

Abstract
Alternative splicing is a major contributor to genomic complexity, disease, and development. Previous studies have captured some of the characteristics that distinguish alternative splicing from constitutive splicing. However, most published work only focuses on skipped exons and/or a single species. Here we take advantage of the highly curated data in the MAASE database (see related paper in this issue) to analyze features that characterize different modes of splicing. Our analysis confirms previous observations about alternative splicing, including weaker splicing signals at alternative splice sites, higher sequence conservation surrounding orthologous alternative exons, shorter exon length, and more frequent reading frame maintenance in skipped exons. In addition, our study reveals potentially novel regulatory principles underlying distinct modes of alternative splicing and a role of a specific class of repeat elements (transposons) in the origin/evolution of alternative exons. These features suggest diverse regulatory mechanisms and evolutionary paths for different modes of alternative splicing.
Keywords: MAASE database, constitutive splicing, alternative splicing, regulatory motifs, transposable elements, evolution of alternative splicing
INTRODUCTION
Alternative splicing affects over half of the genes in the human genome (Croft et al. 2000; Modrek et al. 2001; Brett et al. 2002; Johnson et al. 2003). Other genomes also show evidence of a high frequency of alternative splicing (Brett et al. 2002). Alternative splicing plays many important roles in gene expression, expanding the complexity of the proteome (possibly explaining the unexpectedly small number of genes in advanced organisms) and providing regulatory strategies to control gene expression in both normal and disease states (Dredge et al. 2001; Caceres and Kornblihtt 2002; Baelde et al. 2004a,b; Garcia-Blanco et al. 2004).
Previous studies have detected a number of differences between alternatively and constitutively spliced exons. For example, while the sequences of constitutive splice sites more closely resemble that of the consensus splice site sequence than do those of alternative splice sites, alternatively spliced exons are shorter in length, have a higher degree of sequence conservation between orthologous exons, and maintain transcript reading frame at a higher frequency than do constitutively spliced exons (Senapathy et al. 1990; Stamm et al. 2000; Clark and Thanaraj 2002; Sorek and Ast 2003; Thanaraj and Stamm 2003; Itoh et al. 2004; Sorek et al. 2004b; Sugnet et al. 2004). However, the focus in many of these studies has been on skipped exons and/or one species at a time.
The emphasis on skipped exons is possibly because of the ease in the identification of skipped exons by aligning mRNA/ EST sequences with genomic sequence. Although several alternative splicing database efforts considered different modes of splicing, those analyses suffered from either limited database content or insufficient quality control on the computationally derived information. We have constructed the MAASE database system to support our splicing microarray efforts (Zheng et al. 2004). The MAASE database system consists of a manual/ computational tool for annotating alternative splicing events and a database of highly curated information annotated. In constructing MAASE, we compiled detailed information (i.e., mode of splicing) to assist with microarray oligonucleotide design, as well as to better relate experimental results to specific splicing pathways. In addition, the MAASE database system includes curated information for both human and mouse, thus providing a unique opportunity to systematically characterize different modes of alternative splicing.
In this study, we report features that distinguish alternative and constitutive splicing, and more importantly, features that distinguish distinct modes of alternative splicing. Our results confirm many previously reported differences between alternative and constitutive splicing. When these differences were further studied, we find many of these features also distinguish between different modes of splicing. Furthermore, we detect, for the first time, an association of multiple classes of transposable elements with alternatively spliced regions, suggesting a contribution of these repetitive elements to the origin of alternative splicing. Together, these results will facilitate the development of predictive tools for alternative splicing and uncover potential regulatory mechanisms for different modes of alternative splicing.
Data set
Data sets were retrieved from the Manually Annotated Alternatively Spliced Events (MAASE) Database (Zheng et al. 2004; Zheng et al. 2005). We use the following nomenclature and abbreviations for different splicing modes and their associated intron sequences. Constitutively Spliced exons (CS) are internal exons, which are supported by four or more annotated transcripts in MAASE, and which show no evidence of alternative splicing in MAASE. It is possible that this class may mistakenly include some alternative exons found in mRNAs or ESTs that are found in GenBank or other general databases, but were not selected for annotation in MAASE. Skipped Exons (ES) are those included in one transcript but skipped in another. Retained Introns (IN) are sequences included as a part of an exon in one transcript and excluded as an entire intron in another. Alternative Acceptor exons (AA) have competing 3′ splice sites with the same downstream exon end. Alternative Donor exons (AD) have competing 5′ splice sites with the same upstream exon start. It is important to note that the stringent definition of AA and AD exclude alternative 3′ splice site choice paired with alternative polyadenylation and alternative 5′ splice site choice linked with alternative promoters. This enabled us to focus our statistical analysis on individual splicing modes without confounding effects from other coupling events.
CS introns are sequences that lie between two constitutive exons. ES introns are sequences flanking skipped exons. AA introns are sequences upstream of the alternative 3′ splice sites, whereas AD introns are those downstream of the alternative 5′ splice sites. The combined data set retrieved from MAASE comprises 2641 and 1967 CS, 468 and 340 ES, 246 and 161 IN, 310 and 282 AA, and 176 and 164 AD from human and mouse, respectively. Alternatively Spliced exons (AS) refer to the combination of all modes of alternative splicing within our set. For simplicity, we did not include complex modes of alternative splicing (Zheng et al. 2005) from the MAASE database.
RESULTS
We systematically surveyed different features and characteristics in CS and each mode of AS. Features included (1) descriptive statistics, such as length and reading frame conservation of each exon class, length of each intron class, consensus strength of each acceptor or donor splice site, and conservation of orthologous exons; (2) parameters affecting splice site selection; (3) sequence motifs associated with different modes of splicing; and (4) genomic landscape surrounding CS and AS.
Distinct sequence and structural features between CS and AS
Exon and intron length
It is well known that splice sites bracketing an exon are recognized by an “exon definition” mechanism in mammalian cells (Berget 1995). This mechanism implies the potential effect of exon length on splice site recognition across the exon. If an exon is too short, steric hindrance may interfere with the interaction between the two ends. On the other hand, a long exon has a higher probability of harboring negative elements. Thus, splice site selection can be influenced by exon length.
The constraint on exon length is clearly evident by the similar lengths in CS between human and mouse, which have median lengths of 120 and 122 nt, respectively (Table 1). In contrast, ES have median lengths of 101 and 88 nt, while IN have median lengths of 145 and 179 nt, in human and mouse, respectively. These results agree with previous reports that ES are significantly shorter than CS and that IN are significantly longer than CS (Stamm et al. 2000; Thanaraj and Stamm 2003; Galante et al. 2004). The constitutive portion of AA and AD are similar in length to CS, whereas the alternative portion of both AA and AD are significantly shorter. Therefore, when the constitutive and alternative portions of AA and AD are combined, they are significantly longer than CS. It is interesting to note that all exons or exonic regions involved in alternative splicing show greater length variability than do CS (Table 1). The high degree of variation may reflect a combination of multiple influences on splicing regulation.
TABLE 1.
Median exon lengths
| Human | Mouse | |||||
| Exon classes | Median | SD | P-value | Median | SD | P-value |
| CS | 120 | 94 | 122 | 79 | ||
| ES | 101 | 539 | 3.7 × 10−11 | 88 | 338 | 1.3 × 10−21 |
| IN | 145 | 479 | 3.8 × 10−4 | 179 | 617 | 4.2 × 10−7 |
| AA constitutive | 107 | 250 | 0.076 | 116 | 243 | 0.959 |
| AA alternative | 21 | 80 | 1.0 × 10−58 | 18 | 191 | 4.6 × 10−54 |
| AA both | 144 | 280 | 8.4 × 10−8 | 147 | 353 | 6.2 × 10−9 |
| AD constitutive | 125 | 397 | 0.248 | 120 | 174 | 0.097 |
| AD alternative | 31 | 309 | 2.7 × 10−27 | 22.5 | 163 | 6.1 × 10−33 |
| AD both | 172 | 540 | 1.2 × 10−10 | 143 | 248 | 3.3 × 10−5 |
“AA constitutive” and “AD constitutive” refer to the nonalternative portion; “AA alternative” and “AD alternative” refer to the alternative portion of the exon; “AA both” and “AD both” refer to the combination of the constitutive and alternative portions. (SD) Standard deviation. P-values were determined using the Wilcoxon rank sum test to test the significance of the difference between each alternative class with CS.
In most cases, the length of introns varies dramatically (Table 2). IN are significantly shorter compared with all other classes of introns (Galante et al. 2004). Furthermore, ES are frequently flanked by long introns (median = 2266 nt in human and median = 1736 nt in mouse), which was also previously observed (Clark and Thanaraj 2002). Finally, CS, the least variable exon class, are flanked by the most variable introns in terms of length.
TABLE 2.
Median intron lengths
| Human | Mouse | |||||
| Intron classes | Median | SD | P-value | Median | SD | P-value |
| CS | 1508 | 14622 | 1160 | 14479 | ||
| ES | 2266 | 11633 | 1.6 × 10−13 | 1736 | 8100 | 2.5 × 10 −11 |
| IN | 145 | 479 | 1.8 × 10−81 | 179 | 617 | 2.9 × 10 −44 |
| AA | 1152 | 8744 | 2.1 × 10−2 | 1246 | 13982 | 0.865 |
| AD | 2141 | 6409 | 0.101 | 1502 | 4780 | 0.661 |
(SD) Standard deviation. P-values were determined using the Wilcoxon rank sum test to test the significance of the difference between each alternative class with CS.
Reading frame preservation
The length of ES tends to be a multiple of 3 nt more frequently than does the length of CS. It has been suggested that this helps to retain the integrity of the transcript in an event of ES (Sorek et al. 2004b). In order to determine whether this trend is seen among other AS, we calculated the frequency of maintenance in all AS classes. Results show that the high frequency of reading frame maintenance is only associated with ES (~65%), and not with CS or other modes of AS (~40%) (Table 3). In fact, reading frame preservation is greatly reduced in the alternative portion of both AA and AD (~15%) in human and mouse. This observation is significantly different from a previous report in which Zavolan et al. (2003) analyzed full-length RIKEN mouse cDNAs and observed a much higher level of reading frame maintenance in the alternative portion of both AA and AD. Possible explanations for this discrepancy include differential sampling and splicing mode definitions (e.g., we exclude all AA and AD paired with alternative promoters or polyadenylation sites).
TABLE 3.
Frequency each exon class maintains reading frame
| Human | Mouse | |
| Exon classes | Frequency | Frequency |
| CS | 40% | 41% |
| ES | 57% | 70% |
| IN | 39% | 32% |
| AA constitutive | 41% | 43% |
| AA alternative | 13% | 11% |
| AA both | 39% | 43% |
| AD constitutive | 30% | 41% |
| AD alternative | 17% | 12% |
| AD both | 47% | 43% |
Definitions for each AA and AD exon class are given in Table 1.
Splice site strength
Alternative splice sites generally show a lower level of sequence conservation in comparison to consensus splice sites (Stamm et al. 2000; Zavolan et al. 2003; Itoh et al. 2004). The divergence of alternative splice site sequences from the consensus may be one of the underlying mechanisms for the alternative use of a splice site. To further test this idea, alternative splice sites from each mode of splicing were scored using a log-odds matrix constructed based on CS splice sites. Sequences from each species were separately scored with a species-specific matrix. Indeed, alternative splice sites are significantly weaker (P < 10−4 to P < 10−73) than those of CS splice sites (Table 4). Interestingly, the degree of conservation varies among different modes of splicing with the hierarchical order being as follows: CS donors and acceptors > ES donor and acceptor > AD donor and AA acceptor > IN donor and acceptor. This hierarchy is consistent between human and mouse and with both donors and acceptors, suggesting the presence of evolutionary selective pressures for the conservation of splice site sequences in different modes of splicing.
TABLE 4.
Median acceptor and donor splice site strengths
| Human | Mouse | |||||
| Acceptor splice sites | Median | SD | P-value | Median | SD | P-value |
| CS | 7.17 | 2.07 | 7.07 | 3.10 | ||
| ES | 6.54 | 2.47 | 1.8 × 10−9 | 6.22 | 2.84 | 1.1 × 10−8 |
| IN | 3.8 | 6.38 | 4.8 × 10−45 | 4.81 | 5.87 | 4.2 × 10−7 |
| AA distal | 5.67 | 4.09 | 3.0 × 10−10 | 5.64 | 2.21 | 3.0 × 10−9 |
| AA proximal | 5.62 | 4.38 | 2.7 × 10−14 | 5.16 | 2.91 | 1.8 × 10−14 |
| AD | 7.43 | 4.60 | 0.112 | 7.29 | 2.99 | 0.252 |
| Donor splice sites | ||||||
| CS | 6.89 | 2.33 | 6.64 | 3.04 | ||
| ES | 6.26 | 2.57 | 7.0 × 10−9 | 5.87 | 2.91 | 2.0 × 10−4 |
| IN | 1.74 | 6.19 | 2.5 × 10−9 | 3.28 | 5.45 | 5.0 × 10−25 |
| AA | 6.84 | 5.20 | 0.255 | 6.49 | 5.04 | 0.857 |
| AD distal | 4.75 | 4.82 | 2.0 × 10−17 | 5.47 | 4.66 | 2.7 × 10−6 |
| AD proximal | 4.45 | 5.11 | 5.3 × 10−18 | 5.03 | 3.49 | 5.8 × 10−9 |
(SD) Standard deviation. P-values were determined using the Wilcoxon rank sum test to test the significance of the difference between each alternative class with CS.
Orthologous exons
Orthologous AS have been found to be more conserved than orthologous CS (Modrek and Lee 2003; Sorek and Ast 2003; Philipps et al. 2004; Sorek et al. 2004b; Sugnet et al. 2004). There are 266 orthologous human–mouse genes in our data set. Using BLASTZ (Schwartz et al. 2000), we paired each exon with its orthologous partner and classified the exon pairs into four categories as follows: pairs that are alternatively spliced (AS) (52), pairs that are constitutively spliced (CS) (2320), pairs that are alternatively spliced in human but not in mouse (human-specific AS) (197), and pairs that are alternatively spliced in mouse but not in human (mouse-specific AS) (91). We observed (Table 5) a significantly higher level of sequence conservation in orthologous AS (93%) than in orthologous CS (89%), which agrees with a previous observation (Sorek et al. 2004b). However, species-specific AS (human-specific AS and mouse-specific AS) do not show greater sequence conservation than orthologous CS. Moreover, orthologous AS are significantly shorter than orthologous CS, as previously noted (Sorek et al. 2004b). In contrast, species-specific AS are longer than orthologous CS. These observations reinforce the idea that orthologous AS may belong to a different functional category than species-specific AS (Modrek and Lee 2003).
TABLE 5.
Median sequence conservation and length of orthologous exons
| Conservation | Length | |||||
| Orthologous exons | Median | SD | P-value | Median | SD | P-value |
| CS | 89% | 5.92 | 126 | 139 | ||
| AS | 93% | 9.19 | 2.9 × 10−4 | 103 | 394 | 7.8 × 10−6 |
| Human-specific AS | 89% | 7.38 | 0.719 | 135 | 268 | 1.6 × 10−2 |
| Mouse-specific AS | 89% | 7.78 | 0.442 | 140 | 337 | 5.3 × 10−4 |
CS refers to orthologous exon pairs in which neither is alternatively spliced. AS refers to orthologous exon pairs in which both are alternatively spliced. Human-specific AS and Mouse-specific AS refer to orthologous exon pairs which are alternatively spliced in human or mouse, but not in both species. (SD) Standard deviation. P-values were determined using the Wilcoxon rank sum test to test the significance of the difference between each alternative class with CS.
Parameters affecting splice site selection
AS differ significantly from CS in terms of splicing signal strength and length, suggesting that these parameters play important roles in splice site selection. We measured the EST coverage (as described in the accompanying MAASE database paper, Zheng et al. 2005) of specific spliced junctions to infer the expression level of specific alternative splicing events, keeping in mind the caveats associated with this quantification method (Modrek et al. 2001; Xie et al. 2002).
It has been widely assumed that the stronger a splice site, the more often it is used. This observation forms a basis for computational methods to predict splicing regulatory elements (Fairbrother et al. 2002). To test whether this is indeed the case, we determined the number of ESTs corresponding to the stronger (more conserved) or the weaker (less conserved) competing splice sites in AD and AA and find a positive correlation between the splice site strength and splice site selection in both human and mouse. In human, 63% of AA junctions and 58% of AD junctions show a positive correlation, with the remaining percentage of splicing events showing a negative correlation (perhaps due to other regulator mechanisms that are dominant over the splice site strength). In mouse, 61% for AA junctions and 55% for AD junctions shows the positive correlation. These findings demonstrate that stronger splice sites are preferred in >50% of the cases, thus supporting the contribution of splice site strength to splice site selection in gene expression.
For ES and IN, we could not use the same approach as above because the two alternative splice sites in these two modes are used as a unit. Therefore, we had to investigate the correlation between the combined strength of splice sites and EST frequency. We separated each splicing event into two classes as follows: one showing a higher EST frequency when the alternative splice sites are used rather than skipped (the “more retained” group), and the other with a higher EST frequency when the alternative splice sites are skipped rather than used (the “less retained” group). The splice site strengths within the two groups can then be compared to ask whether splice site strength is associated with splice site selection. Using this strategy, we found that, for ES, the “more retained” group has a median splice site strength significantly stronger than that of the “less retained” group in both human (P <8.8 × 10−6) and mouse (P < 6.6 × 10−4), thus suggesting that splice site strength and exon inclusion are positively correlated. For IN, the “less retained” has a median splice site strength significantly stronger than that of the “more retained” group in both human (P < 1.6 × 10−5) and mouse (P < 1.3 × 10−2), thus suggesting that splice site strength and intron removal are positively correlated.
Next, we investigated the relationship between exon length and splice site choice. For AD and AA, we counted the number of ESTs corresponding to junctions resulting in either the longer (using the proximal splice site) or shorter (using the distal splice site) spliced exon. Results indicate that the proximal sites (thus longer exons) are preferentially used in both AD and AA. In human, 66% of AA junctions and 70% of AD junctions correspond to the use of proximal sites. In mouse, the percentages are 67% for AA junctions and 65% for AD junctions. This bias likely reflects the so-called “proximal rule”, which states that the proximal splice site will be preferentially selected when the two competing sites have equal splice site strengths and similar cis-regulatory elements in flanking exons (Reed and Maniatis 1986; Ibrahim el et al. 2005). Because naturally occurring competing alternative splice sites are not exactly equal, as experimentally arranged, it is likely that splice site selection is influenced by splice site strength and distinct regulatory mechanisms in addition to the positional effect.
For ES and IN, results show an interesting relationship between exon length and splice site usage. With ES, the median length of “more retained” is significantly longer than that of “less retained” in both human (P < 1.4 × −5) and mouse (P < 1.1 × −2), meaning that the longer the exon, the more likely it is to be included. With IN, on the other hand, the median length of “more retained” is significantly shorter than those in “less retained” in both human (P < 2.6 × −5) and mouse (P < 3.7 × −3). This suggests that shorter introns may be more likely to be included than longer introns. However, these results may also be a reflection of the detection bias for shorter introns in sequencing EST clones, because shorter retained introns are more easily detected than longer retained introns.
Together, these results suggest a strong link between the strength of splice sites, the position of splice sites, and the length of alternative exons on the expression of an alternative splicing event. Similar trends in human and mouse strongly argue for the biological importance of these characteristics.
Sequence motifs implicated in splicing regulation
Because alternative splice sites are weaker than constitutive splice sites, it has been speculated that AS may have different frequencies and/or ratios of exonic splicing enhancers (ESEs) and silencers (ESSs) elements compared with CS. However, most of the studies conducted thus far focus on ES (Wang et al. 2004; Zhang and Chasin 2004). To test the generality of this observation in other modes of alternative splicing, we determined the distributions of computationally predicted ESEs and ESSs (Fairbrother et al. 2002; Wang et al. 2004; Zhang and Chasin 2004) onto each exon class. The two sets of ESEs and ESSs from the Burge (RESCUE-ESE and FAS-ESS) and Chasin (PESE and PESS) groups were separately mapped. RESCUE-ESEs are hexamers found to be enriched in exon versus intron sequences and in exon sequences with strong splice sites versus weak splice sites (Fairbrother et al. 2002). FAS-ESSs are hexamers identified as splicing silencers in a functional assay (Wang et al. 2004). PESEs and PESSs are octamers found to be enriched and depleted, respectively, in internal noncoding exons versus unspliced pseudo exons and in internal noncoding exons versus 5′ untranslated regions of intronless genes (Zhang and Chasin 2004). Each element was mapped within each exon sequence in each exon class (Fig. 1). The Wilcoxon rank sum test was then used to compare the distributions of a specific element within CS and an AS class (see Materials and Methods for further details). Comparisons resulting in a P-value ≤ 0.05 [and thus (1-p) ≥ 0.95] are highlighted in red. Individual hexamers are arranged according to their differential distributions in comparison between CS and ES. Clearly, a set of hexamers is over-represented in ES, while a different set is under-represented in ES (thus, over-represented in CS). Interestingly, when different modes of alternative splicing were compared with CS, different sets of over- and under-represented hexamers were identified, indicating that different modes of alternative splicing may have distinct preferences in selecting positive (ESEs) and negative (ESSs) regulatory elements. The most dramatic case can be seen in the comparison between CS and IN. IN have a dramatically higher frequency of ESSs. IN are, in this sense, more similar to introns. Trends are consistent for both human and mouse and for the two sets of previously predicted ESEs and ESSs.
FIGURE 1.

Mapping of previously computationally predicted ESEs and ESSs to different modes of AS. RESCUE-ESEs and FAS-ESSs (Fairbrother et al. 2002; Wang et al. 2004) are hexamers. PESEs and PESSs (Zhang and Chasin 2004) are octamers. Because alternative exons vary greatly in length, we normalized counts of ESEs and ESSs to their frequencies in 100 nt. Each element was mapped onto each exon of each exon class. The distribution of each element from an AS class was compared with the distribution from CS using the Wilcoxon rank sum test to determine the significance of the difference (P-value). To efficiently illustrate the comparison, the value of (1-p) was plotted for each element, a positive value indicates enrichment in CS and a negative value indicates enrichment in AS. Values of (1-p) ≥ 0.95 or ≤ −0.95 are displayed in red. Each bar represents one element and elements are ordered from least to the most frequent in ES. This ES-referenced order was used to display individual bars for each AS mode. Different modes of splicing are associated with different frequencies and distinct identities of ESEs and ESSs. Results from both sets of enhancer and silencer elements and from human and mouse are consistent, suggesting the potential for mode-specific regulation.
The distinct association and frequencies of ESEs and ESSs with individual splicing modes suggests the potential for mode-specific regulation. To uncover exonic regulatory elements associated with different modes of AS, we identified hexamers that are over-represented and under-represented in each AS class compared with CS. To enhance our predictive power, we incorporated comparative genomics and took advantage of two independently annotated data sets to develop a novel approach to motif analysis by using information from both human and mouse. Important regulatory elements are conserved between human and mouse (Sorek and Ast 2003; Kan et al. 2004; Sugnet et al. 2004; Yeo et al. 2004); therefore, including this intergenomic comparison would allow us to focus on biologically important regulatory elements.
Figure 2 shows the intergenomic comparison of each mode of AS with CS. Using the RSA tools (van Helden et al. 2000), frequencies of all possible 4096 hexamers were compared with a third-order Markov background model consisting of CS and AS to obtain a Z-score for each hexamer. Z-scores represent the standard deviation of each hexamer from the background model; however, they are not linearly comparable. Therefore, to directly compare the hexamers in different modes of AS to CS, Z-scores were converted to P-values (based on the standard normal curve). Over- and under-represented hexamers in each AS class were identified by comparing the P-values of hexamers from each AS class with the P-values of corresponding hexamers in the CS class (Δp = −log pCS–−log pAS). A positive Δp represents a hexamer that is under-represented in an AS class, while a negative Δp represents a hexamer that is over-presented in an AS class. The plot shows Δp for each AS exon class for human and mouse. In each plot, hexamers in quadrant H−M− are those under-represented in AS (thus over-represented in CS) in both human and mouse, while hexamers in quadrant H+M+ are those over-represented in a class of AS (thus under-represented in CS) in both human and mouse. The conservation between the two species suggests that the enriched hexamers have a higher potential to be biologically relevant. Hexamers in H−M+ and H+M− represent species-specific differences. Assuming that regulatory mechanisms are conserved between human and mouse (Yeo et al. 2004), the number of species-specific hexamers is expected to be low. Quadrants H−M+ and H−M+ can therefore be used to choose a cut-off for compiling a list of hexamers in quadrants H+M+ and H−M− that are over-represented and under-represented in each class of AS.
FIGURE 2.

Identification of potentially mode-specific regulatory motifs. Panels A–D are analyses of motif distribution between constitutive splicing (CS) and different modes of alternative splicing (ES, IN, AA, and AD). All 4096 possible hexamers were counted for each exon class in both human and mouse obtaining a Z-score (compared with a third-order Markov background model based on the combined CS and AS) describing the standard deviation of each hexamer frequency. P-values were then obtained from the Z-scores using the standard normal curve. Over- and under-represented hexamers in each AS class were identified by comparing the P-values of hexamers from each AS class with the P-values of corresponding hexamers in the CS class (Δp = −log pCS– −log pAS ). A positive Δp represents a hexamer that is under-represented in an AS class, while a negative Δp represents a hexamer that is over-presented in an AS class. The plot shows Δp for each AS exon class for human and mouse. H+M+ represents hexamers that are over-represented in an AS class compared with CS in both human and mouse. H−M− represents hexamers that are under-represented in an AS class compared with CS in both human and mouse. H−M+ and H+M− represent species-specific differences. Because species-specific differences are expected to be low, we made a cut-off based on minimal species-specific differences (shown in red) to retrieve significantly over- and under-represented hexamers in each class of AS. These hexamers were then compared with previously predicted ESEs and ESSs. These hexamers were directly comparable to the RESCUE-ESEs and FAS-ESSs hexamers. However, with the PESE and PESS octamers, the hexamers were compared with hexamers that were represented at least three times within the octamers (Wang et al. 2004). The number of previously predicted motifs is displayed in the bar graphs below each Δp plot. Strikingly, the motifs enriched in different AS modes belong to distinct sets with little overlap, indicating that we may have identified mode-specific cis-acting regulatory elements.
Analysis of these over- and under-represented hexamers in each AS class further supports the potential of mode-specific regulation. When under-represented hexamers from each mode were compared with each other, there was at least a 70% overlap in each comparison (data not shown). These under-represented hexamers in AS are actually over-represented hexamers in CS; thus, this high degree of consistency not only validated our prediction method (ability to consistently identify hexamers associated with CS in multiple comparisons), but also gave strong support for the existence of a unique set of potential regulatory elements associated with CS. Interestingly, when over-represented hexamers from each mode where compared with each other, the highest percentage of overlap was <10% (data not shown). The low degree of overlap between over-represented hexamers in each AS class suggests the existence of potentially mode-specific regulatory elements in each AS class.
Further analysis of these over- and under-represented AS hexamers reveals the presence of previously predicted ESEs and ESSs (Fairbrother et al. 2002; Wang et al. 2004; Zhang and Chasin 2004). Our AS hexamers were directly compared with RESCUE-ESE and FAS-ESS hexamers. Over-represented hexamers, those which are present three or more times within the PESE and PESS octamers, were used to compare with our AS hexamers (Wang et al. 2004). Results reveal strikingly consistent findings in the frequency and distribution of ESEs and ESSs within different modes (Fig. 2). However, because there are a higher number of previously predicted ESEs than ESSs, interpretations concerning these ratios need to be carefully considered. Beside the trend in ratios, it is clear that one set of ESEs are selectively enriched in ES compared with CS (H+M+) and a different set of ESEs are selectively depleted in ES compared with CS (H−M−). In IN, on the other hand, the prominent features are the enrichment of ESSs and the depletion of ESEs. Thus, IN resembles regular introns in terms of the distribution of regulatory motifs. Interestingly, AA and AD seem to have significantly reduced ESEs, suggesting that the depletion of positive regulatory elements may be a key mechanism for these modes of alternative splicing. Together, the observations re-enforce the possibility that different mechanisms are involved in regulating different modes of alternative splicing.
Exonization of multiple types of transposable elements
Mammalian genomes have remarkably high percentages of transposable elements (Lander et al. 2001; Waterston et al. 2002). ALU elements within intronic regions have been shown to be capable of becoming exonized, contributing to ~5% of the skipped exons surveyed (Sorek et al. 2002, 2004a). There are, however, multiple classes of transposable elements, including short interspersed nuclear elements (SINEs), long interspersed nuclear elements (LINEs), long terminal repeats (LTRs), and DNA transposons (DNAs). The tendency for these elements to become exonized remains unknown. In addition, the contribution of transposable elements to the creation of other modes of alternative splicing has yet to be investigated.
Contribution of SINEs to alternative splicing in both human and mouse
SINEs are 100–400 nt retrotransposons, which include ALU elements, and make up ~13% of the human genome and ~8% of the mouse genome (Lander et al. 2001; Waterston et al. 2002). Our analysis shows that SINE elements are abundantly present in introns (Fig. 3). SINE elements are relatively low in abundance near the splice site and become more pronounced farther into the intron. This abundance is seen in both CS and AS junctions. Previous reports indicate that Alu elements in human are also present in exons, but they are only associated with AS (Sorek et al. 2002; Lev-Maor et al. 2003). We have extended the analysis to all classes of SINEs and found that they are associated with AS, not CS. Furthermore, when AS were separately analyzed according to splicing modes, we found that only ES are associated with SINE elements.
FIGURE 3.

Contribution of transposable elements (SINEs, LINEs, LTRs, DNAs) in the evolution of alternative splicing. Exon regions are depicted as black boxes and intron regions are depicted as black lines at the bottom. The 300-nucleotide (nt) regions surrounding each acceptor and donor splice site were divided into 25-nt bins. Each bin represents a collection of sequences at a defined position surrounding the splice sites. The height of each bar indicates the percentage of those sequences overlapping the repeat elements. Blue bars represent the association of individual repeat elements with AS; yellow bars represent the association of individual repeat elements with CS. Results show that only transposable elements, not other classes of repeat elements (i.e., simple repeats and low complexity repeats), are specifically associated with alternative exons.
Since ALU elements are primate specific, we considered whether exonization of SINEs is limited to primates. Strikingly, we found a similar degree of association of SINEs with AS in mouse. SINE-associated AS in mouse is, once again, limited to ES and largely corresponds to B4 and MIR elements (Lander et al. 2001; Waterston et al. 2002). Surprisingly, the B1 elements in mouse, which are the closest relatives to the ALU elements in human (both are derived from 7SL RNA [Quentin 1994]), are not strongly associated with AS. These findings strongly suggest that transposable elements other than ALU may become exonized and contribute to the creation of novel alternative splicing events.
Role of other transposable elements in alternative splicing
To systematically survey the potential involvement of the remaining classes of transposable elements in alternative splicing, we performed a similar analysis on LINEs, LTRs, and DNAs. LINEs are ~6000-nt-long retrotransposons, which make up ~21% of the human genome and ~19% of the mouse genome (Lander et al. 2001; Waterston et al. 2002). As shown in Figure 3, LINEs are preferentially associated with AS, with the L1 type being the major category and ES being the majority of AS. We note a lower occurrence and a lower AS association of LINEs in the mouse, but the explanation is unclear.
LTRs make up ~8.5% of the human genome and ~10% of the mouse genome, whereas DNAs are the least abundant element, making up ~3% in human and ~1% in mouse. Similar to SINEs and LINEs, LTRs and DNAs are both present in AS in human. The majority of the DNAs associated with AS are MERs (Lander et al. 2001; Waterston et al. 2002). We could not find any LTRs or DNAs in either CS or AS in mouse. It is unclear whether this represents a true difference between human and mouse, is because of a limited sample size, or is a reflection of the low abundance of DNAs in mouse.
Lack of association between other genomic repeat classes and alternative splicing
Results described thus far support a specific association of transposons with AS, with ES being the predominant exon class. To determine whether this association is a general theme across all classes of repeated elements or is only restricted to transposable elements, we mapped two additional repeat classes, simple repeats and low complexity repeats. These two repeat elements are observed in both CS and AS (Fig. 3). There appears to be no positional preference of either of these two repeat classes for either AS or CS in either human or mouse. Thus, our findings suggest that transposable elements, but not other types of repeat elements, may play a specific role in the evolution of AS (Kazazian 2004; Miller and Capy 2004).
DISCUSSION
In this study, we used highly curated information from the MAASE database to characterize specific features associated with constitutive and different modes of alternative splicing, taking into account evolutionary conservation between human and mouse. Many of our findings confirm previous reports, indicating that signals at alternative splice sites are weaker, the length of AS are shorter than that of CS, ES tend to preserve the reading frame at a greater frequency than CS, and orthologous AS are more conserved than orthologous CS. These features are shown to be correlated with splice site selection (based on EST frequencies at individual spliced junctions) of AS. The trend indicates that splice site selection is generally governed by the strength of splice sites (preference for stronger sites), the position of splice sites (preference for proximal sites), and the length of alternative exons (preference for longer exons).
We have now significantly extended previous findings by showing that many of these features also differ between different modes of AS. For example, different modes of splicing have distinct splice site strengths, distinct alternatively spliced exon length distributions, and distinct frequencies of reading frame maintenance. Furthermore, different modes of splicing are shown to be associated with different distributions and frequencies of ESEs and ESSs, suggesting that different modes of splicing may be differentially regulated. To investigate this critical issue, we identified over- and under-represented hexamers in each AS class compared with CS. Our analysis revealed distinct hexamers associated with distinct modes of splicing. In addition, we find that distinct modes are also associated with different elements of previously predicted ESEs and ESSs, suggesting the potential for mode-specific regulation. It will be interesting to further study these mode-specific hexamers, ESEs and ESSs.
Finally, analysis of the different modes of splicing also illustrates that different modes may have emerged from distinct evolutionary paths. Distinct evolutionary pathways have been previously postulated for ES and mutually exclusive exons (Kondrashov and Koonin 2001); here we report observations suggesting the same may be true for other modes as well. Throughout our analysis, IN is clearly distinct from other modes of AS; however, differences also exist between ES, AA, and AD. ES are shorter in length and have a higher frequency of reading frame preservation than AA and AD. In addition, only ES show a specific association with transposable elements. This suggests that exonization of transposable sequences is not a general phenomenon of alternative splicing, rather, it is a specific trait associated with the creation of ES. The differences between ES, AA, and AD, in fact, highlight fundamental similarities between CS, AA, and AD. For example, CS and the constitutive portions of AA and AD share similar degrees of reading frame preservation and similar exon-length distributions. These similarities suggest a potential evolutionary mechanism in which the constitutive portions of AA and AD originate as CS, with portions of neighboring introns being later converted into the alternative portions of AA and AD (Kondrashov and Koonin 2003).
In conclusion, annotation of alternative splicing events in both human and mouse has allowed us to make general distinctions between constitutive and alternative splicing events and to identify specific characteristics associated with different modes of alternative splicing. Future enlargement of the MAASE database will facilitate further refinement of the features and motifs associated with AS. Our findings may serve as a foundation for the development of alternative splicing prediction tools for mammalian genomes. Finally, unique sequence features associated with AS may allow for the elucidation of cellular regulatory programs for alternative splicing when combined with information on the expression of trans-acting factors.
MATERIALS AND METHODS
Statistical analysis
Unless noted otherwise, all P-values were determined using the Wilcoxon rank sum test, a statistical test of the null hypothesis that two populations are identical against the alternative hypothesis that the two are not identical. All comparisons were made between CS and a specified class of AS.
Splice site strength calculation
A log-odds matrix was used to calculate the score of each splice site. Separate log-odds matrices were built for human and mouse using their respective CS splice sites. The nucleotide positions scored for acceptor splice sites spanned from the last 13 nt of the intron to the first nucleotide of the exon. The nucleotide positions scored for donor splice sites spanned from the last 3 nt of the exon to the first 7 nt of the intron. The scoring function is defined as:
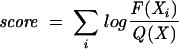 |
where F(Xi) is the frequency of finding X at position i in sequence I (each donor or acceptor sequence) and Q(X) is the frequency of X in the corresponding CS splice site.
Orthologous exons
We identified orthologous human–mouse genes using Homolo-Gene (Wheeler et al. 2004). Between orthologous genes, all human exons were compared with all mouse exons using BLASTZ (Schwartz et al. 2000). The exon pairs scoring the highest sequence similarity were defined to be orthologous exon pairs. Default parameters were used.
The mapping of previously predicted ESEs and ESSs
Previously predicted ESEs and ESSs from Chris Burge’s group (RESCUE-ESEs and FAS-ESSs) and Larry Chasin’s group (PESEs and PESSs) were mapped onto each exon of each exon class. The distribution of elements in an AS class is compared with CS using the Wilcoxon rank sum test.
The identification of potential mode-specific regulatory motifs
Using the RSA tools (van Helden et al. 2000), all 4096 possible hexamers were counted and compared with a background model consisting of a third-order Markov model computed from the joint set of CS and AS from the same species to obtain a Z-score for each hexamer. Z-scores measure the deviation from expectation in units of standard deviations. Each Z-score was converted to a P-value using the standard normal curve. This conversion allowed over- and under-represented hexamers for each AS class to be identified by comparing with hexamers from CS. For each hexamer, the P-value for each AS mode was subtracted from the P-value for CS (Δp = −log pCS – −log pAS). Hexamers with positive Δp values are those that are under-presented in an AS class, while a negative Δp score are those that are over-represented in an AS class. This Δp value for each hexamer was similarly calculated for human and mouse.
Analysis of genomic repeat elements
Human and mouse repeat elements were retrieved from the annotated databases of the UCSC Genome Browser (http://genome.ucsc.edu/index.html). The genomic locations of each repeat class (SINEs, LINEs, LTRs, DNAs, simple repeats, and low complexity repeats) were surveyed for genomic positional overlap with specific CS and AS exon and intron regions surrounding the splice sites. The sequence regions bordering individual splice sites, from −150 to +150, were divided into 25 nucleotide bins. The frequency at which a bin overlapped with a specific repeat element is calculated.
Acknowledgments
This work was supported by a grant from the National Cancer Institute (CA888351) to X-D.F. and M.G, and by the facilities of the National Biomedical Computational Resource (RR-08605) at the San Diego Supercomputer Center, University of California, San Diego. We acknowledge the contribution of Dr. Larry Chasin to the statistical analysis of over- and under-represented motifs in human and mouse and many other constructive comments and suggestions during the review of this manuscript.
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2660805.
REFERENCES
- Baelde, H.J., Eikmans, M., Doran, P.P., Lappin, D.W., de Heer, E., and Bruijn, J.A. 2004a. Gene expression profiling in glomeruli from human kidneys with diabetic nephropathy. Am. J. Kidney Dis. 43: 636–650. [DOI] [PubMed] [Google Scholar]
- Baelde, H.J., Eikmans, M., van Vliet, A.I., Bergijk, E.C., de Heer, E., and Bruijn, J.A. 2004b. Alternatively spliced isoforms of fibronectin in immune-mediated glomerulosclerosis: The role of TGFβ and IL-4. J. Pathol. 204: 248–257. [DOI] [PubMed] [Google Scholar]
- Berget, S.M. 1995. Exon recognition in vertebrate splicing. J. Biol. Chem. 270: 2411–2414. [DOI] [PubMed] [Google Scholar]
- Brett, D., Pospisil, H., Valcarcel, J., Reich, J., and Bork, P. 2002. Alternative splicing and genome complexity. Nat. Genet. 30: 29–30. [DOI] [PubMed] [Google Scholar]
- Caceres, J.F. and Kornblihtt, A.R. 2002. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. 18: 186–193. [DOI] [PubMed] [Google Scholar]
- Clark, F. and Thanaraj, T.A. 2002. Categorization and characterization of transcript-confirmed constitutively and alternatively spliced introns and exons from human. Hum. Mol. Genet. 11: 451–464. [DOI] [PubMed] [Google Scholar]
- Croft, L., Schandorff, S., Clark, F., Burrage, K., Arctander, P., and Mattick, J.S. 2000. ISIS, the intron information system, reveals the high frequency of alternative splicing in the human genome. Nat. Genet. 24: 340–341. [DOI] [PubMed] [Google Scholar]
- Dredge, B.K., Polydorides, A.D., and Darnell, R.B. 2001. The splice of life: Alternative splicing and neurological disease. Nat. Rev. Neurosci. 2: 43–50. [DOI] [PubMed] [Google Scholar]
- Fairbrother, W.G., Yeh, R.F., Sharp, P.A., and Burge, C.B. 2002. Predictive identification of exonic splicing enhancers in human genes. Science 297: 1007–1013. [DOI] [PubMed] [Google Scholar]
- Galante, P.A., Sakabe, N.J., Kirschbaum-Slager, N., and de Souza, S.J. 2004. Detection and evaluation of intron retention events in the human transcriptome. RNA 10: 757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Blanco, M.A., Baraniak, A.P., and Lasda, E.L. 2004. Alternative splicing in disease and therapy. Nat. Biotechnol. 22: 535–546. [DOI] [PubMed] [Google Scholar]
- Ibrahim el, C., Schaal, T.D., Hertel, K.J., Reed, R., and Maniatis, T. 2005. Serine/arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers. Proc. Natl. Acad. Sci. 102: 5002–5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, H., Washio, T., and Tomita, M. 2004. Computational comparative analyses of alternative splicing regulation using full-length cDNA of various eukaryotes. RNA 10: 1005–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J.M., Castle, J., Garrett-Engele, P., Kan, Z., Loerch, P.M., Armour, C.D., Santos, R., Schadt, E.E., Stoughton, R., and Shoemaker, D.D. 2003. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302: 2141–2144. [DOI] [PubMed] [Google Scholar]
- Kan, Z., Castle, J., Johnson, J.M., and Tsinoremas, N.F. 2004. Detection of novel splice forms in human and mouse using cross-species approach. Pac. Symp. Biocomput. 42–53. [DOI] [PubMed]
- Kazazian Jr., H.H 2004. Mobile elements: Drivers of genome evolution. Science 303: 1626–1632. [DOI] [PubMed] [Google Scholar]
- Kondrashov, F.A. and Koonin, E.V. 2001. Origin of alternative splicing by tandem exon duplication. Hum. Mol. Genet. 10: 2661–2669. [DOI] [PubMed] [Google Scholar]
- ———. 2003. Evolution of alternative splicing: Deletions, insertions and origin of functional parts of proteins from intron sequences. Trends Genet. 19: 115–119. [DOI] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B. Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Lev-Maor, G., Sorek, R., Shomron, N., and Ast, G. 2003. The birth of an alternatively spliced exon: 3′ splice site selection in Alu exons. Science 300: 1288–1291. [DOI] [PubMed] [Google Scholar]
- Miller, W.J. and Capy, P. 2004. Mobile genetic elements as natural tools for genome evolution. Methods Mol. Biol. 260: 1–20. [DOI] [PubMed] [Google Scholar]
- Modrek, B. and Lee, C.J. 2003. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat. Genet. 34: 177–180. [DOI] [PubMed] [Google Scholar]
- Modrek, B., Resch, A., Grasso, C., and Lee, C. 2001. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 29: 2850–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipps, D.L., Park, J.W., and Graveley, B.R. 2004. A computational and experimental approach toward a priori identification of alternatively spliced exons. RNA 10: 1838–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quentin, Y. 1994. A master sequence related to a free left Alu monomer (FLAM) at the origin of the B1 family in rodent genomes. Nucleic Acids Res. 22: 2222–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, R. and Maniatis, T. 1986. A role for exon sequences and splice-site proximity in splice-site selection. Cell 46: 681–690. [DOI] [PubMed] [Google Scholar]
- Schwartz, S., Zhang, Z., Frazer, K.A., Smit, A., Riemer, C., Bouck, J., Gibbs, R., Hardison, R., and Miller, W. 2000. PipMaker—a web server for aligning two genomic DNA sequences. Genome Res. 10: 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senapathy, P., Shapiro, M.B., and Harris, N.L. 1990. Splice junctions, branch point sites, and exons: Sequence statistics, identification, and applications to genome project. Methods Enzymol. 183: 252–278. [DOI] [PubMed] [Google Scholar]
- Sorek, R. and Ast, G. 2003. Intronic sequences flanking alternatively spliced exons are conserved between human and mouse. Genome Res. 13: 1631–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek, R., Ast, G., and Graur, D. 2002. Alu-containing exons are alternatively spliced. Genome Res. 12: 1060–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek, R., Lev-Maor, G., Reznik, M., Dagan, T., Belinky, F., Graur, D., and Ast, G. 2004a. Minimal conditions for exonization of intronic sequences: 5′ splice site formation in alu exons. Mol. Cell. 14: 221–231. [DOI] [PubMed] [Google Scholar]
- Sorek, R., Shemesh, R., Cohen, Y., Basechess, O., Ast, G., and Shamir, R. 2004b. A non-EST-based method for exon-skipping prediction. Genome Res. 14: 1617–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm, S., Zhu, J., Nakai, K., Stoilov, P., Stoss, O., and Zhang, M.Q. 2000. An alternative-exon database and its statistical analysis. DNA Cell. Biol. 19: 739–756. [DOI] [PubMed] [Google Scholar]
- Sugnet, C.W., Kent, W.J., Ares M., Jr., and Haussler, D. 2004. Transcriptome and genome conservation of alternative splicing events in humans and mice. Pac. Symp. Biocomput. 66–77. [DOI] [PubMed]
- Thanaraj, T.A. and Stamm, S. 2003. Prediction and statistical analysis of alternatively spliced exons. Prog. Mol. Subcell. Biol. 31: 1–31. [DOI] [PubMed] [Google Scholar]
- van Helden, J., Andre, B., and Collado-Vides, J. 2000. A web site for the computational analysis of yeast regulatory sequences. Yeast 16: 177–187. [DOI] [PubMed] [Google Scholar]
- Wang, Z., Rolish, M.E., Yeo, G., Tung, V., Mawson, M., and Burge, C.B. 2004. Systematic identification and analysis of exonic splicing silencers. Cell 119: 831–845. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E. Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
- Wheeler, D.L., Church, D.M., Edgar, R. Federhen, S., Helmberg, W., Madden, T.L., Pontius, J.U., Schuler, G.D., Schriml, L.M., Sequeira, E., et al. 2004. Database resources of the National Center for Biotechnology Information: Update. Nucleic Acids Res. 32: D35–D40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, H., Zhu, W.Y., Wasserman, A., Grebinskiy, V., Olson, A., and Mintz, L. 2002. Computational analysis of alternative splicing using EST tissue information. Genomics 80: 326–330. [DOI] [PubMed] [Google Scholar]
- Yeo, G., Hoon, S., Venkatesh, B., and Burge, C.B. 2004. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc. Natl. Acad. Sci. 101: 15700–15705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavolan, M., Kondo, S., Schonbach, C., Adachi, J., Hume, D.A., Hayashizaki, Y., and Gaasterland, T. 2003. Impact of alternative initiation, splicing, and termination on the diversity of the mRNA transcripts encoded by the mouse transcriptome. Genome Res. 13: 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.H. and Chasin, L.A. 2004. Computational definition of sequence motifs governing constitutive exon splicing. Genes & Dev. 18: 1241–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, C.L., Nair, T.M., Gribskov, M., Kwon, Y.S., Li, H.R., and Fu, X.D. 2004. A database designed to computationally aid an experimental approach to alternative splicing. Pac. Symp. Biocomput. 78–88. [DOI] [PubMed]
- Zheng, C.L, Kwon, Y.-S., Li, H.-R., Zhang, K., Coutinho-Mansfield, G., Yang, C., Nair T.M., Gribskov, M., and Fu, X.-D. 2005. MAASE: An alternative splicing database designed for supporting splicing microarray applications. RNA (this issue). [DOI] [PMC free article] [PubMed]