGATAGTAAGCTGTCCTCAT
U25D-SP: GATCCCGGGCCTCAGAGATAGTAAGC
RNA oligonucleotides
RNA oligonucleotides (Dharmacon) were deprotected following the manufacturer’s instructions and prepared as described above.
U25Cwt: GGCCAAUGAUGA
U25CA5dA: GGCC(dA)AUGAUGA
U25CU7dU: GGCCAA(dU)GAUGA
U25CU7(P=S): GGCCAA(P=S)UGAUGA
U25CG8I: GGCCAAU(Ino)AUGA
U25CA9P: GGCCAAUG(Pur)UGA
U25CG11I: GGCCAAUGAU(Ino)A
U25Dwt: CUGAGGCCCGGGAUC
U25DG63I: CUGAGGCCCGGGAUC
U25DG63dG: CU(dG)AGGCCCGGGAUC
U25DA64P: CUG(Pur)GGCCCGGGAUC
U25DA64dA: CUG(dA)GGCCCGGGAUC
U25DG65I: CUGA(Ino)GCCCGGGAUC
U25DG65dG: CUGA(dG)GCCCGGGAUC
U25Dwt8: CUGAGGCC
U25DU62(P=S)8: C(P=S)UGAGGCC
U25DA64(P=S)8: CUG(P=S)AGGCC
Plasmids and PCR transcription templates
To generate pUC19.XlU25, the sequence of Xenopus laevis U25 was amplified by PCR from pGEM3Z.XU25 (Tycowski et al. 1996) using primers ET7FU25 and BSRU25. ET7FU25 contains a T7 promoter and a mutation changing the first nucleotide of U25 to a G enabling in vitro transcription and extending the terminal stem by one base pair. Both the purified PCR fragment and the pUC19 cloning vector were digested with EcoRI and BamHI, and subsequently ligated together. The plasmid was linearized by digestion with BamHI for in vitro transcription.
The circularly permuted U25 (cpU25) sequence removes U25 nt 68–75, replacing them with UUC (L-i–L-iii) to close the stem with a tetraloop. The new 5′ and 3′ ends are located between U25 boxes C′ and D′. U25 nt 36–39 were mutated to enable formation of a terminal stem in cpU25 with a 3′-overhang analogous to the U25 transcript: 5′-GGUC-3′ (s-i–s-iv) and 5′-GACCGGAUC-3′ (s-v–s-xiii) (paired nucleotides are underlined).
pUC19.cpU25 (LBS-XV-55/2) was generated by annealing two overlapping DNA oligonucleotides, 5′cpU25 and 3′cpU25, and filling in the overhangs with Klenow fragment. 5′cpU25 contains the T7 promoter sequence. The oligonucleotides (400 pmol each) were annealed in 20 μL containing 50 mM Tris–HCl pH 8 and 50 mM NaCl by heating to 88°C for 2 min followed by cooling for 40 min in a rack at room temperature. To fill in the overhangs, the annealed oligonucleotides (10 μL of annealing reaction) were added to a 25 μL reaction containing 1× Klenow buffer (50 mM Tris–HCl pH 8, 10 mM MgCl2, 0.1 mM DTT), 1× dNTPs (0.1 mM each), and 4 U of Klenow fragment (Promega). The reaction was incubated at room temperature for 35 min and quenched by the addition of EDTA to 28 mM. The reaction mixture was diluted with TE, extracted with PCA, and ethanol precipitated. The resulting duplex DNA was digested with EcoRI and BamHI, and ligated into EcoRI/BamHI digested pUC19. The plasmid was linearized by digestion with BamHI for in vitro transcription.
The terminal stem mutant of cpU25, TSM–cpU25, was generated from pUC19.cpU25 using oligonucleotides cpU25MF and cpU25MR and the QuikChange Site-Directed Mutagenesis kit (Strategene) following the manufacturer’s instructions.
The templates for transcription of U25ΔC and U25ΔD RNAs were generated by amplification from pUC19.XlU25 using primer pairs: U2513–34 and BSRU25 for ΔC and ET7FU25 and U2541–60R for ΔD using Pfu polymerase (Stratagene) with annealing at 58°C.
In vitro transcription
Standard radioactive transcription reactions included 1× transcription buffer (40 mM Tris–HCl pH 7.6, 15 mM MgCl2, 4 mM spermidine, 10 mM DTT, and 0.05% Triton X-100), 0.8 mM ATP, GTP, and CTP, 0.02 mM UTP, 20–40 μCi [32P]-UTP (400 Ci/ mmol), 0.05 μg/μL linearized plasmid, and 50 μL T7 RNAP/mL of reaction. After incubation at 37°C for 2–4 h, the transcripts were diluted with 1 volume of formamide loading buffer (85% ultrapure formamide, 1× TBE, 10 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanole) and gel purified. The RNAs were eluted into TEN (10 mM Tris–HCl pH 7.6, 1 mM EDTA, and 0.25 M NaCl) overnight at 4°C, extracted with PCA, and ethanol precipitated prior to use. Concentrations were calculated based on radiolabel incorporation.
Nucleotide analogs were incorporated into U25 and cpU25 transcripts as described (Szewczak et al. 2002). ATPαS, GTPαS, CTPαS, UTPαS, PurTPαS, InoTPαS, dATPαS, dGTPαS, dCTPαS, and dUTPαS were added to transcription reactions as described (Cochrane and Strobel 2004) except for dCTPαS, which was added at 4 mM to achieve a level of incorporation comparable to the other analogs. Concentrations of unlabeled RNAs were determined spectrophotometrically. For cpU25, guanosine (5 mM) was added to initiate transcription and the amount of GTP was reduced to 200 μM, producing transcripts with free 5′-hydroxyls.
U25ΔC and U25ΔD RNAs were synthesized from reactions containing 1× transcription buffer, 1 mM ATP, GTP, CTP, and UTP, and 50 μL T7 RNAP/mL of reaction. The U25ΔC PCR fragment was digested with BamHI before transcription. In each case, the pellet from a 100 μL PCR reaction was used in a 100 μL transcription reaction. Following incubation at 37°C for 2 h, 1 U of RQ1 DNase (Promega) was added and the reactions incubated for a further 15 min.
Protein preparation and gel mobility shift assays
Human GST–15.5K (Nottrott et al. 1999) was expressed and purified, and the GST tag was removed as described (Szewczak et al. 2002). Mobility shift experiments were performed as described (Szewczak et al. 2002).
Nucleotide analog interference mapping
NAIM assays were performed essentially as described (Szewczak et al. 2002). 5′-End labeled RNAs (U25 or cpU25NαS, 2–8 × 105 cpm) were heated to > 90°C for 1 min, cooled for 15 min in the heat block shifted to room temperature, and added to 8 μL containing buffer A (20 mM HEPES pH 7.9, 150 mM KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% Triton X-100) and 1 mg/mL tRNA. Binding reactions (10 μL total volume) were initiated by adding h15.5K (30 nM for U25, 40 nM for cpU25 RNP1, and 1000 nM for cpU25 RNP2). Following incubation at 4°C for 60 min and addition of glycerol to 8.3%, the complexes were resolved on 10% nondenaturing (80:1 acrylamide: bis-acrylamide) gels. The shifted complexes were visualized by autoradiography, excised from the gel, and eluted overnight at RT into 9 mM Tris–HCl pH 7.5, 0.9 mM EDTA, 0.27 M NaCl, and 0.45% SDS. The eluates were extracted with PCA, precipitated with ethanol, and the resulting pellets dissolved in formamide loading buffer (10 μL).
Each shifted RNA sample was split: 7 μL was treated with 0.5 μL I2/dyes (8 mM I2, 8% ethanol, 92% formamide loading buffer) and 3 μL was reserved for the untreated control. Unselected RNA transcripts were treated in parallel. All of the RNA samples were heated (> 90°C, 3 min) and were resolved by 15% denaturing PAGE. The fragmentation pattern was analyzed using a Storm PhosphorImager (Molecular Dynamics), and normalized phosphorothioate and interference (κ) values were calculated as described (Ryder et al. 2000). Kappa is defined by the equation:
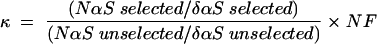 |
where NαS is the peak intensity for each position in the parental nucleotide-containing RNA and δαS is the peak intensity for each position in the nucleotide analog-containing RNA in the unselected and selected lanes. NF is a normalization factor adjusting for lane-to-lane differences in loading and the extent of reaction. NF = 1/(average of all peaks with intensities within three standard deviations of the mean).
HPLC separation of RP and SP phosphorothioates
RNA oligonucleotides containing phosphorothioate (P=S) substitutions (U25CU7(P=S), U25DU62(P=S)8, and U25DA64(P=S)8) were separated into the RP- and SP-containing fractions by reverse-phase HPLC using a Varian Dynamax (Microsorb C18 300 A° ) column. U25CU7(P=S) was separated using a gradient of 2%–15% CH3CN in 0.1 M triethyl ammonium acetate (TEAA) over 80 min to give 1.5 min separation between the peaks. U25DU62(P=S)8 and U25DA64(P=S)8 were resolved using a gradient of 1%–5% CH3CN in 0.1 M TEAA over 5 min followed by 5%–12% over 90 min to give 90-sec separation of U62(P=S)8, and 3-min separation of the A64(P=S)8. The box D phosphorothioate-containing oligonucleotides were 7 nt shorter than the other box D oligonucleotides to facilitate HPLC separation. Solvent was removed from the purified fraction in vacuo, and the oligonucleotides were redissolved in H2O. The presence of a phosphorothioate linkage in each fraction was verified by treatment with I2 in ethanol (data not shown).
Singly substituted U25 RNAs
U25 RNAs containing single functional group substitutions were generated using DNA splint-mediated RNA ligation. RNAs containing substitutions in box C were generated by joining synthetic oligonucleotides (40 pmol) to a 5′-end labeled transcript of U25ΔC (nt 13–75, 20 pmol) in the presence of a DNA splint (U25C–SP, 20 pmol). U25ΔC transcripts were treated with calf intestinal phosphatase (Roche), ethanol precipitated, 5′-end labeled with γ-ATP and polynucleotide kinase (US Biochemicals) and ethanol precipitated to produce a radioactive pellet used directly in ligation reactions. The transcript pellet, substituted oligonucleotide, and splint were annealed in 5 μL containing 50 mM Tris–HCl pH 7.5 and 20 mM KCl by heating to > 90°C and then cooling for 15 min in the heating block shifted to room temperature. The ligation reaction was initiated by the addition of 5 μL containing 1× ligation buffer (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM ATP, 1% polyvinyl alcohol), 20 mM DTT, 20 U RNase Inhibitor (Roche), and 1 U T4 DNA ligase (Roche). After incubation at 37°C for 2 h, RQ1 DNase (1 U) was added and incubated for an additional 15 min. The reactions were quenched with 10 μL formamide loading buffer, and the RNAs were purified from 10% denaturing gels. The ligated RNAs were recovered as described above. Box D substitutions were made using a 5′-end labeled oligonucleotide (20 pmol), U25ΔD transcript (40 pmol) and U25D–SP (20 pmol), and followed the same protocol.
Determination of apparent binding constants for singly substituted RNAs
A double membrane filter binding assay (Wong and Lohman 1993; Batey et al. 2001; Kuhn et al. 2002) was used to determine the binding affinities of RNAs containing single nucleotide substitutions. Ligated U25 RNAs, dissolved in water, were preheated to > 90°C and then cooled for 15 min in the heating block removed from the heat source. The RNAs (final conc. < 0.2 nM) were diluted into 40 μL reactions containing 1× binding buffer (30 mM HEPES pH 7.9, 50 mM KCl, 4.5 mM MgCl2, 1 mM DTT, 0.1 mM EDTA, and 10% glycerol) and 1 mg/mL tRNA. The reactions were initiated by addition of varying amounts of GST–15.5K or buffer D (no protein control), and incubated at 4°C for 60–90 min. BA-85 nitrocellulose (Schleicher and Schuell) and positively charged Zeta Probe GT (Bio-Rad) membranes were washed in 1× binding buffer for 1 h and then assembled in a Minifold apparatus (Schleicher and Schuell). Each well was washed with 1× binding buffer (40 μL) prior to sample application (40 μL), and washed twice (40 μL, 1× binding buffer) afterward. The appropriate wild-type ligated RNA was included. RNA–protein titrations were performed in duplicate or triplicate in each experiment.
The amounts of bound and free RNA were quantified using a Storm PhosphorImager (Molecular Dynamics). The fraction bound in the no protein well was subtracted from the values determined for the protein-containing wells to correct for the minimal retention of counts on the nitrocellulose membrane. Corrected values were used in all subsequent calculations. Apparent binding constants (Kd) were calculated using the equation: FB = (FBmax × [protein])/(appKd × [protein]), where FB is the fraction RNA bound, FBmax is the maximum fraction RNA bound, and Kd is the apparent equilibrium binding constant (Bevilacqua and Cech 1996).
Changes in the free energy of binding due to atomic substitutions were calculated using the equation ΔG = −RT(ln Kd), where R = 1.987 cal·mol−1K−1 and T = 277 K. ΔΔG values were calculated within each experiment, and then subsequently averaged to produce the values displayed in Figure 5.
Acknowledgments
We thank Kazio Tycowski and Alex Szewczak for comments on the manuscript, Jesse Cochrane for advice on filter binding experiments, Amy Seila and Anne Kosek for heroic efforts with the HPLC, members of the Steitz and Strobel labs for lively discussion, and Dan Klein for his analysis of K-turns. S.A.S. is the recipient of NSF Grant CHE-0100057. J.A.S. is an Investigator of the Howard Hughes Medical Institute and recipient of NIH Grant GM 26154.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2830905.
REFERENCES
- Batey, R.T., Sagar, M.B., and Doudna, J.A. 2001. Structural and energetic analysis of RNA recognition by a universally conserved protein from the signal recognition particle. J. Mol. Biol. 307: 229–246. [DOI] [PubMed] [Google Scholar]
- Bevilacqua, P.C. and Cech, T.R. 1996. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry 35: 9983–9994. [DOI] [PubMed] [Google Scholar]
- Cahill, N.M., Friend, K., Speckman, W., Li, Z.-H., Terns, R., Terns, M., and Steitz, J.A. 2002. Site-specific crosslinking analyses reveal an asymmetric protein distribution for a box C/D snoRNP. EMBO J. 21: 3816–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane, J.C. and Strobel, S.A. 2004. Probing RNA structure and function by nucleotide analog interference mapping. Curr. Protocols Nucleic Acid Chem. 6.9: 6.9.1–6.9.21. [DOI] [PubMed] [Google Scholar]
- Cojocaru, V., Nottrott, S., Klement, R., and Jovin, T.M. 2005. The snRNP 15.5K protein folds its cognate K-turn RNA: A combined theoretical and biochemical study. RNA 11: 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq, X. and Kiss, T. 2000. Processing of intron-encoded box C/D small nucleolar RNAs lacking a 5′,3′-terminal stem structure. Mol. Cell. Biol. 20: 4522–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decatur, W.A. and Fournier, M.J. 2003. RNA-guided nucleotide modification of ribosomal and other RNAs. J. Biol. Chem. 278: 695–698. [DOI] [PubMed] [Google Scholar]
- Galardi, S., Fatica, A., Bachi, A., Scaloni, A., Presutti, C., and Bozzoni, I. 2002. Purified box C/D snoRNPs are able to reproduce site-specific 2′-O-methylation of target RNA in vitro. Mol. Cell. Biol. 22: 6663–6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goody, T.A., Melcher, S.E., Norman, D.G., and Lilley, D.M. 2004. The kink-turn motif in RNA is dimorphic, and metal ion-dependent. RNA 10: 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henras, A.K., Dez, C., and Henry, Y. 2004. RNA structure and function in C/D and H/ACA s(no)RNPs. Curr. Opin. Struct. Biol. 14: 335–343. [DOI] [PubMed] [Google Scholar]
- Hirose, T., Shu, M.D., and Steitz, J.A. 2003. Splicing-dependent and -independent modes of assembly for intron-encoded box C/D snoRNPs in mammalian cells. Mol. Cell 12: 113–123. [DOI] [PubMed] [Google Scholar]
- Kiss-László , Z., Henry, Y., Bachellerie, J.-P., Caizergues-Ferrer, M., and Kiss, T. 1996. Site-specific ribose methylation of preribosomal RNA: A novel function for small nucleolar RNAs. Cell 85: 1077–1088. [DOI] [PubMed] [Google Scholar]
- Kiss-László , Z., Henry, Y., and Kiss, T. 1998. Sequence and structural elements of methylation guide snoRNAs essential for site-specific ribose methylation of pre-rRNA. EMBO J. 17: 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, D.J., Schmeing, T.M., Moore, P.B., and Steitz, T.A. 2001. The kink-turn: A new RNA secondary structure motif. EMBO J. 20: 4214–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, J.F., Tran, E.J., and Maxwell, E.S. 2002. Archaeal ribosomal protein L7 is a functional homolog of the eukaryotic 15.5kD/ Snu13p snoRNP core protein. Nucleic Acids Res. 30: 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura, S., Ikawa, Y., and Inoue, T. 2003. Biochemical characterization of the kink-turn RNA motif. Nucleic Acids Res. 31: 5544– 5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, M.J. and Sharp, P.A. 1992. Site-specific modification of pre-mRNA: The 2′-hydroxyl groups at the splice sites. Science 256: 992–997. [DOI] [PubMed] [Google Scholar]
- Moore, T., Zhang, Y., Fenley, M.O., and Li, H. 2004. Molecular basis of box C/D RNA-protein interactions; Cocrystal structure of archaeal L7Ae and a box C/D RNA. Structure 12: 807–818. [DOI] [PubMed] [Google Scholar]
- Nicoloso, M., Qu, L.-H., Michot, B., and Bachellerie, J.-P. 1996. Intron-encoded, antisense small nucleolar RNAs: The characterization of nine novel species points to their direct role as guides for the 2′-O-ribose methylation of rRNAs. J. Mol. Biol. 260: 178–195. [DOI] [PubMed] [Google Scholar]
- Niewmierzycka, A. and Clarke, S. 1999. S-Adenosylmethionine-dependent methylation in Saccharomyces cerevisiae. Identification of a novel protein arginine methyltransferase. J. Biol. Chem. 274: 814–824. [DOI] [PubMed] [Google Scholar]
- Nissen, P., Ippolito, J.A., Ban, N., Moore, P.B., and Steitz, T.A. 2001. RNA tertiary interactions in the large ribosomal subunit: The A-minor motif. Proc. Natl. Acad. Sci. 98: 4899–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottrott, S., Hartmuth, K., Fabrizio, P., Urlaub, H., Vidovic, I., Ficner, R., and Lührmann, R. 1999. Functional interaction of a novel 15.5kD [U4/U6.U5] tri-snRNP protein with the 5′ stem-loop of U4 snRNA. EMBO J. 18: 6119–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs, R.L., Lischwe, M.A., Spohn, W.H., and Busch, H. 1985. Fibrillarin: A new protein of the nucleolus identified by autoimmune sera. Biol. Cell 54: 123–133. [DOI] [PubMed] [Google Scholar]
- Omer, A.D., Ziesche, S., Ebhardt, H., and Dennis, P.P. 2002. In vitro reconstitution and activity of a C/D box methylation guide ribonucleoprotein complex. Proc. Natl. Acad. Sci. 99: 5289–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid, R., Aittaleb, M., Chen, Q., Spiegel, K., Demeler, B., and Li, H. 2003. Functional requirement for symmetric assembly of archaeal box C/D small ribonucleoprotein particles. J. Mol. Biol. 333: 295–306. [DOI] [PubMed] [Google Scholar]
- Ryder, S.P., Ortoleva-Donnelly, L., Kosek, A.B., and Strobel, S.A. 2000. Chemical probing of RNA by nucleotide analog interference mapping. Methods Enzymol. 317: 92–109. [DOI] [PubMed] [Google Scholar]
- Strobel, S.A. and Shetty, K. 1997. Defining the chemical groups essential for Tetrahymena group I intron function by nucleotide analog interference mapping. Proc. Natl. Acad. Sci. 94: 2903–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczak, A.A., Ortoleva-Donnelly, L., Ryder, S.P., Moncoeur, E., and Strobel, S.A. 1998. A minor groove RNA triple helix within the catalytic core of a group I intron. Nat. Struct. Biol. 5: 1037–1042. [DOI] [PubMed] [Google Scholar]
- Szewczak, L.B.W., DeGregorio, S.J., Strobel, S.A., and Steitz, J.A. 2002. Exclusive interaction of the 15.5 kD protein with the terminal box C/D motif of a methylation guide snoRNP. Chem. Biol. 9: 1095–1107. [DOI] [PubMed] [Google Scholar]
- Tollervey, D., Lehtonen, H., Jansen, R., Kern, H., and Hurt, E.C. 1993. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell 72: 443–457. [DOI] [PubMed] [Google Scholar]
- Tran, E.J., Zhang, X., and Maxwell, E.S. 2003. Efficient RNA 2′-O-methylation requires juxtaposed and symmetrically assembled archaeal box C/D and C′/D′ RNPs. EMBO J. 22: 3930–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycowski, K.T., Smith, C.M., Shu, M.-D., and Steitz, J.A. 1996. A small nucleolar RNA requirement for site-specific ribose methylation of rRNA in Xenopus. Proc. Natl. Acad. Sci. 93: 14480–14485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycowski, K.T., You, Z.-H., Graham, P.J., and Steitz, J.A. 1998. Modification of U6 spliceosomal RNA is guided by other small RNAs. Mol. Cell 2: 629–638. [DOI] [PubMed] [Google Scholar]
- Vidovic, I., Nottrott, S., Hartmuth, K., Lührmannn, R., and Ficner, R. 2000. Crystal structure of the spliceosomal 15.5kD protein bound to a U4 snRNA fragment. Mol. Cell 6: 1331–1342. [DOI] [PubMed] [Google Scholar]
- Villa, T., Ceradini, F., Presutti, C., and Bozzoni, I. 1998. Processing of the intron-encoded U18 small nucleolar RNA in the yeast Saccharomyces cerevisiae relies on both exo- and endonucleolytic activities. Mol. Cell. Biol. 18: 3376–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins, N.J., Ségault, V., Charpentier, B., Nottrott, S., Fabrizio, P., Bachi, A., Wilm, M., Rosbash, M., Branlant, C., and Lührmann, R. 2000. A common core RNP structure shared between the small nucleolar box C/D RNPs and the spliceosomal U4 snRNP. Cell 103: 457–466. [DOI] [PubMed] [Google Scholar]
- Watkins, N.J., Dickmanns, A., and Luhrmann, R. 2002. Conserved stem II of the box C/D motif is essential for nucleolar localization and is required, along with the 15.5K protein, for the hierarchical assembly of the box C/D snoRNP. Mol. Cell. Biol. 22: 8342–8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein, L.B. and Steitz, J.A. 1999. Guided tours: From precursor snoRNA to functional snoRNP. Curr. Opin. Cell Biol. 11: 378–384. [DOI] [PubMed] [Google Scholar]
- Wong, I. and Lohman, T.M. 1993. A double-filter method for nitro-cellulose- filter binding: Application to protein–nucleic acid interactions. Proc. Natl. Acad. Sci. 90: 5428–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, L., Watkins, N.J., and Maxwell, E.S. 1997. Identification of specific nucleotide sequences and structural elements required for intronic U14 snoRNA processing. RNA 3: 17–26. [PMC free article] [PubMed] [Google Scholar]