

Abstract
The polypyrimidine tract binding (PTB) protein is a potent regulator of alternative mRNA splicing. It also participates in other essential cellular functions, including translation initiation and polyadenylation. Several published reports have suggested that the protein forms a dimer in solution, a feature that has been widely incorporated into mechanistic models of protein function. However, recent studies have provided indications that full-length PTB is a monomer. Here we present new biophysical and biochemical evidence supporting the monomeric status of the protein. By use of blue-native polyacrylamide gel electrophoresis and size-exclusion chromatography, PTB was observed as a single molecular species under native reducing environments, though in oxidizing conditions, a larger protein species was also detected. Further analyses of wild-type and mutant PTB molecules with SDS-PAGE and time-of-flight electrospray ionization mass spectroscopy confirmed these observations. They also identified the single reduced species as monomeric PTB and the higher-molecular-weight nonreduced species as disulphide-linked PTB dimer mediated by Cys23. Our results indicate that the use of oxidizing environments in previous studies is likely to have contributed to the misassignment of PTB as a dimer. Although purified PTB may form disulphide-linked dimers under these conditions, in the reducing intracellular environment the protein will be monomeric. These findings have implications for the construction of models of PTB function in regulating mRNA metabolism.
Keywords: polypyrimidine tract binding protein, PTB, monomer, disulphide, dimer
INTRODUCTION
The polypyrimidine tract binding protein (PTB) is an RNA-binding protein with an extensive repertoire of cellular roles. It has been demonstrated to play a crucial role in the negative regulation of alternative splicing, via the repression of exon inclusion, for a growing number of genes (see Izquierdo et al. 2005; Sharma et al. 2005; and references therein). Indeed, PTB has been shown to regulate its own alternative splicing (Wollerton et al. 2004). PTB also promotes translation initiation from a variety of viral (Hellen et al. 1993; Kaminski et al. 1995; Pilipenko et al. 2001) and cellular (Mitchell et al. 2001, 2005; Pickering et al. 2003) internal ribosome entry sites and is reported to regulate the polyadenylation (Luo 1999; Castelo-Branco et al. 2004), localization (Cote et al. 1999), and stabilization (Knoch et al. 2004) of mRNA.
PTB contains four RNA recognition motifs (RRMs), the most common motif found in RNA-binding proteins (Maris et al. 2005). In PTB, both RRMs 1 and 4 display a classic βαββαβ topology, while RRMs 2 and 3 possess a C-terminal extension incorporating a fifth β-strand, which enlarges the RNA-binding β-sheet surface (Conte et al. 2000; Simpson et al. 2004). Each RRM in PTB contributes to RNA binding, with specificity for short polypyrimidine sequences, although the protein has also recently been reported to associate with double-stranded RNA (Mitchell et al. 2005).
Initial investigations of the oligomerization state of PTB were interpreted as indicating that PTB is a homodimer in solution, with dimerization being postulated to result from interactions predominantly involving RRM2 (Perez et al. 1997; Oh et al. 1998). The assumption of homodimer formation was widely incorporated, both explicitly and implicitly, into models of PTB function (Wagner and Garcia-Blanco 2001; Mitchell et al. 2003; Pickering et al. 2004). However, more recent analytical ultracentrifugation analysis of PTB and a range of recombinant subfragments suggest that the protein may in fact be monomeric (Simpson et al. 2004; Amir-Ahmady et al. 2005) and have therefore challenged the existing consensus on PTB dimerization. The methods used in these recent studies rely on an accurate determination of the PTB concentration, a nontrivial measurement given the low extinction coefficients of the constructs used (Simpson et al. 2004; Amir-Ahmady et al. 2005).
We report here new concentration-independent biochemical and biophysical analyses of wild-type and mutant PTB. Our results show that PTB dimerization only occurs in oxidizing conditions and is mediated by a single intermolecular disulphide bridge involving Cys23. This finding indicates that some of the previous reports of PTB dimerization are due to experiments being performed under oxidizing conditions. Under reducing conditions, which is the redox state relevant to the intracellular milieu, PTB is found to be monomeric.
RESULTS
PTB dimerization requires an oxidizing environment
Comparison of the range of published results on PTB oligomerization suggested that the assignment of PTB as a monomer or dimer might correlate with the use of reducing or oxidizing conditions in the experiment (Perez et al. 1997; Oh et al. 1998; Simpson et al. 2004). We therefore decided to investigate this possibility explicitly.
Blue native polyacrylamide gel electrophoresis (BN-PAGE), a “charge shift” method of electrophoresis (Schagger et al. 1994), was used to determine the molecular mass and native oligomeric status of purified recombinant PTB1. In oxidizing conditions, both monomeric and dimeric forms were observed (Fig. 1, lane 1); no species larger than a dimer was identified. However, in a reducing environment, PTB1 dimerization was completely abrogated (Fig. 1, lane 2). Interestingly, the PTB1 monomer band appeared as a doublet in the absence of DTT but as a single band following reduction (Fig. 1, cf. lanes 1 and 2).
FIGURE 1.
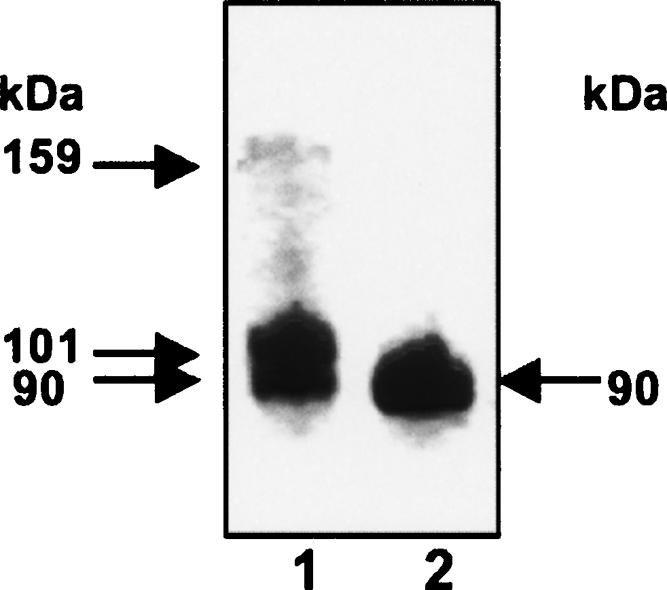
Blue-native polyacrylamide gel electrophoresis (BN-PAGE). Ten micrograms of PTB1 was separated under native conditions using BN-PAGE. (Lane 1) Oxidizing environment, (lane 2) reducing environment (+10 mM DTT). Distinct protein bands are marked by arrows and their calculated molecular weight listed.
The apparent molecular weight of PTB1 was calculated from triplicate experiments using a linear plot of Mr against electrophoretic migration of known protein standards (see Materials and Methods). The calculated molecular weights were as follows: PTB1 dimer, 159 kDa; nonreduced PTB1 monomer, 101 and 90 kDa; and reduced PTB1 monomer, 90 kDa. While these values are larger than are the theoretical masses (monomer, 58,316 Da; dimer, 116,633 Da), the monomeric values are broadly consistent with those obtained previously by using size-exclusion chromatography (Simpson et al. 2004). It is likely that that the molecular mass is overestimated by this technique due to the extended rod-shaped structure of PTB1, which was revealed by small-angle X-ray scattering (Simpson et al. 2004).
We next used size-exclusion chromatography to examine PTB oligomerization. Under reducing conditions (2 mM DTT), PTB1 eluted with an apparent molecular weight in the region of 100 kDa, with minimal additional protein signal outside this major peak (Fig. 2, dotted line; Simpson et al. 2004). This is likely to correspond to monomeric PTB given the asymmetric shape of the protein. In the absence of any reducing agent, although most of the protein eluted at ~100 kDa, the peak was significantly broader and a second elution peak was observed at a predicted molecular mass of 239 kDa (Fig. 2, solid line). This elution profile is broadly consistent with that obtained under oxidizing conditions by Perez et al. (1997). The 239-kDa peak observed in our elution under oxidizing conditions is presumably due to disulphide-linked dimers and probably corresponds to the protein shoulder previously observed at ~200 kDa (Perez et al. 1997). The predicted mass of this dimer peak is over twice that of the eluted monomeric protein and suggests that formation of an intermolecular disulphide bridge does not lead to a more compact, globular protein conformation.
FIGURE 2.

Size-exclusion chromatography of PTB1. Size-exclusion chromatography was performed on PTB1 under both oxidizing (solid line) and reducing conditions (dashed line). The estimated molecular weights of the elution peaks are labeled.
Cys23 mediates intermolecular disulphide bridge formation
The change in oligomeric profile for PTB1 upon variation in reducing environment provided strong evidence that the observed dimers resulted from intermolecular disulphide bridge formation. PTB contains three cysteines: at residue 23, in the N-terminal sequence preceding RRM1, and at residues 250 and 251 within RRM2. The N-terminal sequence of PTB is likely to be flexible since it is proteolytically sensitive (Simpson et al. 2004), and Cys23 is therefore potentially accessible for participation in disulphide bridges. The three-dimensional structure of RRM2 (PDB ID 1sjr) reveals that Cys250 and Cys251 are found close to the protein surface at the C-terminal end of the loop between helix-2 and strand β4 (Simpson et al. 2004). Although Cys251 appears inaccessible to solvent, Cys250 is partially surface-exposed and may also be able to participate in disulphide bridge formation.
To clarify whether PTB could form disulphide bridge–mediated dimers, site-directed mutagenesis was used to generate the mutant constructs PTB1-C23S and PTB1-C250S/C251S. The formation of disulphide-mediated dimers was assayed by SDS-PAGE using either an oxidizing or a reducing sample loading buffer. Under reducing conditions, wild-type PTB1 and both mutants (PTB1-C23S, PTB1-C250S/C251S) migrated solely as monomers (Fig. 3, lanes 1–3). In contrast, in an oxidizing environment, both PTB1 and PTB1-C250S/C251S migrated at sizes consistent with the presence of both monomer and dimer (Fig. 3, lanes 4,6). However, PTB1-C23S remained entirely monomeric (Fig. 3, lane 5). These results clearly demonstrated not only that PTB1 can form disulphide-mediated dimers but that these are mediated solely through intermolecular interactions involving Cys23.
FIGURE 3.

SDS-PAGE analysis of PTB1 and cysteine mutant derivatives. One microliter of 10 μM protein samples was analyzed in either reducing (lanes 1–3) or oxidizing (lanes 4– 6) environments. (Lanes 1,4) PTB1, (lanes 2,5) PTB1-C23S, (lanes 3,6) PTB1-C250S/C251S, (lane 7) Benchmark protein ladder (Invitrogen). The arrow indicates the presence of disulphide-mediated dimers.
The lack of formation of intermolecular disulphide bonds by cysteines C250 and C251 within RRM2 is consistent with the observation by NMR that they readily form an intramolecular connection. When exposed to oxidizing conditions (either the absence or the depletion of 10 mM DTT), chemical shift changes are observed in both cysteines and also in residues immediately adjacent in the structure, which are entirely consistent with disulphide bond formation (Fig. 4A,B). The chemical shift changes are reversible by the addition of fresh reducing agent and are not accompanied by any change in NMR line width, indicating that the domain remains monomeric. Together these data indicate that the cysteine residues in RRM2 readily form an intramolecular disulphide bond.
FIGURE 4.

NMR analysis of intramolecular disulphide formation by C250 and C251. (A) Expanded region from the two-dimensional 1H-15N HSQC spectrum of PTB RRM2 domain in the presence (black) and the absence (red) of 10 mM reduced DTT. Some residues exhibiting a change in chemical shift upon oxidation are labeled. A question mark indicates the presumed shift of C251, which was not determined unambiguously. (B) Cartoon representation of PTB RRM2. Residues associated with significant shifts in A are shown in a stick representation (side chains only). The structure is colored according to the change in chemical shift upon oxidation (gray, no shift; pink, small shift; red, large shift). The figure was generated using Pymol (DeLano Scientific).
To probe the oligomerization state of PTB and the impact of the Cys residues under more native conditions, we used nanospray ElectroSpray Ionisation Mass Spectrometry (ESI-MS), a technique that is well adapted to the analysis of both covalent and noncovalent protein–protein interactions (Hernandez and Robinson 2001; Hanson and Robinson 2004). In particular, we compared the oligomerization states of wild-type PTB1, the PTB1-C23S mutant, and PTB1–1234, a truncated construct that lacks the first 54 amino acids (and therefore lacks Cys23). Proteins were analyzed at a concentration of 10 μM in 50 mM ammonium acetate (pH 6.5).
Under oxidizing conditions, analysis of PTB1 identified mass/charge peaks consistent with the presence of both monomeric (58,328 ± 7 Da) and dimeric species (116,661 ± 15 Da) (Fig. 5A). For the monomer, two conformations were indicated by the presence of two charge state distributions in the spectra. Increasing instrument pressure in the source/transfer region, thereby favoring noncovalent dimer formation, produced minimal detectable changes in the observed spectra. In addition, MS/MS analysis failed to dissociate the PTB1 dimer peaks into monomers, even with increasing collision energy (data not shown). However, the presence of 5 mM DTT eliminated the dimer peaks; it also resulted in the appearance of a peak at 5.7 kDa, most likely a minor proteolytic contaminant (Fig. 5B). The presence of dimers could not be restored with increasing pressure.
FIGURE 5.

Electrospray ionization mass spectroscopy analysis of PTB proteins. Samples containing 2 μL of 10 μM protein in 50 mM ammonium acetate (pH 6.5) were analyzed by nanospray electrospray ionization mass spectroscopy under native conditions using a modified QToF2 instrument fitted with a standard Z-spray nanoflow electrospray source. The mass/charge peaks were identified using MassLynx, and the corresponding masses are listed to the right of each panel. (A) PTB1, (B) reduced PTB1 (buffer supplemented with 5 mM DTT), (C) PTB1-1234, (D) PTB1-C23S.
To confirm that these dimers were disulphide mediated, we performed ESI-MS analysis of the constructs PTB1-1234 and PTB1-C23S, in which Cys23 has been deleted and mutated, respectively. Only monomeric protein could be identified for PTB1-1234 (53,108 ± 5 Da) and PTB1-C23S (58,349 ± 17 Da) under both oxidizing (Fig. 5C,D) and reducing (data not shown) conditions. Together these data strongly indicate that the PTB1 dimers formed in oxidizing conditions are the result of covalent disulphide interactions between the cysteine residue at position 23 on each protein molecule.
DISCUSSION
This report provides new biophysical and biochemical evidence that PTB exists as a monomer in solution, confirming recent findings derived from analytical ultracentrifugation experiments with PTB (Simpson et al. 2004; Amir-Ahmady et al. 2005). Moreover, in conjunction with results from NMR and small-angle X-ray scattering experiments (Simpson et al. 2004), the present observations provide an explanation for the previous misconception that PTB exists as a homodimer in solution. Firstly, the asymmetrical rod-shaped conformation of PTB1 leads to a significant overestimation of the molecular weight of PTB monomers when native gels or size-exclusion techniques are used. Secondly, we have shown that although PTB dimers can form under oxidizing conditions, these are entirely due to the formation of an intermolecular disulphide bridge involving Cys23 on both partners. We suggest that both of these factors contributed to the apparent observation of PTB dimerization in previous analyses of PTB oligomerization, which used gel filtration chromatography, in vitro pull-down assays, nondenaturing PAGE, and glutaraldehyde cross-linking, and were all performed under oxidizing conditions (Perez et al. 1997; Oh et al. 1998). In experiments performed under reducing conditions (Figs. 1, 2, 3, 5), PTB behaved as a monomer; we found no evidence for noncovalent dimerization.
In vitro pull-down assays (Perez et al. 1997) and glutaraldehyde cross-linking experiments (Oh et al. 1998) using recombinant PTB fragments lacking Cys23 provided evidence for self-association of PTB that cannot be attributed to the formation of disulphide bridges and led to the suggestion that RRM2 mediated noncovalent association of PTB monomers. However, in these studies, the self-association behavior was principally observed for constructs in which RRM2 was located either at the N or the C terminus of the truncated protein, an exposed position that is not found in full-length PTB. Subsequent analyses using both NMR and analytical ultracentrifugation have shown that these types of fragment aggregate nonspecifically (Simpson et al. 2004).
The formation of disulphide-mediated PTB dimers cannot account for self-association behavior deduced from yeast two-hybrid experiments (Oh et al. 1998), since the reducing environment of the yeast nucleus would have prevented disulphide bridge formation. However, it remains possible that the apparent PTB–PTB interaction observed by this technique could have been mediated by interactions with other proteins or with RNA. Yeast two-hybrid data need to be supported by more direct analyses of protein–protein interactions, but the balance of the data now suggests that PTB is indeed monomeric.
Until recently it has been widely accepted that human PTB exists as a homodimer in solution. It is now apparent that under reducing conditions PTB exists as a monomer and that this is the physiologically relevant state of the protein in the reducing environment of the cell. This study provides a revealing example of the care that needs to be taken in determining the oligomeric state of intracellular proteins. The findings will also help to clarify analyses of the likely stoichiometry of PTB–RNA complexes and the formulation of models of PTB function. Of course, it remains possible that multimers of PTB will form when the protein associates with RNA targets or other proteins in vivo (Chou et al. 2000; Polydorides et al. 2000; Huttelmaier et al. 2001; Amir-Ahmady et al. 2005).
MATERIALS AND METHODS
Plasmid construction and protein expression
The plasmids PTB1 (residues 1–531) and PTB1-1234 (residues 55–531) expressing human PTB have been described previously (Simpson et al. 2004). Mutant constructs PTB1-C23S and PTB1-C250S/C251S were created by QuikChange site-directed mutagenesis of PTB1 using the following primer pairs, in which the mutated codons are underlined: C23S, GACGAGCTCTTCTCTACTAGTGTCACTAACGGACC and GGTCCGTTAGTGACACTAGTAGAGAAGAGCTCGTC; C250S/C251S, AGAACATCTACAACGCCTCCTCCACGCTGCGCATC and GATGCGCAGCGTTGGAGGAGGCGTTGTAGATGTTCT. Mutations were confirmed by sequencing. Proteins were expressed essentially as described (Conte et al. 2000) by using SG13009 cells and were purified by using TALON (Clontech) affinity resin.
BN-PAGE
BN-PAGE analysis was performed based on the method of Schagger and von Jagow (1991) using 10 μg of protein in 20 mM Tris (pH 7.7), 200 mM NaCl, and 10% glycerol. To create a reducing environment, this was supplemented with 10 mM DTT. Electrophoresis was performed by using prefocused (100 V, 60 min, 4°C) 4%–15% Tris-HCl gradient gels (Biorad) for 2–4 h at 15 mA and 4°C. The cathode buffer was 50 mM Tricine, 15 mM BisTris-HCl (pH 7.0), and 0.02% (w/v) Coomassie blue G; the anode buffer, 50 mM BisTris-HCl (pH 7.0). To enhance final visualization of protein bands, gels were completely destained (40% EtOH, 10% acetic acid) and then restained (50% EtOH, 10% acetic acid, 0.1% [w/v] Coomassie blue). A linear plot of Mr against distance migrated was obtained for each gel by using the following proteins: β-amylase (200 kDa), alcohol dehydrogenase and its dissociated subunits (150 kDa and 50 kDa), HSA monomer and disulphide-linked dimer (66 kDa and 132 kDa), and chicken albumin (45 kDa).
SDS-PAGE
One microliter of 10 μM protein stocks was subjected to standard electrophoresis in 12% SDS-polyacrylamide gels using both reducing and nonreducing sample loading buffers. Two times reducing sample loading buffer is 125 mM Tris-HCl (pH 6.8), 2% SDS, 20% glycerol, 0.02% Bromophenol blue, and 730 mM β-mercaptoethanol. Oxidizing sample loading buffer did not contain β-mercaptoethanol. Samples were not boiled prior to electrophoresis.
Size-exclusion chromatography
Size-exclusion chromatography was performed as described previously (Simpson et al. 2004) by using either a reducing (25 mM Tris at pH 7.2, 250 mM NaCl, 2 mM DTT), or a nonreducing (25 mM Tris at pH 7.2, 250 mM NaCl) buffer.
NMR spectroscopy
Two-dimensional 1H-15N NMR spectra were recorded at 303 K on a 500-MHz four-channel Bruker DRX500 spectrometer equipped with a z-shielded gradient triple resonance cryoprobe. Samples of reduced 15N-labeled PTB1 RRM2 were ~100–200 μM (pH 6.5) in 50 mM Na phosphate buffer, 150 mM NaCl, 10 mM DTT, and 1 mM NaN3. Spectra of oxidized protein were acquired both by allowing samples containing DTT to oxidize in air and by preparing fresh protein in identical buffer but with the omission of DTT. Chemical shift changes upon oxidation were judged significant by using a weighted sum of 10(ΔδH) + ΔδN (ppm) as described previously (Simpson et al. 1999).
Time-of-flight electrospray ionization MS
For analysis under native conditions, purified protein was buffer exchanged into 50 mM ammonium acetate (pH 6.5) and diluted to a final concentration of 10 μM in 50 mM ammonium acetate (pH 6.5). Spectra were also recorded on samples supplemented with 2–5 mM DTT. Data were acquired by using a modified QToF2 instrument (Waters) fitted with a standard Z-spray nano-flow electrospray source. Sample solutions were sprayed from borosilicate glass capillaries (1 mm outer diameter × 0.5 mm inner diameter) that had been drawn down to a closed taper using a Model P-97 Flaming/Brown micropipette puller (Sutter Instrument) and coated with gold using a SEM coater (Polaron). The tapered end of the capillary was cut under a microscope. A low backing pressure of nitrogen gas was used to initiate and maintain flow through the capillary. All data were acquired in positive ion mode using a nitrogen cone gas flow to aid desolvation. Throttling of the source rotary pump was used to control instrument pressures for collisional cooling. Spectra were acquired with the following parameters: capillary voltage, 1.8 kV; cone voltage, 200 V; extractor voltage, 0 V; 1 × 10−2 mbar indicated source pressure; and 6.9 × 10−7 mbar indicated ToF analyser pressure. All data were processed by using MassLynx software, and cesium iodide solution was employed for calibration.
Acknowledgments
This work was funded by grant support from the Wellcome Trust.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2214405.
REFERENCES
- Amir-Ahmady, B., Boutz, P.L., Markovtsov, V., Phillips, M.L., and Black, D.L. 2005. Exon repression by polypyrimidine tract binding protein. RNA 11: 699–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo-Branco, P., Furger, A., Wollerton, M., Smith, C., Moreira, A., and Proudfoot, N. 2004. Polypyrimidine tract binding protein modulates efficiency of polyadenylation. Mol. Cell. Biol. 24: 4174–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, M.Y., Underwood, J.G., Nikolic, J., Luu, M.H., and Black, D.L. 2000. Multisite RNA binding and release of polypyrimidine tract binding protein during the regulation of c-src neural-specific splicing. Mol. Cell 5: 949–957. [DOI] [PubMed] [Google Scholar]
- Conte, M.R., Grune, T., Ghuman, J., Kelly, G., Ladas, A., Matthews, S., and Curry, S. 2000. Structure of tandem RNA recognition motifs from polypyrimidine tract binding protein reveals novel features of the RRM fold. EMBO J. 19: 3132–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote, C.A., Gautreau, D., Denegre, J.M., Kress, T.L., Terry, N.A., and Mowry, K.L. 1999. A Xenopus protein related to hnRNP I has a role in cytoplasmic RNA localization. Mol. Cell 4: 431–437. [DOI] [PubMed] [Google Scholar]
- Hanson, C.L. and Robinson, C.V. 2004. Protein–nucleic acid interactions and the expanding role of mass spectrometry. J. Biol. Chem. 279: 24907–24910. [DOI] [PubMed] [Google Scholar]
- Hellen, C.U., Witherell, G.W., Schmid, M., Shin, S.H., Pestova, T.V., Gil, A., and Wimmer, E. 1993. A cytoplasmic 57-kDa protein that is required for translation of picornavirus RNA by internal ribosomal entry is identical to the nuclear pyrimidine tract-binding protein. Proc. Natl. Acad. Sci. 90: 7642–7646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, H. and Robinson, C.V. 2001. Dynamic protein complexes: Insights from mass spectrometry. J. Biol. Chem. 276: 46685–46688. [DOI] [PubMed] [Google Scholar]
- Huttelmaier, S., Illenberger, S., Grosheva, I., Rudiger, M., Singer, R.H., and Jockusch, B.M. 2001. Raver1, a dual compartment protein, is a ligand for PTB/hnRNPI and microfilament attachment proteins. J. Cell. Biol. 155: 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo, J.M., Majos, N., Bonnal, S., Martinez, C., Castelo, R., Guigo, R., Bilbao, D., and Valcarcel, J. 2005. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol. Cell 19: 475–484. [DOI] [PubMed] [Google Scholar]
- Kaminski, A., Hunt, S.L., Patton, J.G., and Jackson, R.J. 1995. Direct evidence that polypyrimidine tract binding protein (PTB) is essential for internal initiation of translation of encephalomyocarditis virus RNA. RNA 1: 924–938. [PMC free article] [PubMed] [Google Scholar]
- Knoch, K.P., Bergert, H., Borgonovo, B., Saeger, H.D., Altkruger, A., Verkade, P., and Solimena, M. 2004. Polypyrimidine tract-binding protein promotes insulin secretory granule biogenesis. Nat. Cell Biol. 6: 207–214. [DOI] [PubMed] [Google Scholar]
- Luo, G. 1999. Cellular proteins bind to the poly(U) tract of the 3′ untranslated region of hepatitis C virus RNA genome. Virology 256: 105–118. [DOI] [PubMed] [Google Scholar]
- Maris, C., Dominguez, C., and Allain, F.H. 2005. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 272: 2118–2131. [DOI] [PubMed] [Google Scholar]
- Mitchell, S.A., Brown, E.C., Coldwell, M.J., Jackson, R.J., and Willis, A.E. 2001. Protein factor requirements of the Apaf-1 internal ribosome entry segment: Roles of polypyrimidine tract binding protein and upstream of N-ras. Mol. Cell. Biol. 21: 3364–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, S.A., Spriggs, K.A., Coldwell, M.J., Jackson, R.J., and Willis, A.E. 2003. The Apaf-1 internal ribosome entry segment attains the correct structural conformation for function via interactions with PTB and unr. Mol. Cell 11: 757–771. [DOI] [PubMed] [Google Scholar]
- Mitchell, S.A., Spriggs, K.A., Bushell, M., Evans, J.R., Stoneley, M., Le Quesne, J.P., Spriggs, R.V., and Willis, A.E. 2005. Identification of a motif that mediates polypyrimidine tract-binding protein-dependent internal ribosome entry. Genes & Dev. 19: 1556–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, Y.L., Hahm, B., Kim, Y.K., Lee, H.K., Lee, J.W., Song, O., Tsukiyama-Kohara, K., Kohara, M., Nomoto, A., and Jang, S.K. 1998. Determination of functional domains in polypyrimidine-tract-binding protein. Biochem. J. 331: 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, I., McAfee, J.G., and Patton, J.G. 1997. Multiple RRMs contribute to RNA binding specificity and affinity for polypyrimidine tract binding protein. Biochemistry 36: 11881–11890. [DOI] [PubMed] [Google Scholar]
- Pickering, B.M., Mitchell, S.A., Evans, J.R., and Willis, A.E. 2003. Polypyrimidine tract binding protein and poly r(C) binding protein 1 interact with the BAG-1 IRES and stimulate its activity in vitro and in vivo. Nucleic Acids Res. 31: 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering, B.M., Mitchell, S.A., Spriggs, K.A., Stoneley, M., and Willis, A.E. 2004. Bag-1 internal ribosome entry segment activity is promoted by structural changes mediated by poly(rC) binding protein 1 and recruitment of polypyrimidine tract binding protein 1. Mol. Cell. Biol. 24: 5595–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilipenko, E.V., Viktorova, E.G., Guest, S.T., Agol, V.I., and Roos, R.P. 2001. Cell-specific proteins regulate viral RNA translation and virus-induced disease. EMBO J. 20: 6899–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydorides, A.D., Okano, H.J., Yang, Y.Y., Stefani, G., and Darnell, R.B. 2000. A brain-enriched polypyrimidine tract-binding protein antagonizes the ability of Nova to regulate neuron-specific alternative splicing. Proc. Natl. Acad. Sci. 97: 6350–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger, H. and von Jagow, G. 1991. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199: 223–231. [DOI] [PubMed] [Google Scholar]
- Schagger, H., Cramer, W.A., and von Jagow, G. 1994. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217: 220–230. [DOI] [PubMed] [Google Scholar]
- Simpson, P.J., Bolam, D.N., Cooper, A., Ciruela, A., Hazlewood, G.P., Gilbert, H.J., and Williamson, M.P. 1999. A family IIb xylan-binding domain has a similar secondary structure to a homologous family IIa cellulose-binding domain but different ligand specificity. Structure Fold. Des. 7: 853–864. [DOI] [PubMed] [Google Scholar]
- Simpson, P.J., Monie, T.P., Szendroi, A., Davydova, N., Tyzack, J.K., Conte, M.R., Read, C.M., Cary, P.D., Svergun, D.I., Konarev, P.V., et al. 2004. Structure and RNA interactions of the N-terminal RRM domains of PTB. Structure (Camb.) 12: 1631–1643. [DOI] [PubMed] [Google Scholar]
- Wagner, E.J. and Garcia-Blanco, M.A. 2001. Polypyrimidine tract binding protein antagonizes exon definition. Mol. Cell. Biol. 21 3281–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollerton, M.C., Gooding, C., Wagner, E.J., Garcia-Blanco, M.A., and Smith, C.W. 2004. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol. Cell 13: 91–100. [DOI] [PubMed] [Google Scholar]