


Abstract
In vitro bypass of damaged DNA by replicative DNA polymerases is usually blocked by helix-distorting or bulky DNA lesions. In this study, we report that substitution of the divalent metal ion Mg2+ with Mn2+ promotes quantitative replication of model DNA substrates containing the major cisplatin or N-2-acetylaminofluorene adducts by the catalytic subunit (UL30) of the replicative DNA polymerase of herpes simplex virus. The ability of Mn2+ ions to confer bypass of bulky lesions was not observed with other replicative DNA polymerases of the B family, such as bacteriophage T4 or δ polymerases. However, for these enzymes, manganese induced the incorporation of one nucleotide opposite the first (3′) guanine of the d(GpG) intrastrand cisplatin lesion. Translesion replication of the cisplatin adduct by UL30 led to the incorporation of mismatched bases, with the preferential incorporation of dAMP opposite the 3′ guanine of the lesion. Furthermore, substitution of MgCl2 with MnCl2 greatly inhibited the 3′ to 5′ exonuclease of UL30 but had a far lesser effect on that of T4 DNA polymerase. Finally, manganese induced a conformational change in the structure of UL30 bound to the platinated substrate. Taken together, the latter findings suggest a mechanism by which manganese might allow UL30 to efficiently promote translesion DNA synthesis in vitro.
INTRODUCTION
The survival and evolution of organisms depend critically on their ability to faithfully replicate their DNA. However, DNA is continually subjected to damaging agents and a variety of DNA repair pathways have evolved to repair DNA lesions. Despite this, some lesions can escape repair and thus impede the progress of a growing replication fork (1).
DNA polymerases can be classified into six main families based upon phylogenetic relationships with Escherichia coli pol I (Class A), E.coli pol II (Class B), E.coli pol III (Class C), euryarchaeotic pol II (Class D), human pol β (Class X), and E.coli pol UmuC/Din B and eukaryotic RAD 30/XP-V (Class Y) (2). The B family includes replicative DNA polymerases such as herpes simplex virus type-1 (HSV-1) polymerase, bacteriophage T4 polymerase and eukaryotic pol α, δ and ε polymerases. In vitro elongation catalyzed by replicative DNA polymerases appears to be blocked by helix-distorting or bulky DNA lesions (1). Examples of such lesions are the intrastrand adduct between two adjacent guanines produced by the antitumor compound cisplatin [Pt-d(GpG)] or the modification at the C-8 position of a guanine produced by the rat liver carcinogen N-2-acetylaminofluorene (AAF). On the other hand, some polymerases of the X family, like pol β, or the recently discovered DNA polymerases belonging to the Y family, can replicate through at least some DNA structure distorting lesions (3). Translesion synthesis proficient DNA polymerases of the X or Y families share some common properties including very limited processivity and lack of associated 3′ to 5′ exonuclease ‘proofreading’ activity. In addition, the crystal structures of three DNA polymerases of the Y family indicate that, compared with replicative polymerases, they possess both a more ‘open’ active site and the capacity to establish limited, non-specific contacts with the base pair at the primer terminus (4–6). Taken together, these characteristics provide possible mechanisms whereby Y family polymerases can bypass bulky DNA lesions.
Structural and mutagenesis studies have revealed the existence of two crucial acidic amino acid residues in the polymerase active site. These residues chelate a pair of divalent metal ions to promote catalysis. Their proposed role is to enable nucleophilic attack by the 3′-OH on the α-phosphate of the incoming dNTP (7). In vivo, it is probably Mg2+ that is used for catalysis (8). Previous experiments with Mg2+ have shown that replicative DNA polymerases are largely incapable of performing translesion DNA synthesis. In general, these studies revealed that replicative polymerases stopped at the base immediately 3′ to the lesion (9–11). Other studies have revealed that substitution of Mg2+ with Mn2+ induces DNA polymerases to make errors during in vitro DNA replication of undamaged DNA (12) and to insert one additional nucleotide opposite a bulky DNA lesion (13). However, at the concentrations of Mn2+ used in those studies, no significant extension beyond the lesions was detected.
In this study, we have examined the effect of MnCl2 on the capacity of the catalytic subunit (UL30) of the HSV-1 DNA polymerase, together with other polymerases belonging to the same family, to elongate templates containing two unique, well defined DNA lesions. We also investigated the effect of manganese on both the proofreading activity associated with UL30 and T4 DNA polymerases and on eventual conformational changes in the structure of UL30 bound to a platinated substrate.
MATERIALS AND METHODS
Proteins and chemicals
Recombinant HSV-1 UL30 was purified to near homogeneity as described (14). Calf thymus DNA polymerases δ and ε, purified as described (15), were a generous gift from Drs G. Maga and U. Hubscher (University of Zurich). For both enzymes, one unit of activity incorporates 1 nmol of dTMP into acid-precipitable material in 60 min at 37°C in a standard assay containing 0.5 µg (nucleotides) of poly(dA)/oligo(dT) and 20 µM dTTP. T4 DNA polymerase, T4 polynucleotide kinase and AciI restriction endonuclease were from New England Biolabs. [γ-32P]ATP (4500 Ci/mmol) was from Du Pont/NEN. Unlabeled deoxyribonucleotide-triphosphates were from Amersham Life Science. Sequencing grade, TPCK-treated, trypsin was obtained from Promega.
DNA substrates
DNA substrates used in this study are depicted in Figure 1. 60/17mer substrate (Fig. 1A), 60/39mer substrate (Fig. 1B), 60/40mer cytosine (Fig. 1D) and 60/40mer guanine (Fig. 1E), either unmodified or containing a single Pt-d(GpG) intrastrand adduct, were constructed as described (10). 44/15mer substrate (Fig. 1C), either unmodified or containing a single AAF adduct, was prepared as described (16).
Figure 1.

DNA substrates used in this study. Positions of the Pt-d(GpG) and AAF adducts are as indicated. (A) 60/17mer substrate, (B) 60/39mer substrate, (C) 44/15mer substrate, (D) 60/40mer cytosine and (E) 60/40mer guanine.
Primer extension assays
Reactions were performed for the times indicated at 37°C in 25 mM HEPES–NaOH pH 7.5, 1 mM dithiothreitol, 200 µg/ml bovine serum albumin, 500 µM dNTPs, 5% glycerol, 3 mM ATP and 80 mM NaCl with the indicated concentrations of enzymes and substrates. Divalent cations (Mg2+ or Mn2+) were present as indicated. Reactions were terminated by the addition of stop buffer (90% formamide, 0.1% xylene cyanol, 0.1% bromophenol blue, 1 mM EDTA). Samples were heated for 5 min at 90°C and analyzed on 15% polyacrylamide–7 M urea–30% formamide gels followed by autoradiography. Reactions were quantified by storage phosphor analysis with a Molecular Dynamics Storm 840 using the ImageQuant software. Full-length synthesis (60 and 44mers) is expressed as a percentage of all primer extension products. Bona fide replication of platinated template was determined based on the resistance of reaction products to digestion with AciI which is inhibited by the lesion (10,17).
Exonuclease assays
Reaction conditions were identical to those of the primer extension reactions except that deoxyribonucleotide triphosphates were omitted. Reactions were terminated and analyzed as described for the primer extension reactions.
Partial proteolysis assays
Reactions were performed in 25 mM HEPES–NaOH pH 7.5, 5% glycerol, 0.1 M NaCl and 1 mM dithiothreitol containing 90 nM UL30 and 250 nM platinated substrate B with either 10 mM MgCl2 or 4 mM MnCl2, as indicated. Reactions were assembled on ice and incubated at 30°C. Where indicated, trypsin was added to 7.5 µg/ml and incubation continued for 2 min. The reactions were terminated by the addition of 200 µM N-α-tosyl-l-lysine chloromethyl ketone, 0.5 µg/ml leupeptin, 0.7 µg/ml pepstatin A, 1 mM phenylmethylsulfonyl fluoride and 20 mM EDTA. Samples were denatured in SDS sample buffer and resolved by 12% polyacrylamide–0.1% SDS gel electrophoresis. Proteins were visualized either by silver staining or by immuno-blotting using an anti-UL30 rabbit serum.
RESULTS
Substitution of MgCl2 with MnCl2 leads to quantitative bypass of Pt-d(GpG) and AAF adducts by the HSV-1 DNA polymerase
The UL30 gene encodes the catalytic subunit of the HSV-1 DNA polymerase and possesses 3′ to 5′ proofreading exonuclease and RNase H activities in addition to its DNA polymerase activity (18). In order to assess the translesion replication capacity of UL30, we investigated its capacity to elongate a 5′ 32P-labeled 17mer primer annealed to untreated or cisplatin damaged 60mer template A depicted in Figure 1A. The newly synthesized DNA products were resolved by denaturing polyacrylamide gel electrophoresis and visualized by autoradiography. Figure 2A shows that UL30 efficiently replicated the untreated template in the presence of MgCl2 ranging from 5 to 40 mM. When the replication of the cisplatin-modified template was examined, we found that the vast majority (>90%) of the reaction products migrated as a 39mer, indicating arrest opposite the base immediately preceding the Pt-d(GpG) adduct. Quantification of the data shows that the extent of blockage was constant at all concentrations of MgCl2 examined (Fig. 2B). The faint band corresponding to full-length product detectable with the platinated template was sensitive to digestion with AciI (data not shown), indicating the presence of a small percentage (7–10%) of unplatinated DNA in our template preparation (see Materials and Methods). Substitution of MgCl2 with MnCl2 allowed efficient bypass replication of the Pt-d(GpG) lesion by UL30 (Fig. 2C). The data show that quantitative translesion synthesis was achieved with increasing concentrations of MnCl2, concomitant with the disappearance of the band at the arrest site. It should be noted that at a MnCl2 concentration of 0.5 mM, which does not yet enable UL30 to extend beyond the lesion, UL30 inserted one nucleotide opposite the first G of the Pt-d(GpG) adduct, in contrast to that observed with MgCl2. Significant full-length translesion synthesis products were obtained at 1 mM MnCl2, while at 4 mM up to 80% of the platinated substrate was replicated (Fig. 2D). This is in striking contrast to the products obtained following replication of the same substrate in the presence of comparable or higher concentrations of MgCl2 (compare lanes 12 and 13 of Fig. 2C). Full-length products obtained in the presence of MnCl2 were resistant to AciI digestion, indicating that the cisplatin lesion was still present in the final product following DNA synthesis by UL30 (data not shown).
Figure 2.

Replication of unplatinated and platinated substrate A by UL30 DNA polymerase in the presence of MgCl2 or MnCl2. (A) Autoradiogram of the reaction products. Reactions were performed as described in Materials and Methods with 10 nM substrate A and 100 nM UL30 for 60 min at the indicated concentrations of MgCl2. Lane 1, no enzyme. Lanes 2–6, replication of unplatinated substrate. Lanes 7–11, replication of platinated substrate. The positions of the 17mer (primer) and 60mer (full-length product) are indicated by arrows. The printed sequence on the right side corresponds to the part of the substrate containing the Pt-d(GpG) adduct (>Pt). (B) Quantification of the data shown in (A). Open circles, unplatinated substrate. Closed circles, platinated substrate. (C) Autoradiogram of the reaction products. Reactions were performed as described in Materials and Methods with 10 nM substrate A and 100 nM UL30 for 60 min at the indicated concentrations of MnCl2 or MgCl2. Lane 1, no enzyme. Lanes 2–7, replication of unplatinated substrate. Lanes 8–13, replication of platinated substrate. The positions of the 17mer (primer) and 60mer (full-length product) are indicated by arrows. The printed sequence on the right side corresponds to the part of the substrate containing the Pt-d(GpG) adduct (>Pt). (D) Quantification of the data shown in (C). Open circles, unmodified substrate. Closed circles, platinated substrate.
In subsequent experiments, we found that MnCl2 allowed UL30 to efficiently bypass the cisplatin lesion after only 5 min of incubation both at high polymerase/substrate ratios (10:1, Fig. 2C) and at an equimolar protein/DNA ratio (data not shown). Next, we investigated whether the capacity of Mn2+ to allow Pt-d(GpG) translesion synthesis by UL30 could be translated to another bulky DNA lesion. For this purpose we chose a template containing an AAF modification at the C8 position of a specific guanine (template C, Fig. 1C). This modification induces a severe conformational change in the vicinity of the adduct (19). As can be seen in Figure 3A, DNA synthesis by UL30 in the presence of Mg2+ arrests predominantly at the base preceding the lesion, albeit some low level incorporation was observed opposite the modified guanine. This pattern did not change for concentrations of MgCl2 up to 40 mM (data not shown). However, similar to the results obtained with the cisplatin lesion, substituting 10 mM MgCl2 with 4 mM MnCl2 led to quantitative bypass of the adduct (Fig. 3B).
Figure 3.

Replication of AAF-modified substrate C by UL30 DNA polymerase in the presence of MgCl2 or MnCl2. (A) Autoradiogram of the reaction products. Reactions were performed as described in Materials and Methods with 10 nM substrate C and 25 nM UL30 for the times indicated. Lanes 1–3, replication of AAF-modified substrate with MgCl2. Lanes 4–6, replication of AAF-modified substrate with MnCl2. The position of the 15mer (primer) and 44mer (full-length product) are indicated by arrows on the left. The printed sequence on the right side corresponds to the part of the substrate containing the AAF adduct (–AAF). (B) Quantification of the data shown in (A). Open triangles, AAF-modified substrate with MgCl2. Closed triangles, AAF-modified substrate with MnCl2.
Taken together, these data show that Mn2+ induces UL30 to promote translesion synthesis which is not restricted to a specific DNA lesion.
Mn2+ does not enable T4 or calf thymus δ DNA polymerases to elongate past the Pt-d(GpG) adduct, but allows incorporation of one nucleotide opposite the lesion
In addition to the HSV-1 polymerase, bacteriophage T4 and eukaryotic δ and ε polymerases belong to the class B family of DNA polymerases. Therefore, we asked whether Mn2+ could also influence the capacity of these enzymes to promote translesion synthesis. Cisplatin lesions have been found to block elongation in vitro by T4 polymerase with MgCl2 as catalytic cation (9). We have confirmed this finding and show that at a concentration of 4 mM MnCl2, which induces robust translesion synthesis by UL30, T4 polymerase could not elongate past the cisplatin (Fig. 4A) or AAF (data not shown) adducts. Quantification of the small fraction of full-length products (7%) indicates that these are likely to be a result of extension of undamaged DNA present in the template preparation. However, in contrast to what was observed with MgCl2, MnCl2 allowed T4 polymerase to incorporate one nucleotide opposite the 3′ guanine of the Pt-d(GpG) adduct (Fig. 4A, compare lanes 5 and 6 with 7 and 8).
Figure 4.

Replication of unplatinated and platinated substrate A by T4 and δ DNA polymerases in the presence of MgCl2 or MnCl2. (A) Autoradiogram of the reaction products. Reactions were performed as described in Materials and Methods with 10 nM substrate A, 45 nM T4 polymerase and either MgCl2 or MnCl2 for the times indicated. Lanes 1–4, replication of unplatinated substrate. Lanes 5–8, replication of platinated substrate. (B) Autoradiogram of the reaction products. Reactions were performed as described in Materials and Methods except that NaCl was omitted, with 10 nM substrate A, 0.4 U δ polymerase and either MgCl2 or MnCl2 for the times indicated. Lanes 1–4, replication of unplatinated substrate. Lanes 5–8, replication of platinated substrate. The positions of the 17mer (primer) and 60mer (full-length product) are indicated by arrows. The printed sequences on the right side of (A) and (B) correspond to the part of the substrate containing the Pt-d(GpG) adduct (>Pt).
We previously reported that, using the same platinated DNA as in this study, DNA synthesis catalyzed by calf thymus DNA polymerases δ and ε in the presence of MgCl2 arrested at the base preceding the Pt-d(GpG) lesion (10). Here, we found that 4 mM MnCl2 was also unable to promote translesion synthesis by these polymerases (Fig. 4B and data not shown). However, as for T4 polymerase, we found that Mn2+ allowed δ polymerase to incorporate one nucleotide opposite the 3′ guanine of the Pt-d(GpG) adduct (Fig. 4B, compare lanes 5 and 6 with 7 and 8).
Taken together, these data indicate that among the B class replicative DNA polymerases tested, the capacity of MnCl2 to induce synthesis past bulky lesions is restricted to UL30. However, Mn2+ allowed the insertion of one nucleotide opposite the 3′ guanine of the Pt-d(GpG) adduct by T4 and δ polymerases.
Translesion synthesis by the HSV-1 DNA polymerase is error prone
Translesion synthesis of the Pt-d(GpG) adduct by UL30 in the presence of manganese could be mutagenic or non-mutagenic depending on whether the correct or incorrect nucleotide is incorporated opposite the lesion. To obtain qualitative information about possible mis-incorporation opposite the cisplatin adduct, primer extension assays were performed with individual deoxynucleotides. These experiments were performed with platinated template B (Fig. 1B) annealed to a 5′ 32P-labeled 39mer to prime DNA synthesis immediately 3′ to the Pt-d(GpG) lesion. As expected, in the presence of MgCl2 no incorporation was detected opposite the lesion, with the exception of marginal incorporation of dCMP due to the presence of low amounts of unplatinated template (Fig. 5). With MnCl2, in addition to the incorporation of dCMP as the correct base, incorporation of all three other dNMPs was observed opposite the 3′ guanine of the Pt-d(GpG) adduct, indicating that significant misincorporation had occurred (Fig. 5). Quantification of the data indicates that MnCl2 directed the incorporation of deoxynucleotides in the following order: dCMP = dAMP > dTMP > dGMP.
Figure 5.
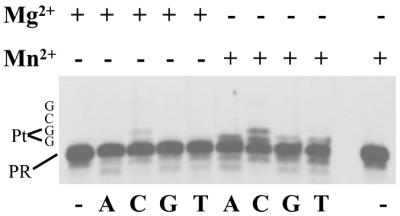
Single nucleotide incorporation by UL30 DNA polymerase on platinated substrate B in the presence of MgCl2 or MnCl2. Autoradiogram of the reaction products. Reactions were performed as described in Materials and Methods with 20 nM platinated substrate B, 10 nM UL30 and either 10 mM MgCl2 or 4 mM MnCl2 for 15 min. Single nucleotides were present at 500 µM as indicated. First and last lanes are with no enzyme. The sequence of the first four nucleotides of the template following the primer is indicated on the left (PR, primer).
These data indicate that translesion synthesis of the Pt-d(GpG) adduct by the HSV-1 polymerase is error prone.
MnCl2 inhibits the 3′ to 5′ exonuclease of the HSV-1 DNA polymerase on platinated DNA
An early model (20) suggested that proofreading exonuclease activity might play a role in preventing chain elongation caused by bulky lesions and that successful translesion synthesis might require suppression of the proofreading activity during lesion bypass.
Consequently, we examined whether MnCl2 has an effect on the proofreading exonuclease associated with the HSV-1 DNA polymerase. To test this possibility we used platinated substrates D and E depicted in Figure 1D and E, in which either a cytosine or guanine was inserted opposite the 3′ guanine of the Pt-d(GpG) adduct. We found that 3′ to 5′ exonuclease digestion by UL30 was significantly lower with MnCl2 than with MgCl2 (Fig. 6A, compare lanes 1–5 and 11–15 with lanes 6–10 and 16–20). Quantification of the data indicates that upon completion of the reaction <5% of the labeled primer remained with MgCl2, while up to 26% of the primers resisted exonuclease degradation with MnCl2.
Figure 6.

Effect of MnCl2 on the 3′ to 5′ exonuclease activities of UL30 and T4 DNA polymerases. (A) Autoradiogram of the reaction products with UL30 DNA polymerase. Reactions were performed as described in Materials and Methods with 20 nM platinated substrates D or E and 20 nM UL30 and either 10 mM MgCl2 or 4 mM MnCl2 for the times indicated. Lanes 1–10, substrate D. Lanes 11–20, substrate E. (B) Autoradiogram of the reaction products with T4 and UL30 DNA polymerases. Reactions were performed as described in Materials and Methods with 20 nM platinated substrate D and 20 nM UL30 or T4 polymerase and either 10 mM MgCl2 or 4 mM MnCl2 for the times indicated.
Since translesion synthesis by the T4 DNA polymerase was not induced by MnCl2, we examined whether substitution of magnesium with manganese also affected the 3′ to 5′ exonuclease associated with this enzyme. Figure 6B shows that, in contrast to UL30, the 3′ to 5′ exonuclease of T4 DNA polymerase on substrate D was barely affected by substituting magnesium with manganese (compare lanes 2–4 with lanes 5–7).
These data provide evidence for differential inhibition of the proofreading exonuclease activities of the HSV-1 and T4 DNA polymerases by manganese.
MnCl2 induces a conformational change in the HSV-1 DNA polymerase
Structural alterations imposed by helix-distorting DNA adducts are likely to influence the interaction of such DNA with a DNA polymerase. Substituting manganese for magnesium at the polymerase active site may affect the conformational transition required for the insertion of a base opposite the adduct. To test this idea, we used limited proteolysis to investigate conformational changes upon binding of UL30 to a Pt-d(GpG)-containing primer template in the presence of either MgCl2 or MnCl2. The substrate for this experiment was platinated substrate B (Fig. 1B), such that the polymerase is positioned to incorporate the next nucleotide opposite the lesion. Figure 7 shows the results of partial proteolysis of UL30 with trypsin analyzed by silver staining and immuno-blotting. The tryptic cleavage pattern is similar to what has previously been reported for UL30 (21). There was no difference in the cleavage pattern of UL30 with MgCl2 or MnCl2 in the absence of DNA substrate (compare Fig. 7A, lanes 1 and 2 and Fig. 7B, lanes 2 and 3). Likewise, addition of DNA substrate did not significantly affect the cleavage pattern in the presence of MgCl2 (Fig. 7A, lane 3 and Fig. 7B, lane 4). However, the cleavage pattern with DNA substrate in the presence of MnCl2 showed significantly diminished levels of the species at ∼66 kDa and elevated levels of the species at ∼100 kDa (Fig. 7A, lane 4 and Fig. 7B, lane 5).
Figure 7.

Partial proteolysis of UL30 DNA polymerase in the presence of MgCl2 or MnCl2. Reactions were performed as described in Materials and Methods. The positions of UL30, trypsin and molecular weight markers are as indicated. The asterisks indicate the proteolytic species affected by substitution of MgCl2 (10 mM) with MnCl2 (4 mM). (A) Silver stain of reaction products. Lane 1, trypsin digest of UL30 with MgCl2. Lane 2, trypsin digest of UL30 with MnCl2. Lane 3, as lane 1 with platinated substrate B. Lane 4, as lane 2 with platinated substrate B. Lane 5, untreated UL30. Lane 6, trypsin. (B) Immuno-blotting using an anti-UL30 rabbit serum followed by chemiluminescent analysis. Lane 1, untreated UL30. Lane 2, trypsin digest of UL30 with MgCl2. Lane 3, trypsin digest of UL30 with MnCl2. Lane 4, as lane 2 with platinated substrate B. Lane 5, as lane 3 with platinated substrate B.
These data indicate that MnCl2 induces a conformational change upon interaction of UL30 with a primer template which parallels the inhibition of its 3′ to 5′ exonuclease activity.
DISCUSSION
Recent studies indicate that specialized, low fidelity DNA polymerases are recruited to replicate across DNA lesions, where they temporarily substitute for the high fidelity and processive replicative polymerases (3,22–24). In agreement with this picture, replicative DNA polymerases appear to be far more selective than specialized lesion bypass enzymes and they are unable to transit many types of chemical modifications that severely distort the structure of DNA (1,25). Very likely, the fact that replicative enzymes possess both an associated proofreading exonuclease activity and an active site that can accommodate only the correct nucleotide matching the template (25,26) is instrumental in their inability to replicate damaged DNA.
All known DNA polymerases possess two crucial amino acid residues in their active site, which bind a pair of divalent metal ions and promote catalysis. Mg2+ is probably the divalent metal ion utilized by most polymerases for catalysis in vivo (8). Mn2+ can substitute for Mg2+ as a required cofactor but it is clear that its presence at millimolar concentrations influences both the fidelity and lesion bypass capacity of a number of DNA polymerases (12,13).
In this study, we have compared the ability of a number of replicative DNA polymerases of the B family to replicate model templates containing two unique and well defined DNA lesions as a function of increasing concentrations of MgCl2 or MnCl2. We found that only increasing concentrations of MnCl2 induced quantitative translesion replication of the Pt-d(GpG) and AAF adducts by the catalytic subunit of the HSV-1 DNA polymerase (Figs 2 and 3). For two other polymerases of the B family, eukaryotic pol δ and T4 DNA polymerases, MnCl2 did not permit elongation past the platinum and AAF lesions, but allowed incorporation of a single nucleotide opposite the 3′ guanine of the Pt-d(GpG) adduct (Fig. 4). A similar observation has been reported following replication by E.coli polymerase I of a template containing a pyrimidine dimer, where incorporation was detected opposite the 3′ thymine of the lesion only in the presence of MnCl2 (27).
Translesion replication of the Pt-d(GpG) adduct by UL30 in the presence of manganese was mutagenic, as shown by the detectable incorporation of all three mismatched bases opposite the 3′ guanine of the lesion, dAMP being the preferred one (Fig. 5). This finding is in agreement with the known mutagenic properties of manganese ions (12).
Early experiments suggested that inhibition of the proofreading activity may enable bypass replication by a DNA polymerase (20). According to this model, insertion of any nucleotide opposite the damaged site triggers the proofreading function because the lesion does not allow correct base pair formation, thus leading to a continuous cycle of nucleotide insertion and excision, which would prevent chain elongation. Recent data demonstrating that proofreading-deficient mutants of E.coli DNA polymerase III were able to replicate past pyrimidine dimers and AAF adducts have strengthened this possibility (28,29). Therefore, we decided to test if the MnCl2-induced capacity of UL30 to replicate across bulky DNA lesions was accompanied by a reduction in its proofreading activity. Our data indicate that MnCl2, at a concentration which allows quantitative bypass of cisplatin and AAF adducts, inhibited the enzyme’s 3′ to 5′ exonuclease activity on a platinated substrate bearing two different nucleotide residues opposite the 3′ guanine of the Pt-d(GpG) adduct (Fig. 6A). Interestingly, such inhibition is barely detectable on the 3′ to 5′ exonuclease activity associated with the T4 polymerase (Fig. 6B). These data suggest a correlation between the extent of exonuclease inhibition and the capacity of UL30 and T4 polymerase to elongate past the cisplatin lesion. The crystal structure of the NH2-terminal 388-residue fragment of T4 polymerase, which contains the 3′ to 5′ exonuclease active site, revealed the presence of two aspartate residues that serve as ligands for two metal ions (30). Similarly, a survey of the region containing the 3′ to 5′ exonuclease activity of UL30, which is located in the N-terminal half of the polypeptide, revealed the presence of three aspartate residues which are critical to 3′ to 5′ exonuclease activity (31). It is possible that, to account for the differences between the two enzymes, Mn2+ ions interact differently with these residues and cause differential inhibition of exonuclease activity.
However, early studies on polymerase termination indicated that even enzymes with no exonuclease activity were unable to efficiently catalyze in vitro synthesis past bulky lesions, suggesting that the mere inhibition of the proofreading activity might not be sufficient per se for increased translesion replication (32). Furthermore, because DNA lesions are expected to disturb the structure of the active site of a given polymerase, it is reasonable to expect that they may interfere with conformational rearrangements of the enzyme. Therefore, we envisaged the possibility that binding of Mn2+ instead of Mg2+ ions to the catalytic diad of UL30 may affect the conformational transition which occurs prior to the chemical step. To test this idea, we used limited proteolysis which has previously been used to study conformational changes in response to DNA damage in the Klenow fragment of E.coli DNA polymerase I (33). Our data (Fig. 7) indicate that Mn2+ induces a conformational change in UL30 in response to binding the damaged template primer. It should be noted that the Mn2+-induced conformational changes were also observed with undamaged template primer (data not shown). Therefore, our data support the notion that Mn2+ ions affect a conformational transition that might be required, together with the inhibition of the 3′ to 5′ exonuclease activity, for replication across a bulky DNA adduct. Interestingly, DNA polymerases of the B family exhibit functional coupling between their 3′ to 5′ exonuclease and polymerase activities. The crystal structures of two members of this family (34,35) suggest that this coupling may be due to the concerted movement of the exonuclease and finger domains relative to the catalytic region of the palm domain. This model raises the possibility that the inhibition of the 3′ to 5′ exonuclease activity by Mn2+ ions is directly related to the alteration in the conformation of UL30 (Fig. 7).
In conclusion, we have presented data showing that substituting MnCl2 for MgCl2 as catalytic cofactor led to quantitative translesion replication of bulky DNA adducts in vitro by the catalytic subunit (UL30) of the replicative HSV-1 polymerase. In contrast, in reactions catalyzed by other replicative polymerases of the B family, such as eukaryotic pol δ and T4 phage polymerases, MnCl2 only led to the insertion of an extra base opposite the first guanine of the Pt-d(GpG) adduct and did not permit further extension. We also found that MnCl2 inhibited to a greater extent the 3′ to 5′ exonuclease activity associated with UL30 than that of T4 polymerase. Finally, we detected some specific, MnCl2 driven conformational changes in the structure of UL30 upon binding to both unmodified and damaged (platinated) substrate.
Several mechanisms for Mn2+ mutagenesis have been proposed. It has been suggested, based on results obtained with T4 DNA polymerase, that the mutagenic effects of Mn2+ are caused by increasing the binding of a mispaired nucleotide (36). Recently, the effect of metal ion substitution on the dynamics of translesion DNA synthesis catalyzed by an exonuclease deficient form of T4 polymerase opposite an abasic site was quantitatively evaluated by steady-state and transient kinetics techniques (37). The Mn2+-dependent enhancement in translesion DNA synthesis was attributed to a substantial increase in the rate of conformational change preceding phosphoryl transfer. In the case of DNA polymerase β, it has been shown that Mn2+ increases catalysis compared with Mg2+, thereby allowing nucleotidyl transfer to take place with little or no regard to instructions from the template (38).
We would like to propose that Mn2+ can influence the translesion capacity of the HSV-1 replicative polymerase both by reducing its proofreading activity and by inducing conformational changes in its structure when bound to DNA.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr Neil Johnson for critical reading of the manuscript. This work was supported in part by grants 4373 from the Association pour la Recherche sur le Cancer (to G.V.) and GM62643 from the National Institutes of Health and BM 022 from the Florida Biomedical Research Program (to P.E.B.).
REFERENCES
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. American Society for Microbiology, Washington, DC.
- 2.Burgers P.M., Koonin,E.V., Bruford,E., Blanco,L., Burtis,K.C., Christman,M.F., Copeland,W.C., Friedberg,E.C., Hanaoka,F., Hinkle,D.C., Lawrence,C.W., Nakanishi,M., Ohmori,H., Prakash,L., Prakash,S., Reynaud,C.A., Sugino,A., Todo,T., Wang,Z., Weill,J.C. and Woodgate,R. (2001) Eukaryotic DNA polymerases: proposal for a revised nomenclature. J. Biol. Chem., 276, 43487–43490. [DOI] [PubMed] [Google Scholar]
- 3.Friedberg E.C., Fischhaber,P.L. and Kisker,C. (2001) Error-prone DNA polymerases: novel structures and the benefits of infidelity. Cell, 107, 9–12. [DOI] [PubMed] [Google Scholar]
- 4.Trincao J., Johnson,R.E., Escalante,C.R., Prakash,S., Prakash,L. and Aggarwal,A.K. (2001) Structure of the catalytic core of S. cerevisiae DNA polymerase eta: implications for translesion DNA synthesis. Mol. Cell, 8, 417–426. [DOI] [PubMed] [Google Scholar]
- 5.Zhou B.L., Pata,J.D. and Steitz,T.A. (2001) Crystal structure of a DinB lesion bypass DNA polymerase catalytic fragment reveals a classic polymerase catalytic domain. Mol. Cell, 8, 427–437. [DOI] [PubMed] [Google Scholar]
- 6.Ling H., Boudsocq,F., Woodgate,R. and Yang,W. (2001) Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell, 107, 91–102. [DOI] [PubMed] [Google Scholar]
- 7.Steitz T.A. (1999) DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem., 274, 17395–17398. [DOI] [PubMed] [Google Scholar]
- 8.Kornberg A. and Baker,T. (1991) DNA Replication. W.H. Freeman and Co., New York, NY.
- 9.Comess K.M., Burstyn,J.N., Essigmann,J.M. and Lippard,S.J. (1992) Replication inhibition and translesion synthesis on templates containing site-specifically placed cis-diamminedichloroplatinum(II) DNA adducts. Biochemistry, 31, 3975–3990. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann J.S., Pillaire,M.J., Maga,G., Podust,V., Hubscher,U. and Villani,G. (1995) DNA polymerase beta bypasses in vitro a single d(GpG)-cisplatin adduct placed on codon 13 of the HRAS gene. Proc. Natl Acad. Sci. USA, 92, 5356–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belguise-Valladier P., Maki,H., Sekiguchi,M. and Fuchs,R.P. (1994) Effect of single DNA lesions on in vitro replication with DNA polymerase III holoenzyme. Comparison with other polymerases. J. Mol. Biol., 236, 151–164. [DOI] [PubMed] [Google Scholar]
- 12.Sirover M.A. and Loeb,L.A. (1976) Infidelity of DNA synthesis in vitro: screening for potential metal mutagens or carcinogens. Science, 194, 1434–1436. [DOI] [PubMed] [Google Scholar]
- 13.Moore P.D., Bose,K.K., Rabkin,S.D. and Strauss,B.S. (1981) Sites of termination of in vitro DNA synthesis on ultraviolet- and N-acetylaminofluorene-treated phi X174 templates by prokaryotic and eukaryotic DNA polymerases. Proc. Natl Acad. Sci. USA, 78, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boehmer P.E. (1996) Expression, purification and characterization of the herpes simplex virus type-1 DNA polymerase. Methods Enzymol., 275, 16–35. [DOI] [PubMed] [Google Scholar]
- 15.Weiser T., Gassmann,M., Thommes,P., Ferrari,E., Hafkemeyer,P. and Hubscher,U. (1991) Biochemical and functional comparison of DNA polymerases alpha, delta and epsilon from calf thymus. J. Biol. Chem., 266, 10420–10428. [PubMed] [Google Scholar]
- 16.Lindsley J.E. and Fuchs,R.P. (1994) Use of single-turnover kinetics to study bulky adduct bypass by T7 DNA polymerase. Biochemistry, 33, 764–772. [DOI] [PubMed] [Google Scholar]
- 17.Villani G., Cazaux,C., Pillaire,M.J. and Boehmer,P. (1993) Effects of a single intrastrand d(GpG) platinum adduct on the strand separating activity of the Escherichia coli proteins RecB and RecA. FEBS Lett., 333, 89–95. [DOI] [PubMed] [Google Scholar]
- 18.Crute J.J. and Lehman,I.R. (1989) Herpes simplex-1 DNA polymerase. Identification of an intrinsic 5′ to 3′ exonuclease with ribonuclease H activity. J. Biol. Chem., 264, 19266–19270. [PubMed] [Google Scholar]
- 19.Fuchs R.P., Lefevre,J.F., Pouyet,J. and Daune,M.P. (1976) Comparative orientation of the fluorene residue in native DNA modified by N-acetoxy-N-2-acetylaminofluorene and two 7-halogeno derivatives. Biochemistry, 15, 3347–3351. [DOI] [PubMed] [Google Scholar]
- 20.Villani G., Boiteux,S. and Radman,M. (1978) Mechanism of ultraviolet-induced mutagenesis: extent and fidelity of in vitro DNA synthesis on irradiated templates. Proc. Natl Acad. Sci. USA, 75, 3037–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weisshart K., Kuo,A.A., Hwang,C.B., Kumura,K. and Coen,D.M. (1994) Structural and functional organization of herpes simplex virus DNA polymerase investigated by limited proteolysis. J. Biol. Chem., 269, 22788–22796. [PubMed] [Google Scholar]
- 22.Woodgate R. (1999) A plethora of lesion-replicating DNA polymerases. Genes Dev., 13, 2191–2195. [DOI] [PubMed] [Google Scholar]
- 23.Goodman M.F. and Tippin,B. (2000) Sloppier copier DNA polymerases involved in genome repair. Curr. Opin. Genet. Dev., 10, 162–168. [DOI] [PubMed] [Google Scholar]
- 24.Johnson R.E., Washington,M.T., Prakash,S. and Prakash,L. (1999) Bridging the gap: a family of novel DNA polymerases that replicate faulty DNA. Proc. Natl Acad. Sci. USA, 96, 12224–12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellenberger T. and Silvian,L.F. (2001) The anatomy of infidelity. Nature Struct. Biol., 8, 827–828. [DOI] [PubMed] [Google Scholar]
- 26.Doublie S., Tabor,S., Long,A.M., Richardson,C.C. and Ellenberger,T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature, 391, 251–258. [DOI] [PubMed] [Google Scholar]
- 27.Rabkin S.D., Moore,P.D. and Strauss,B.S. (1983) In vitro bypass of UV-induced lesions by Escherichia coli DNA polymerase I: specificity of nucleotide incorporation. Proc. Natl Acad. Sci. USA, 80, 1541–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vandewiele D., Borden,A., O’Grady,P.I., Woodgate,R. and Lawrence,C.W. (1998) Efficient translesion replication in the absence of Escherichia coli Umu proteins and 3′-5′ exonuclease proofreading function. Proc. Natl Acad. Sci. USA, 95, 15519–15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuchs R.P. and Napolitano,R.L. (1998) Inactivation of DNA proofreading obviates the need for SOS induction in frameshift mutagenesis. Proc. Natl Acad. Sci. USA, 95, 13114–13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J., Yu,P., Lin,T.C., Konigsberg,W.H. and Steitz,T.A. (1996) Crystal structures of an NH2-terminal fragment of T4 DNA polymerase and its complexes with single-stranded DNA and with divalent metal ions. Biochemistry, 35, 8110–8119. [DOI] [PubMed] [Google Scholar]
- 31.Hwang Y.T., Liu,B.Y., Coen,D.M. and Hwang,C.B. (1997) Effects of mutations in the Exo III motif of the herpes simplex virus DNA polymerase gene on enzyme activities, viral replication and replication fidelity. J. Virol., 71, 7791–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strauss B.S. (2002) The ‘A’ rule revisited: polymerases as determinants of mutational specificity. DNA Repair, 1, 125–135. [DOI] [PubMed] [Google Scholar]
- 33.Dzantiev L. and Romano,L.J. (2000) Differential effects of N-acetyl-2-aminofluorene and N-2-aminofluorene adducts on the conformational change in the structure of DNA polymerase I (Klenow fragment). Biochemistry, 39, 5139–5145. [DOI] [PubMed] [Google Scholar]
- 34.Wang J., Sattar,A.K., Wang,C.C., Karam,J.D., Konigsberg,W.H. and Steitz,T.A. (1997) Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell, 89, 1087–1099. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez A.C., Park,H.W., Mao,C. and Beese,L.S. (2000) Crystal structure of a pol alpha family DNA polymerase from the hyperthermophilic archaeon Thermococcus sp. 9 degrees N-7. J. Mol. Biol., 299, 447–462. [DOI] [PubMed] [Google Scholar]
- 36.Goodman M.F., Keener,S., Guidotti,S. and Branscomb,E.W. (1983) On the enzymatic basis for mutagenesis by manganese. J. Biol. Chem., 258, 3469–3475. [PubMed] [Google Scholar]
- 37.Hays H. and Berdis,A.J. (2002) Manganese substantially alters the dynamics of translesion DNA synthesis. Biochemistry, 41, 4771–4778. [DOI] [PubMed] [Google Scholar]
- 38.Pelletier H., Sawaya,M.R., Wolfle,W., Wilson,S.H. and Kraut,J. (1996) A structural basis for metal ion mutagenicity and nucleotide selectivity in human DNA polymerase beta. Biochemistry, 35, 12762–12777. [DOI] [PubMed] [Google Scholar]